
DNA Methylation of Genes Participating in Hepatic Metabolisms and Function in Fetal Calf Liver Is Altered by Maternal Undernutrition during Gestation

 ,
,
Abstract
1. Introduction
2. Results
2.1. Phenotypic Effect of Maternal Nutrient Restriction on Fetuses
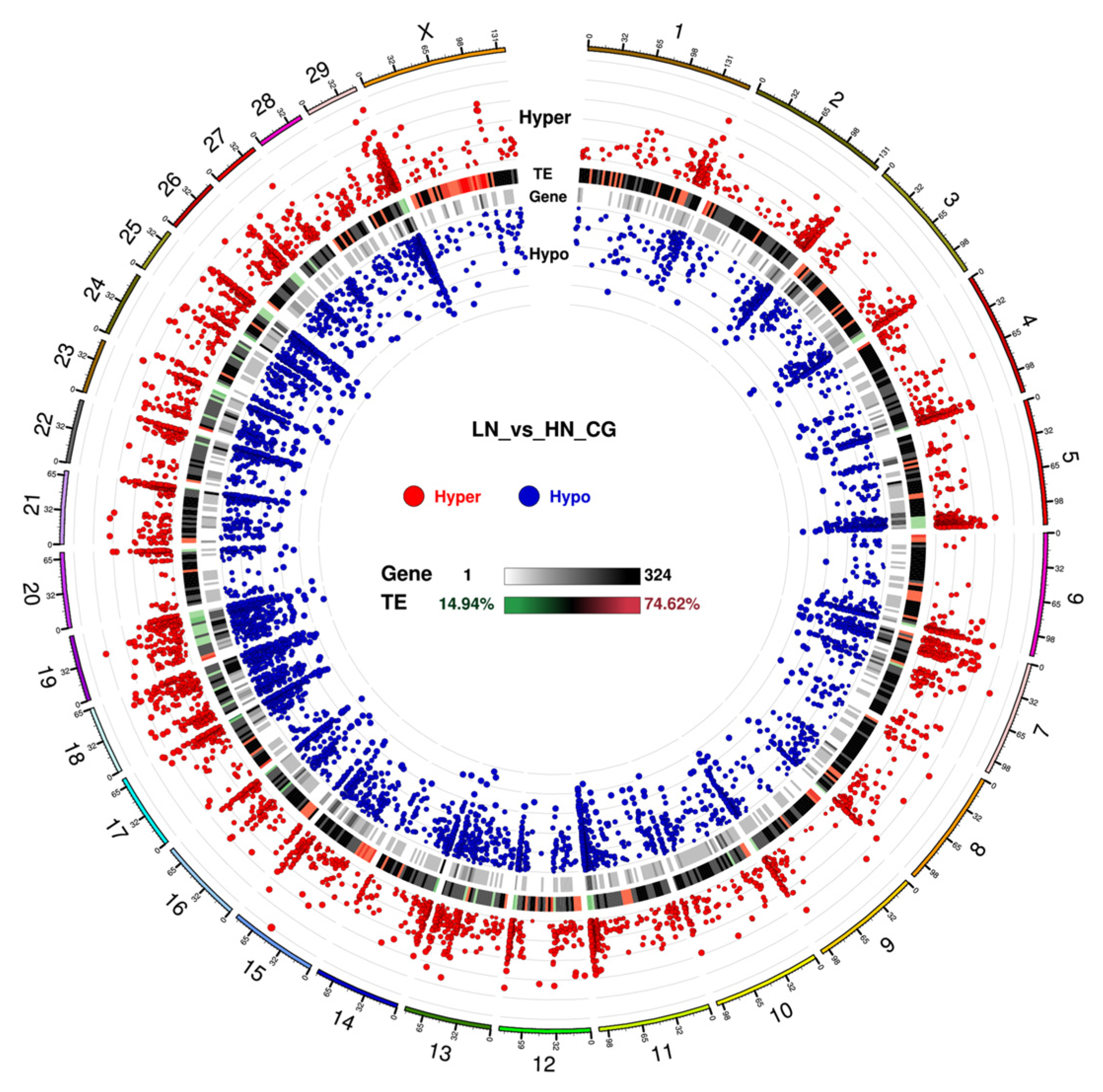
2.2. Distribution of DNA Methylation in LN and HN Fetal Liver Genome
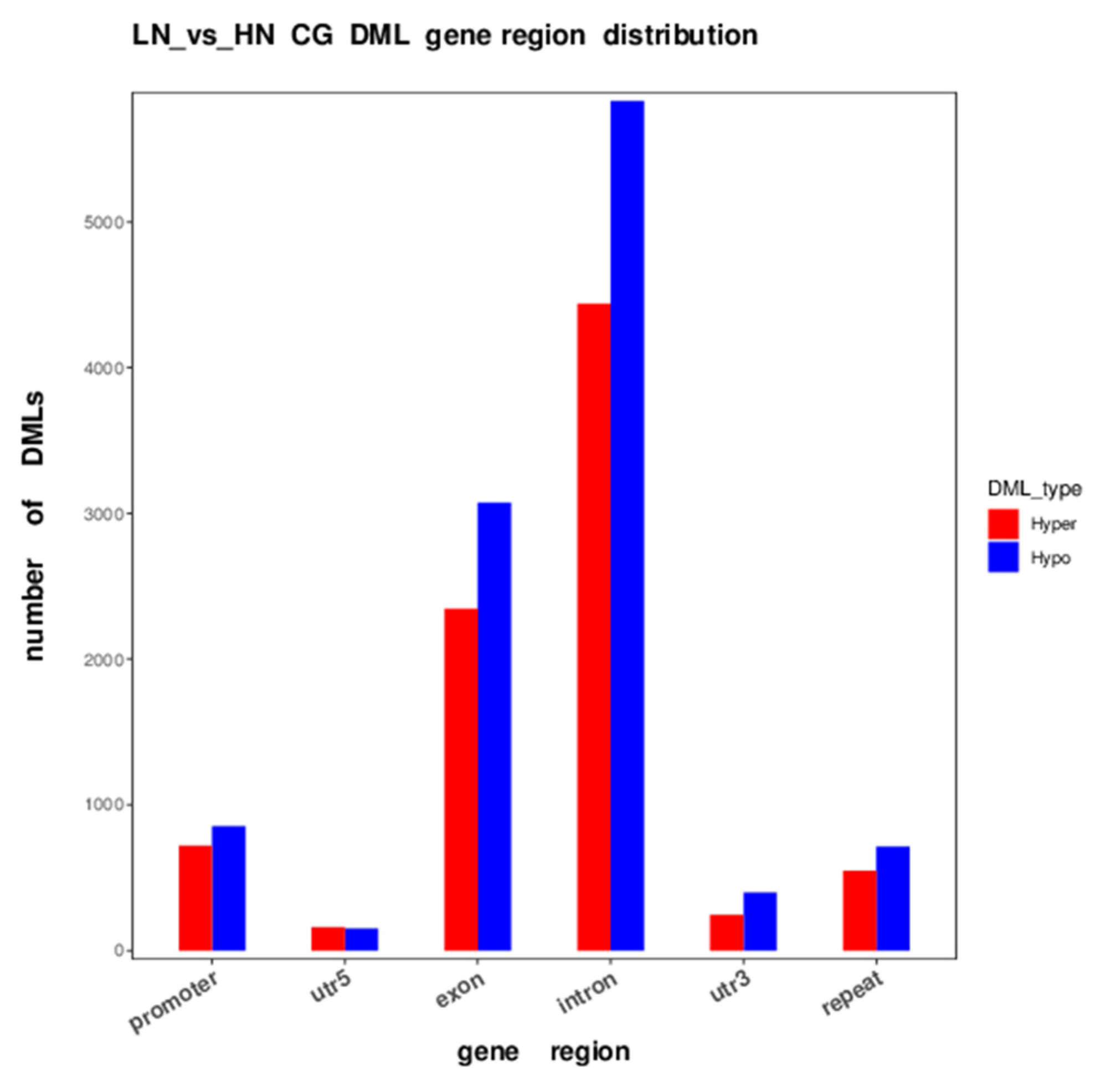
2.3. Differential DNA Methylation between LN and HN Fetal Liver
2.4. Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways Associated with Differentially Methylated Genes
2.5. Genes with Differentially Methylated Loci in Each Genomic Feature
2.6. Alteration in CG Methylation and Gene Expression Is Associated with Energy Metabolism, Steroid Synthesis, and IGF2 Signaling Pathway
3. Discussion
3.1. Impact of MUN on the Hepatic Genome and Molecular Networks in Fetal Calves
3.2. Hyper/Hypomethylated Genes Essential for Hepatic Metabolisms and Function
4. Materials and Methods
4.1. Animals and Dietary Treatment
4.2. Sample Collection
4.3. Genomic DNA Preparation
4.4. Reduced Representation Bisulfite Sequencing (RRBS)
4.5. Analyses of DMR and DML
4.6. Pathway Enrichment Analyses for Hyper- or Hypo-Methylated Genes
4.7. Microarray Analysis
4.8. Gene Expression Analysis in Quantitative PCR
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Govoni, K.E.; Reed, S.A.; Zinn, S.A. Cell biology symposium: Metabolic responses to stress: From animal to cell: Poor maternal nutrition during gestation: Effects on offspring whole-body and tissue-specific metabolism in livestock species. J. Anim. Sci. 2019, 97, 3142–3152. [Google Scholar] [CrossRef] [PubMed]
- Devaskar, S.U.; Chu, A. Intrauterine Growth Restriction: Hungry for an Answer. Physiology 2016, 31, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Muroya, S.; Zhang, Y.; Kinoshita, A.; Otomaru, K.; Oshima, K.; Gotoh, Y.; Oshima, I.; Sano, M.; Roh, S.; Oe, M.; et al. Maternal Undernutrition during Pregnancy Alters Amino Acid Metabolism and Gene Expression Associated with Energy Metabolism and Angiogenesis in Fetal Calf Muscle. Metabolites 2021, 11, 582. [Google Scholar] [CrossRef] [PubMed]
- Osgerby, J.C.; Wathes, D.C.; Howard, D.; Gadd, T.S. The effect of maternal undernutrition on ovine fetal growth. J. Endocrinol. 2002, 173, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Vonnahme, K.A.; Hess, B.W.; Hansen, T.R.; McCormick, R.J.; Rule, D.C.; Moss, G.E.; Murdoch, W.J.; Nijland, M.J.; Skinner, D.C.; Nathanielsz, P.W.; et al. Maternal undernutrition from early- to mid-gestation leads to growth retardation, cardiac ventricular hypertrophy, and increased liver weight in the fetal sheep. Biol. Reprod. 2003, 69, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, L.J.; Foster, T.; Rhodes, P.; Rhind, S.M.; Gardner, D.S. Protein-energy malnutrition during early gestation in sheep blunts fetal renal vascular and nephron development and compromises adult renal function. J. Physiol. 2012, 590, 377–393. [Google Scholar] [CrossRef]
- George, L.A.; Zhang, L.; Tuersunjiang, N.; Ma, Y.; Long, N.M.; Uthlaut, A.B.; Smith, D.T.; Nathanielsz, P.W.; Ford, S.P.; Lie, S.; et al. Early maternal undernutrition programs increased feed intake, altered glucose metabolism and insulin secretion, and liver function in aged female offspring. Am. J. Physiol. Integr. Comp. Physiol. 2012, 302, R795–R804. [Google Scholar] [CrossRef]
- Zhang, Y.; Otomaru, K.; Oshima, K.; Goto, Y.; Oshima, I.; Muroya, S.; Sano, M.; Saneshima, R.; Nagao, Y.; Kinoshita, A.; et al. Effects of low and high levels of maternal nutrition consumed for the entirety of gestation on the development of muscle, adipose tissue, bone, and the organs of Wagyu cattle fetuses. Anim. Sci. J. 2021, 92, e13600. [Google Scholar] [CrossRef]
- Morrison, J.L. Sheep models of intrauterine growth restriction: Fetal adaptations and consequences. Clin. Exp. Pharmacol. Physiol. 2008, 35, 730–743. [Google Scholar] [CrossRef]
- Godfrey, K.M.; Barker, D.J. Fetal programming and adult health. Public Health Nutr. 2001, 4, 611–624. [Google Scholar] [CrossRef]
- Lee, H.S. Impact of Maternal Diet on the Epigenome during In Utero Life and the Developmental Programming of Diseases in Childhood and Adulthood. Nutrients 2015, 7, 9492–9507. [Google Scholar] [CrossRef]
- Goyal, D.; Limesand, S.W.; Goyal, R. Epigenetic responses and the developmental origins of health and disease. J. Endocrinol. 2019, 242, T105–T119. [Google Scholar] [CrossRef]
- Sandoval, C.; Askelson, K.; Lambo, C.A.; Dunlap, K.A.; Satterfield, M.C. Effect of maternal nutrient restriction on expression of glucose transporters (SLC2A4 and SLC2A1) and insulin signaling in skeletal muscle of SGA and Non-SGA sheep fetuses. Domest. Anim. Endocrinol. 2021, 74, 106556. [Google Scholar] [CrossRef]
- Desai, M.; Crowther, N.; Lucas, A.; Hales, C.N. Organ-selective growth in the offspring of protein-restricted mothers. Br. J. Nutr. 1996, 76, 591–603. [Google Scholar] [CrossRef]
- Chandel, N.S. Navigating Metabolism; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2015. [Google Scholar]
- Preidis, G.A.; Keaton, M.A.; Campeau, P.M.; Bessard, B.C.; Conner, M.E.; Hotez, P.J. The Undernourished Neonatal Mouse Metabolome Reveals Evidence of Liver and Biliary Dysfunction, Inflammation, and Oxidative Stress. J. Nutr. 2014, 144, 273–281. [Google Scholar] [CrossRef]
- Gruppuso, P.A.; Boylan, J.M.; Anand, P.; Bienieki, T.C. Effects of Maternal Starvation on Hepatocyte Proliferation in the Late Gestation Fetal Rat. Pediatr. Res. 2005, 57, 185–191. [Google Scholar] [CrossRef]
- Kind, K.L.; Roberts, C.T.; Sohlstrom, A.I.; Katsman, A.; Clifton, P.M.; Robinson, J.S.; Owens, J.A. Chronic maternal feed restriction impairs growth but increases adiposity of the fetal guinea pig. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R119–R126. [Google Scholar] [CrossRef]
- Hyatt, M.A.; Gopalakrishnan, G.S.; Bispham, J.; Gentili, S.; McMillen, I.C.; Rhind, S.M.; Rae, M.T.; Kyle, C.E.; Brooks, A.N.; Jones, C.; et al. Maternal nutrient restriction in early pregnancy programs hepatic mRNA expression of growth-related genes and liver size in adult male sheep. J. Endocrinol. 2007, 192, 87–97. [Google Scholar] [CrossRef]
- Gao, F.; Liu, Y.; Li, L.; Li, M.; Zhang, C.; Ao, C.; Hou, X. Effects of maternal undernutrition during late pregnancy on the development and function of ovine fetal liver. Anim. Reprod. Sci. 2014, 147, 99–105. [Google Scholar] [CrossRef]
- Muroya, S.; Zhang, Y.; Otomaru, K.; Oshima, K.; Oshima, I.; Sano, M.; Roh, S.; Ojima, K.; Gotoh, T. Maternal Nutrient Re-striction Disrupts Gene Expression and Metabolites Associated with Urea Cycle, Steroid Synthesis, Glucose Homeostasis, and Glucuronidation in Fetal Calf Liver. Metabolites 2022, 12, 203. [Google Scholar] [CrossRef]
- Wesolowski, S.R.; El Kasmi, K.C.; Jonscher, K.R.; Friedman, J.E. Developmental origins of NAFLD: A womb with a clue. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.A.; Phillips, E.S.; Jackson, A.A.; Hanson, M.A.; Burdge, G.C. Dietary Protein Restriction of Pregnant Rats Induces and Folic Acid Supplementation Prevents Epigenetic Modification of Hepatic Gene Expression in the Offspring. J. Nutr. 2005, 135, 1382–1386. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.A.; Phillips, E.S.; Torrens, C.; Hanson, M.A.; Jackson, A.A.; Burdge, G.C. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPAR alpha promoter of the off-spring. Br. J. Nutr. 2008, 100, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Slater-Jefferies, J.; Torrens, C.; Phillips, E.S.; Hanson, M.A.; Lillycrop, K.A. Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. Br. J. Nutr. 2007, 97, 435–439. [Google Scholar] [CrossRef]
- Gong, L.; Pan, Y.-X.; Chen, H. Gestational low protein diet in the rat mediates Igf2 gene expression in male offspring via altered hepatic DNA methylation. Epigenetics 2010, 5, 619–626. [Google Scholar] [CrossRef]
- Altmann, S.; Murani, E.; Schwerin, M.; Metges, C.C.; Wimmers, K.; Ponsuksili, S. Dietary protein restriction and excess of pregnant German Landrace sows induce changes in hepatic gene expression and promoter methylation of key metabolic genes in the offspring. J. Nutr. Biochem. 2013, 24, 484–495. [Google Scholar] [CrossRef]
- Van Straten, E.M.; Bloks, V.W.; Huijkman, N.C.; Baller, J.F.; van Meer, H.; Lutjohann, D.; Kuipers, F.; Plosch, T. The liver X-receptor gene promoter is hypermethylated in a mouse model of prenatal protein restriction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R275–R282. [Google Scholar] [CrossRef]
- Zhu, M.J.; Ford, S.P.; Means, W.J.; Hess, B.W.; Nathanielsz, P.W.; Du, M. Maternal nutrient restriction affects properties of skeletal muscle in offspring. J. Physiol. 2006, 575 Pt 1, 241–250. [Google Scholar] [CrossRef]
- National Agriculture and Food Research Organization. Japanese Feeding Standard for Beef Cattle 2008 edn; Japan Livestock Industry Association: Tokyo, Japan, 2009. (In Japanese) [Google Scholar]
- Devailly, G.; Joshi, A. Comprehensive analysis of epigenetic signatures of human transcription control. Mol. Omics 2021, 17, 692–705. [Google Scholar] [CrossRef]
- Brock, G.J.; Charlton, J.; Bird, A. Densely methylated sequences that are preferentially localized at telomere-proximal regions of human chromosomes. Gene 1999, 240, 269–277. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, J.; Tian, G.; Li, N.; Li, Q.; Ye, M.; Zheng, H.; Yu, J.; Wu, H.; Sun, J.; et al. The DNA Methylome of Human Peripheral Blood Mononuclear Cells. PLoS Biol. 2010, 8, e1000533. [Google Scholar] [CrossRef]
- Anastasiadi, D.; Esteve-Codina, A.; Piferrer, F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenet. Chromatin 2018, 11, 37. [Google Scholar] [CrossRef]
- Anand, P.; Boylan, J.M.; Ou, Y.; Gruppuso, P.A. Insulin signaling during perinatal liver development in the rat. Am. J. Physiol. Metab. 2002, 283, E844–E852. [Google Scholar] [CrossRef][Green Version]
- Sunilkumar, S.; Kimball, S.R.; Dennis, M.D. Glucagon transiently stimulates mTORC1 by activation of an EPAC/Rap1 signaling axis. Cell Signal. 2021, 84, 110010. [Google Scholar] [CrossRef]
- Assinder, S.J. The Importance of Experimental Investigation of the Peripheral Oxytocin System. Methods Mol. Biol. 2022, 2384, 1–27. [Google Scholar] [CrossRef]
- Morgan, S.A.; Hassan-Smith, Z.K.; Lavery, G.G. Mechanisms in endocrinology: Tissue-specific activation of cortisol in Cushing’s syndrome. Eur. J. Endocrinol. 2016, 175, R81–R87. [Google Scholar] [CrossRef]
- Christ-Crain, M.; Kola, B.; Lolli, F.; Fekete, C.; Seboek, D.; Wittmann, G.; Feltrin, D.; Igreja, S.C.; Ajodha, S.; Harvey-White, J.; et al. AMP-activated protein kinase mediates glucocorticoid-induced metabolic changes: A novel mechanism in Cushing’s syndrome. FASEB J. 2008, 22, 1672–1683. [Google Scholar] [CrossRef]
- Byrnes, K.; Blessinger, S.; Bailey, N.T.; Scaife, R.; Liu, G.; Khambu, B. Therapeutic regulation of autophagy in hepatic metabolism. Acta Pharm. Sin. B 2022, 12, 33–49. [Google Scholar] [CrossRef]
- Lv, S.-Y.; Cui, B.; Chen, W.-D.; Wang, Y.-D. Apelin/APJ system: A key therapeutic target for liver disease. Oncotarget 2017, 8, 112145–112151. [Google Scholar] [CrossRef][Green Version]
- Dong, X.C.; Copps, K.D.; Guo, S.; Li, Y.; Kollipara, R.; DePinho, R.A.; White, M.F. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008, 8, 65–76. [Google Scholar] [CrossRef]
- Zvibel, I.; Atias, D.; Phillips, A.; Halpern, Z.; Oren, R. Thyroid hormones induce activation of rat hepatic stellate cells through increased expression of p75 neurotrophin receptor and direct activation of Rho. Lab. Investig. 2010, 90, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Moore, G.E.; Ishida, M.; Demetriou, C.; Al-Olabi, L.; Leon, L.J.; Thomas, A.C.; Abu-Amero, S.; Frost, J.M.; Stafford, J.L.; Chaoqun, Y.; et al. The role and interaction of imprinted genes in human fetal growth. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2015, 370, 20140074. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, J.; Kolte, A.M.; Hansen, T.; Nielsen, F.C. IGF2 mRNA-binding protein 2: Biological function and putative role in type 2 diabetes. J. Mol. Endocrinol. 2009, 43, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Dai, N.; Rapley, J.; Angel, M.; Yanik, M.F.; Blower, M.D.; Avruch, J. mTOR phosphorylates IMP2 to promote IGF2 mRNA translation by internal ribosomal entry. Genes Dev. 2011, 25, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Willison, K. Opposite imprinting of the mouse Igf2 and Igf2r genes. Trends Genet. TIG 1991, 7, 107–109. [Google Scholar] [CrossRef]
- Charalambous, M.; Smith, F.M.; Bennett, W.R.; Crew, T.E.; Mackenzie, F.; Ward, A. Disruption of the imprinted Grb10 gene leads to disproportionate overgrowth by an Igf2-independent mechanism. Proc. Natl. Acad. Sci. USA 2003, 100, 8292–8297. [Google Scholar] [CrossRef]
- Kwok, A.; Zvetkova, I.; Virtue, S.; Luijten, I.; Huang-Doran, I.; Tomlinson, P.; Bulger, D.A.; West, J.; Murfitt, S.; Griffin, J.; et al. Truncation of Pik3r1 causes severe insulin resistance uncoupled from obesity and dyslipidaemia by increased energy expenditure. Mol. Metab. 2020, 40, 101020. [Google Scholar] [CrossRef]
- Wang, Z.; Li, V.; Chan, G.C.K.; Phan, T.; Nudelman, A.S.; Xia, Z.; Storm, D.R. Adult Type 3 Adenylyl Cyclase-Deficient Mice Are Obese. PLoS ONE 2009, 4, e6979. [Google Scholar] [CrossRef]
- Liang, C.; Bu, S.; Fan, X. Suppressive effect of microRNA-29b on hepatic stellate cell activation and its crosstalk with TGF-beta1/Smad3. Cell Biochem. Funct. 2016, 34, 326–333. [Google Scholar] [CrossRef]
- Chen, M.; Gavrilova, O.; Liu, J.; Xie, T.; Deng, C.; Nguyen, A.T.; Nackers, L.M.; Lorenzo, J.; Shen, L.; Weinstein, L.S. Alternative Gnas gene products have opposite effects on glucose and lipid metabolism. Proc. Natl. Acad. Sci. USA 2005, 102, 7386–7391. [Google Scholar] [CrossRef]
- Wang, X.J.; Chamberlain, M.; Vassieva, O.; Henderson, C.J.; Wolf, C.R. Relationship between hepatic phenotype and changes in gene expression in cytochrome P450 reductase (POR) null mice. Biochem. J. 2005, 388 Pt 3, 857–867. [Google Scholar] [CrossRef]
- Mutch, D.M.; Klocke, B.; Morrison, P.; Murray, C.A.; Henderson, C.J.; Seifert, M.; Williamson, G. The disruption of hepatic cytochrome p450 reductase alters mouse lipid metabolism. J. Proteome Res. 2007, 6, 3976–3984. [Google Scholar] [CrossRef]
- Porter, T.D. New insights into the role of cytochrome P450 reductase (POR) in microsomal redox biology. Acta Pharm. Sin. B 2012, 2, 102–106. [Google Scholar] [CrossRef][Green Version]
- Riddick, D.S.; Ding, X.; Wolf, C.R.; Porter, T.D.; Pandey, A.V.; Zhang, Q.Y.; Gu, J.; Finn, R.D.; Ronseaux, S.; McLaughlin, L.A.; et al. NADPH-cytochrome P450 oxidoreductase: Roles in physiology, pharmacology, and toxicology. Drug Metab. Dispos. Biol. Fate Chem. 2013, 41, 12–23. [Google Scholar] [CrossRef]
- Sabbagha, N.G.A.A.-A.; Kao, H.-J.; Yang, C.-F.; Huang, C.-C.; Lin, W.-D.; Tsai, F.-J.; Chen, T.-H.; Tarn, W.-Y.; Wu, J.-Y.; Chen, Y.-T. Alternative Splicing in Acad8 Resulting a Mitochondrial Defect and Progressive Hepatic Steatosis in Mice. Pediatr. Res. 2011, 70, 31–36. [Google Scholar] [CrossRef]
- Sekiya, M.; Kainoh, K.; Sugasawa, T.; Yoshino, R.; Hirokawa, T.; Tokiwa, H.; Nakano, S.; Nagatoishi, S.; Tsumoto, K.; Takeuchi, Y.; et al. The transcriptional corepressor CtBP2 serves as a metabolite sensor orchestrating hepatic glucose and lipid homeostasis. Nat. Commun. 2021, 12, 6315. [Google Scholar] [CrossRef]
- Moore, P.S.; Koontz, J.W. Regulation of Tyrosine Aminotransferase by Insulin and Cyclic AMP: Similar Effects on Activity but Opposite Effects on Transcription. Mol. Endocrinol. 1989, 3, 1724–1732. [Google Scholar] [CrossRef]
- Valera, A.; Rodriguez-Gil, J.E.; Yun, J.S.; McGrane, M.M.; Hanson, R.W.; Bosch, F. Glucose metabolism in transgenic mice containing a chimeric P-enolpyruvate carboxykinase/bovine growth hormone gene. FASEB J. 1993, 7, 791–800. [Google Scholar] [CrossRef]
- Jitrapakdee, S.; Wallace, J.C. Structure, function and regulation of pyruvate carboxylase. Biochem. J. 1999, 340 Pt 1, 1–16. [Google Scholar]
- Jitrapakdee, S.; Vidal-Puig, A.; Wallace, J.C. Anaplerotic roles of pyruvate carboxylase in mammalian tissues. Cell. Mol. Life Sci. 2006, 63, 843–854. [Google Scholar] [CrossRef]
- Choubey, V.; Zeb, A.; Kaasik, A. Molecular Mechanisms and Regulation of Mammalian Mitophagy. Cells 2021, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Liu, G.; Zhu, M.; Wang, S.; Jiang, Q.; Loor, J.J.; Yu, H.; Hao, X.; Chen, M.; Gao, W.; et al. Low abundance of mitophagy markers is associated with reactive oxygen species overproduction in cows with fatty liver and causes reactive oxygen species overproduction and lipid accumulation in calf hepatocytes. J. Dairy Sci. 2022, 105, 7829–7841. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson Kvissberg, M.E.; Hu, G.; Chi, L.; Bourdon, C.; Ling, C.; ChenMi, Y.; Germain, K.; van Peppel, I.P.; Weise, L.; Zhang, L.; et al. Inhibition of mTOR improves malnutrition induced hepatic metabolic dysfunction. Sci. Rep. 2022, 12, 19948. [Google Scholar] [CrossRef] [PubMed]
- King, A.L.; Polhemus, D.J.; Bhushan, S.; Otsuka, H.; Kondo, K.; Nicholson, C.K.; Bradley, J.M.; Islam, K.N.; Calvert, J.W.; Tao, Y.-X.; et al. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc. Natl. Acad. Sci. USA 2014, 111, 3182–3187. [Google Scholar] [CrossRef] [PubMed]
- Szijártó, I.A.; Markó, L.; Filipovic, M.R.; Miljkovic, J.L.; Tabeling, C.; Tsvetkov, D.; Wang, N.; Rabelo, L.A.; Witzenrath, M.; Diedrich, A.; et al. Cystathionine γ-Lyase-Produced Hydrogen Sul-fide Controls Endothelial NO Bioavailability and Blood Pressure. Hypertension 2018, 71, 1210–1217. [Google Scholar] [CrossRef]
- Ishii, I.; Akahoshi, N.; Yamada, H.; Nakano, S.; Izumi, T.; Suematsu, M. Cystathionine gamma-Lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J. Biol. Chem. 2010, 285, 26358–26368. [Google Scholar] [CrossRef]
- Wallerath, T.; Gödecke, A.; Molojavyi, A.; Li, H.; Schrader, J.; Förstermann, U. Dexamethasone lacks effect on blood pressure in mice with a disrupted endothelial NO synthase gene. Nitric Oxide 2004, 10, 36–41. [Google Scholar] [CrossRef]
- Zhao, C.X.; Xu, X.; Cui, Y.; Wang, P.; Wei, X.; Yang, S.; Edin, M.L.; Zeldin, D.C.; Wang, D.W. Increased endothelial nitric-oxide synthase expression reduces hypertension and hyperinsulinemia in fructose-treated rats. J. Pharmacol. Exp. Ther. 2009, 328, 610–620. [Google Scholar] [CrossRef]
- Yoshida, D.; Akahoshi, T.; Kawanaka, H.; Yamaguchi, S.; Kinjo, N.; Taketomi, A.; Tomikawa, M.; Shirabe, K.; Maehara, Y.; Hashizume, M. Roles of vascular endothelial growth factor and endothelial nitric oxide synthase during revascularization and regeneration after partial hepatectomy in a rat model. Surg. Today 2011, 41, 1622–1629. [Google Scholar] [CrossRef]
- Gu, J.; Weng, Y.; Zhang, Q.Y.; Cui, H.; Behr, M.; Wu, L.; Yang, W.; Zhang, L.; Ding, X. Liver-specific deletion of the NADPH-cytochrome P450 reductase gene: Impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J. Biol. Chem. 2003, 278, 25895–25901. [Google Scholar] [CrossRef]
- Marshall, S.M.; Gromovsky, A.D.; Kelley, K.L.; Davis, M.A.; Wilson, M.D.; Lee, R.G.; Crooke, R.M.; Graham, M.J.; Rudel, L.L.; Brown, J.M.; et al. Acute Sterol O-Acyltransferase 2 (SOAT2) Knockdown Rapidly Mobilizes Hepatic Cholesterol for Fecal Excretion. PLoS ONE 2014, 9, e98953. [Google Scholar] [CrossRef]
- Pramfalk, C.; Ahmed, O.; Pedrelli, M.; Minniti, M.E.; Luquet, S.; Denis, R.G.; Olin, M.; Härdfeldt, J.; Vedin, L.L.; Steffensen, K.R.; et al. Soat2 ties cholesterol metabolism to β-oxidation and glucose tolerance in male mice. J. Intern. Med. 2022, 292, 296–307. [Google Scholar] [CrossRef]
- Briolay, A.; Bessueille, L.; Magne, D. TNAP: A New Multitask Enzyme in Energy Metabolism. Int. J. Mol. Sci. 2021, 22, 10470. [Google Scholar] [CrossRef]
- Giancotti, A.; Monti, M.; Nevi, L.; Safarikia, S.; D’ambrosio, V.; Brunelli, R.; Pajno, C.; Corno, S.; Di Donato, V.; Musella, A.; et al. Functions and the Emerging Role of the Foetal Liver into Regenerative Medicine. Cells 2019, 8, 914. [Google Scholar] [CrossRef]
- Cabezas-Wallscheid, N.; Buettner, F.; Sommerkamp, P.; Klimmeck, D.; Ladel, L.; Thalheimer, F.B.; Pastor-Flores, D.; Roma, L.P.; Renders, S.; Zeisberger, P.; et al. Vitamin A-Retinoic Acid Signaling Regulates Hematopoietic Stem Cell Dormancy. Cell 2017, 169, 807–823.e19. [Google Scholar] [CrossRef]
- Faria, T.N.; Mendelsohn, C.; Chambon, P.; Gudas, L.J. The targeted disruption of both alleles of RARbeta(2) in F9 cells results in the loss of retinoic acid-associated growth arrest. J. Biol. Chem. 1999, 274, 26783–26788. [Google Scholar] [CrossRef]
- Idres, N.; Marill, J.; Flexor, M.A.; Chabot, G.G. Activation of Retinoic Acid Receptor-dependent Transcription by All-trans-retinoic Acid Metabolites and Isomers. J. Biol. Chem. 2002, 277, 31491–31498. [Google Scholar] [CrossRef]
- Schönberger, K.; Obier, N.; Romero-Mulero, M.C.; Cauchy, P.; Mess, J.; Pavlovich, P.V.; Zhang, Y.W.; Mitterer, M.; Rettkowski, J.; Lalioti, M.E.; et al. Multilayer omics analysis reveals a non-classical retinoic acid signaling axis that regulates hematopoietic stem cell identity. Cell Stem Cell 2022, 29, 131–148.e10. [Google Scholar] [CrossRef]
- Hall, R.K.; Sladek, F.M.; Granner, D.K. The orphan receptors COUP-TF and HNF-4 serve as accessory factors required for induction of phosphoenolpyruvate carboxykinase gene transcription by glucocorticoids. Proc. Natl. Acad. Sci. USA 1995, 92, 412–416. [Google Scholar] [CrossRef]
- Rodríguez, J.C.; Ortiz, J.A.; Hegardt, F.G.; Haro, D. Chicken ovalbumin upstream-promoter transcription factor (COUP-TF) could act as a transcriptional activator or repressor of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene. Biochem. J. 1997, 326 Pt 2, 587–592. [Google Scholar] [CrossRef]
- Park, J.-I.; Tsai, S.Y.; Tsai, M.-J. Molecular mechanism of chicken ovalbumin upstream promoter-transcription factor (COUP-TF) actions. Keio J. Med. 2003, 52, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Habibi, E.; Brinkman, A.B.; Arand, J.; Kroeze, L.I.; Kerstens, H.H.; Matarese, F.; Lepikhov, K.; Gut, M.; Brun-Heath, I.; Hubner, N.C.; et al. Whole-Genome Bisulfite Sequencing of Two Distinct Interconvertible DNA Methylomes of Mouse Embryonic Stem Cells. Cell Stem Cell 2013, 13, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Gifford, C.A.; Ziller, M.J.; Gu, H.; Trapnell, C.; Donaghey, J.; Tsankov, A.; Shalek, A.K.; Kelley, D.R.; Shishkin, A.A.; Issner, R.; et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 2013, 153, 1149–1163. [Google Scholar] [CrossRef]
- Feng, H.; Conneely, K.N.; Wu, H. A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res. 2014, 42, e69. [Google Scholar] [CrossRef]
- Wu, H.; Xu, T.; Feng, H.; Chen, L.; Ben Li, B.; Yao, B.; Qin, Z.; Jin, P.; Conneely, K.N. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res. 2015, 43, e141. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | HN (n = 3) | LN (n = 3) | p-Value | ||
|---|---|---|---|---|---|
| Mean | SEM 1 | Mean | SEM 1 | ||
| Age (d) | 261.33 | 2.03 | 257.67 | 0.88 | 0.173 |
| BW (kg) | 33.31 | 0.42 | 23.5 | 1.72 | 0.005 |
| Liver (g) | 659.37 | 4.49 | 485.15 | 20.98 | 0.001 |
| %BW | 1.98 | 0.01 | 2.08 | 0.15 | 0.529 |
| Genomic Region | Hyper | Hypo | ||
|---|---|---|---|---|
| Loci | % | Loci | % | |
| Promoter | 718 | 8.50 | 854 | 7.75 |
| UTR5 | 159 | 1.88 | 150 | 1.36 |
| Exon | 2344 | 27.73 | 3075 | 27.90 |
| Intron | 4440 | 52.53 | 5832 | 52.92 |
| UTR3 | 244 | 2.89 | 397 | 3.60 |
| Repeat | 547 | 6.47 | 712 | 6.46 |
| Total | 8452 | 100.00 | 11,020 | 100.00 |
| Type of Difference | KEGG Pathway | FDR |
|---|---|---|
| Hypermethylation | ||
| Apelin signaling pathway | 0.000156 | |
| mTOR signaling pathway | 0.000525 | |
| AGE-RAGE signaling pathway in diabetic complications | 0.000548 | |
| Inositol phosphate metabolism | 0.000651 | |
| Wnt signaling pathway | 0.000862 | |
| Amoebiasis | 0.000884 | |
| Hypomethylation | ||
| Adrenergic signaling in cardiomyocytes | 0.000008 | |
| Inflammatory mediator regulation of TRP channels | 0.000011 | |
| Parathyroid hormone synthesis, secretion, and action | 0.000012 | |
| VEGF signaling pathway | 0.000029 | |
| cGMP-PKG signaling pathway | 0.000043 | |
| Gastric acid secretion | 0.000043 | |
| Signaling pathways regulating pluripotency of stem cells | 0.000046 | |
| Platelet activation | 0.000066 | |
| Phospholipase D signaling pathway | 0.000142 | |
| Cortisol synthesis and secretion | 0.000167 | |
| GnRH signaling pathway | 0.000167 | |
| Type II diabetes mellitus | 0.000167 | |
| Relaxin signaling pathway | 0.000184 | |
| Sphingolipid signaling pathway | 0.000275 | |
| Insulin signaling pathway | 0.000306 | |
| Yersinia infection | 0.000370 | |
| Ras signaling pathway | 0.000370 | |
| Dopaminergic synapse | 0.000390 | |
| Human papillomavirus infection | 0.000390 | |
| Vascular smooth muscle contraction | 0.000417 | |
| Insulin resistance | 0.000444 | |
| Tight junction | 0.000552 | |
| PI3K-Akt signaling pathway | 0.000615 | |
| Neurotrophin signaling pathway | 0.000662 | |
| Human cytomegalovirus infection | 0.000767 | |
| Thyroid hormone signaling pathway | 0.000988 |
| Chr. No. 1 | Position | Methylation Difference | Gene Name | Genomic Region | FDR | Microarray | |
|---|---|---|---|---|---|---|---|
| LN/HN 2 | p-Value | ||||||
| 29 | 49,625,542 | 0.425 | LSP1 | promoter | 0.017 | 0.661 | 0.010 |
| 25 | 34,197,023 | 0.342 | POR | promoter | 0.059 | 0.873 | 0.056 |
| 22 | 59,839,258 | 0.335 | ABTB1 | promoter | 0.003 | 0.796 | 0.092 |
| 13 | 57,484,897 | −0.147 | GNAS | promoter | 0.002 | 1.410 | 0.003 |
| 10 | 42,629,559 | −0.366 | RPS29 | promoter | 0.001 | 1.312 | 0.060 |
| 19 | 55,784,207 | −0.394 | UNC13D | exon | 0.000 | 0.699 | 0.003 |
| 5 | 26,877,080 | −0.456 | SOAT2 | intron | 0.003 | 0.692 | 0.031 |
| 5 | 26,877,156 | −0.470 | SOAT2 | exon | 0.001 | 0.692 | 0.031 |
| 19 | 58,319,907 | −0.566 | SLC39A11 | intron | 0.001 | 0.798 | 0.090 |
| 29 | 49,403,320 | −0.571 | IGF2 | intron | 0.000 | 0.851 | 0.056 |
| 29 | 49,403,299 | −0.571 | IGF2 | promoter | 0.001 | 0.851 | 0.056 |
| 25 | 1,592,534 | −0.594 | NTHL1 | intron | 0.000 | 0.888 | 0.040 |
| Chr. No. 1 | Position | Methylation Difference | Gene Name | Genomic Region | FDR | PCR | |
|---|---|---|---|---|---|---|---|
| LN/HN 2 | p-Value | ||||||
| 4 | 5,187,337 | 0.358 | GRB10 | promoter | 0.025 | 0.341 | 0.074 |
| 18 | 39,490,208 | −0.197 | TAT | intron | 0.023 | 0.554 | 0.043 |
| 9 | 96,221,893 | −0.436 | IGF2R | intron | 0.000 | 0.555 | 0.058 |
| 11 | 74,335,256 | −0.205 | ADCY3 | promoter | 0.050 | 0.581 | 0.086 |
| 27 | 40,235,976 | −0.226 | RARB | intron | 0.065 | 0.592 | 0.076 |
| 29 | 44,889,960 | 0.191 | PC | intron | 0.078 | 0.623 | 0.070 |
| 2 | 131,986,576 | −0.382 | PINK1 | exon/utr3 | 0.000 | 0.717 | 0.049 |
| 15 | 83,530,698 | −0.588 | ACAD8 | intron | 0.000 | 0.797 | 0.100 |
| 3 | 74,775,896 | −0.423 | CTH | intron | 0.067 | 1.488 | 0.059 |
| 4 | 113,578,206 | 0.286 | NOS3 | intron | 0.006 | 1.751 | 0.079 |
| 7 | 93,056,076 | 0.205 | NR2F1 | intron | 0.000 | 2.035 | 0.097 |
| 20 | 11,399,276 | −0.489 | PIK3R1 | exon/utr3 | 0.020 | 2.043 | 0.027 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muroya, S.; Otomaru, K.; Oshima, K.; Oshima, I.; Ojima, K.; Gotoh, T. DNA Methylation of Genes Participating in Hepatic Metabolisms and Function in Fetal Calf Liver Is Altered by Maternal Undernutrition during Gestation. Int. J. Mol. Sci. 2023, 24, 10682. https://doi.org/10.3390/ijms241310682
Muroya S, Otomaru K, Oshima K, Oshima I, Ojima K, Gotoh T. DNA Methylation of Genes Participating in Hepatic Metabolisms and Function in Fetal Calf Liver Is Altered by Maternal Undernutrition during Gestation. International Journal of Molecular Sciences. 2023; 24(13):10682. https://doi.org/10.3390/ijms241310682
Chicago/Turabian StyleMuroya, Susumu, Konosuke Otomaru, Kazunaga Oshima, Ichiro Oshima, Koichi Ojima, and Takafumi Gotoh. 2023. "DNA Methylation of Genes Participating in Hepatic Metabolisms and Function in Fetal Calf Liver Is Altered by Maternal Undernutrition during Gestation" International Journal of Molecular Sciences 24, no. 13: 10682. https://doi.org/10.3390/ijms241310682
APA StyleMuroya, S., Otomaru, K., Oshima, K., Oshima, I., Ojima, K., & Gotoh, T. (2023). DNA Methylation of Genes Participating in Hepatic Metabolisms and Function in Fetal Calf Liver Is Altered by Maternal Undernutrition during Gestation. International Journal of Molecular Sciences, 24(13), 10682. https://doi.org/10.3390/ijms241310682