
A Theoretical Study of Organotin Binding in Aromatase


Abstract
1. Introduction
2. Results and Discussion
2.1. Molecular Docking and Clustering
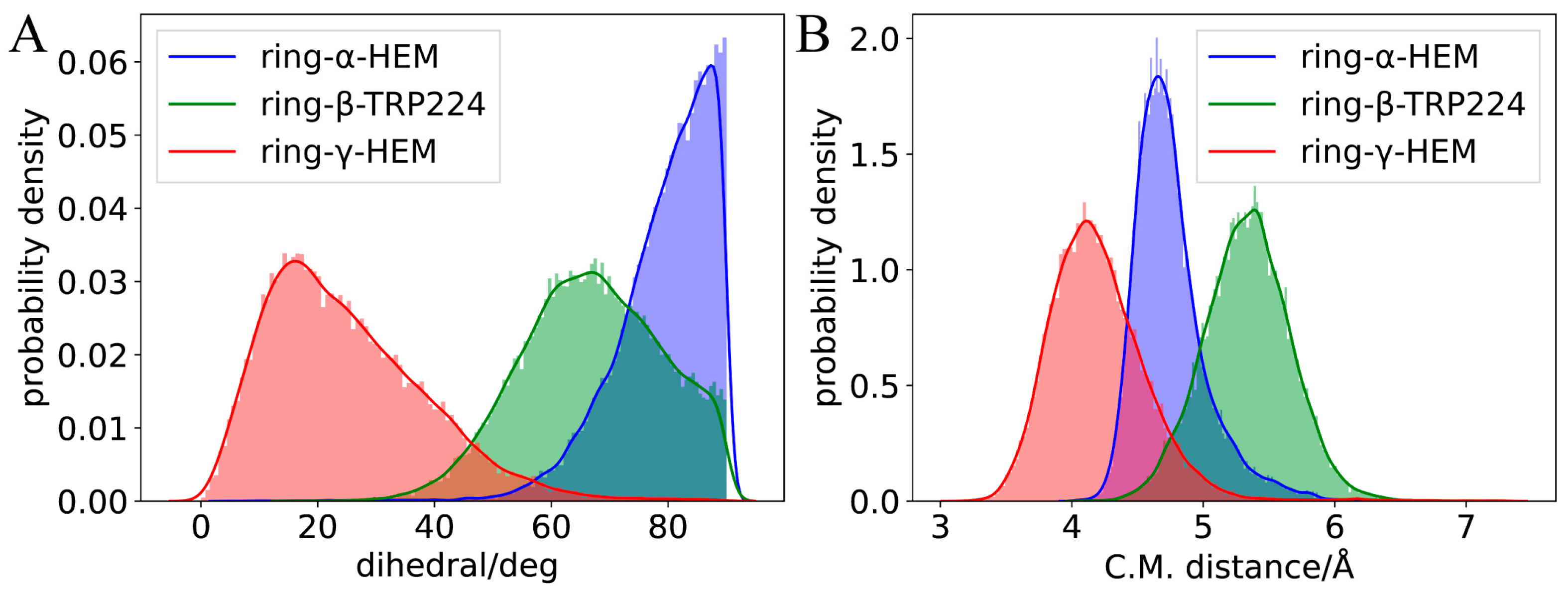
2.2. Molecular Dynamics Simulation of Binding Modes
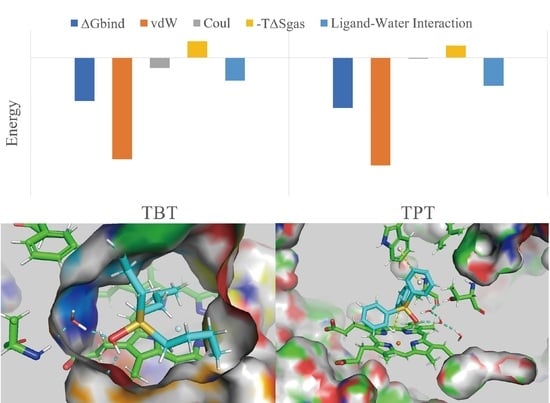
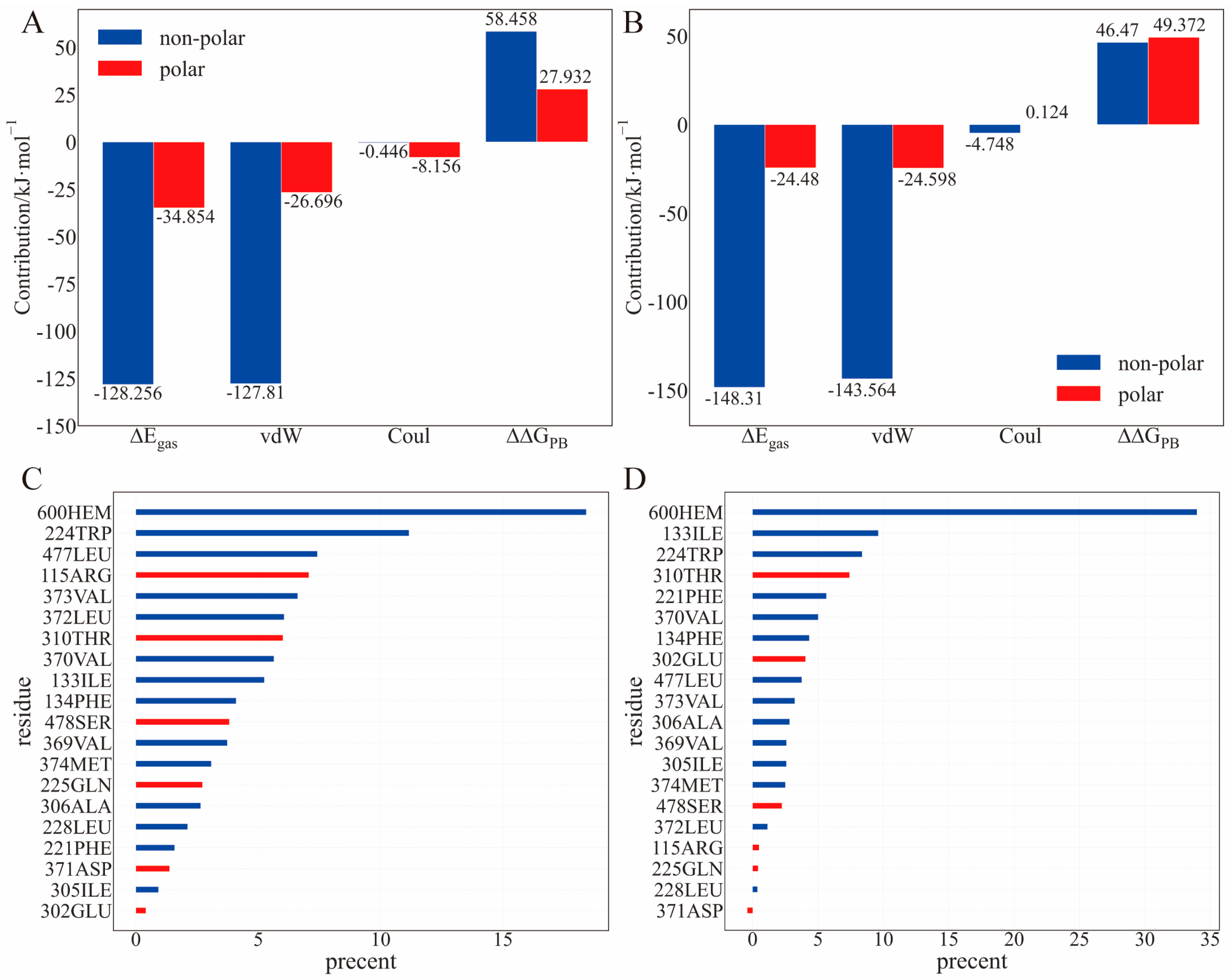
2.3. Binding Energy Analysis
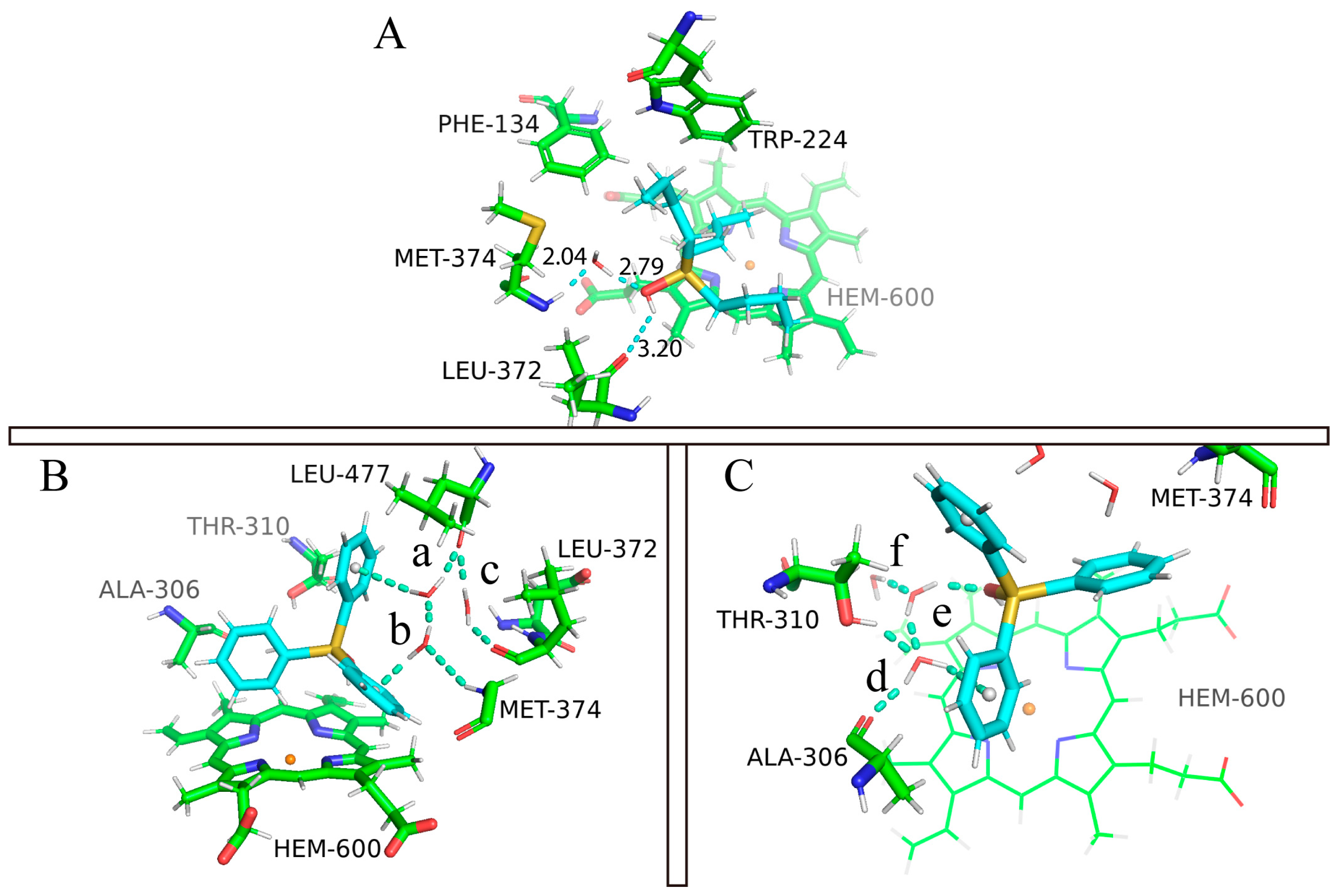
2.4. The Role of Interfacial Water Molecules in OT Binding
3. Methods and Materials
3.1. Model Preparation and Molecular Docking
3.2. System Construction and Topology Preparation
3.3. Molecular Dynamics Simulations
3.4. Binding Free Energy Calculations
3.5. Trajectory Analysis
3.6. Quantum Chemistry Calculation for The π-π Interaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kotake, Y. Molecular Mechanisms of Environmental Organotin Toxicity in Mammals. Biol. Pharm. Bull. 2012, 35, 1876–1880. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Li, P.; Li, Z.-H. Review on endocrine disrupting toxicity of triphenyltin from the perspective of species evolution: Aquatic, amphibious and mammalian. Chemosphere 2021, 269, 128711. [Google Scholar] [CrossRef] [PubMed]
- Golub, M.; Doherty, J. Triphenyltin as a potential human endocrine disruptor. J. Toxicol. Environ. Health B Crit. Rev. 2004, 7, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Delgado Filho, V.S.; Lopes, P.F.; Podratz, P.L.; Graceli, J.B. Triorganotin as a compound with potential reproductive toxicity in mammals. Braz. J. Med. Biol. Res. 2011, 44, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Lo, J.; Egbuta, C. Recent Progress in the Discovery of Next Generation Inhibitors of Aromatase from the Structure-Function Perspective. J. Med. Chem. 2016, 59, 5131–5148. [Google Scholar] [CrossRef]
- Sgrignani, J.; Magistrato, A. Influence of the membrane lipophilic environment on the structure and on the substrate access/egress routes of the human aromatase enzyme. A computational study. J. Chem. Inf. Model. 2012, 52, 1595–1606. [Google Scholar] [CrossRef]
- Park, J.; Czapla, L.; Amaro, R.E. Molecular Simulations of Aromatase Reveal New Insights Into the Mechanism of Ligand Binding. J. Chem. Inf. Model. 2013, 53, 2047–2056. [Google Scholar] [CrossRef]
- Heidrich, D.D.; Steckelbroeck, S.; Klingmuller, D. Inhibition of human cytochrome P450 aromatase activity by butyltins. Steroids 2001, 66, 763–769. [Google Scholar] [CrossRef]
- Cooke, G.M. Effect of organotins on human aromatase activity in vitro. Toxicol. Lett. 2002, 126, 121–130. [Google Scholar] [CrossRef]
- Lo, S.; Alléra, A.; Albers, P.; Heimbrecht, J.; Jantzen, E.; Klingmüller, D.; Steckelbroeck, S. Dithioerythritol (DTE) prevents inhibitory effects of triphenyltin (TPT) on the key enzymes of the human sex steroid hormone metabolism. J. Steroid Biochem. Mol. Biol. 2003, 84, 569–576. [Google Scholar] [CrossRef]
- Kimmel, E.C.; Fish, R.H.; Casida, J.E. Bioorganotin chemistry. Metabolism of organotin compounds in microsomal monooxygenase systems and in mammals. J. Agric. Food Chem. 1977, 25, 1–9. [Google Scholar] [CrossRef]
- Ohhira, S.; Enomoto, M.; Matsui, H. In vitro metabolism of tributyltin and triphenyltin by human cytochrome P-450 isoforms. Toxicology 2006, 228, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Ohhira, S.; Enomoto, M.; Matsui, H. Sex difference in the principal cytochrome P-450 for tributyltin metabolism in rats. Toxicol. Appl. Pharmacol. 2006, 210, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Fish, R.H. A bioorganometallic chemistry perspective: Organometallic chemistry at the interface with biology. J. Organomet. Chem. 2015, 782, 3–16. [Google Scholar] [CrossRef]
- Çevik, U.A.; Celik, I.; Mella, J.; Mellado, M.; Özkay, Y.; Kaplancıklı, Z.A. Design, Synthesis, and Molecular Modeling Studies of a Novel Benzimidazole as an Aromatase Inhibitor. ACS Omega 2022, 7, 16152–16163. [Google Scholar] [CrossRef]
- Tiwari, N.; Pandey, A.; Kumar, A.; Mishra, A. Computational models reveal the potential of polycyclic aromatic hydrocarbons to inhibit aromatase, an important enzyme of the steroid biosynthesis pathway. Comput. Toxicol. 2021, 19, 100176. [Google Scholar] [CrossRef]
- Zhang, C.; Schilirò, T.; Gea, M.; Bianchi, S.; Spinello, A.; Magistrato, A.; Gilardi, G.; Di Nardo, G. Molecular Basis for Endocrine Disruption by Pesticides Targeting Aromatase and Estrogen Receptor. Int. J. Environ. Res. Public Health 2020, 17, 5664. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, J.; Gu, H.; Wei, D.; Xu, Y.-C.; Fu, W.; Yu, Z. Conformational Preferences of π–π Stacking Between Ligand and Protein, Analysis Derived from Crystal Structure Data Geometric Preference of π–π Interaction. Interdiscip. Sci. Comput. Life Sci. 2015, 7, 211–220. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Hubbard, R.E.; Kamran Haider, M. Hydrogen Bonds in Proteins: Role and Strength. In Encyclopedia of Life Sciences (ELS); John Wiley & Sons, Ltd: Chichester, UK, 2010. [Google Scholar] [CrossRef]
- Salonen, L.M.; Ellermann, M.; Diederich, F. Aromatic rings in chemical and biological recognition: Energetics and structures. Angew. Chem. Int. Ed. Engl. 2011, 50, 4808–4842. [Google Scholar] [CrossRef]
- Gao, W.; Jiao, J.; Feng, H.; Xuan, X.; Chen, L. Natures of benzene-water and pyrrole-water interactions in the forms of σ and π types: Theoretical studies from clusters to liquid mixture. J. Mol. Model. 2013, 19, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, G.; Breitner, M.; Bandino, A.; Ghosh, D.; Jennings, G.K.; Hackett, J.C.; Gilardi, G. Evidence for an Elevated Aspartate pKa in the Active Site of Human Aromatase. J. Biol. Chem. 2015, 290, 1186–1196. [Google Scholar] [CrossRef] [PubMed]
- Spinello, A.; Pavlin, M.; Casalino, L.; Magistrato, A. A Dehydrogenase Dual Hydrogen Abstraction Mechanism Promotes Estrogen Biosynthesis: Can We Expand the Functional Annotation of the Aromatase Enzyme? Chem. Eur. 2018, 24, 10840–10849. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Griswold, J.; Erman, M.; Pangborn, W. Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature 2009, 457, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating p Ka s and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Myers, J.; Grothaus, G.; Narayanan, S.; Onufriev, A. A simple clustering algorithm can be accurate enough for use in calculations of pKs in macromolecules. Proteins 2006, 63, 928–938. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Jorgensen, W.; Chandrasekhar, J.; Madura, J.; Impey, R.; Klein, M. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Spinello, A.; Ritacco, I.; Magistrato, A. The Catalytic Mechanism of Steroidogenic Cytochromes P450 from All-Atom Simulations: Entwinement with Membrane Environment, Redox Partners, and Post-Transcriptional Regulation. Catalysts 2019, 9, 81. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Shahrokh, K.; Orendt, A.; Yost, G.S.; Cheatham, T.E. Quantum mechanically derived AMBER-compatible heme parameters for various states of the cytochrome P450 catalytic cycle. J. Comput. Chem. 2012, 33, 119–133. [Google Scholar] [CrossRef]
- Li, P.; Roberts, B.P.; Chakravorty, D.K.; Merz, K.M. Rational Design of Particle Mesh Ewald Compatible Lennard-Jones Parameters for +2 Metal Cations in Explicit Solvent. J. Chem. Theory Comput. 2013, 9, 2733–2748. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Zheng, S.; Tang, Q.; He, J.; Du, S.; Xu, S.; Wang, C.; Xu, Y.; Lin, F. VFFDT: A New Software for Preparing AMBER Force Field Parameters for Metal-Containing Molecular Systems. J. Chem. Inf. Model. 2016, 56, 811–818. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Gunsteren, W.F.v.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Duan, L.; Liu, X.; Zhang, J.Z.H. Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein–Ligand Binding Free Energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Li, J. Gmxtools. Zenodo. Available online: https://doi.org/10.5281/zenodo.6408973 (accessed on 20 March 2022).
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.-P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Parker, T.M.; Burns, L.A.; Parrish, R.M.; Ryno, A.G.; Sherrill, C.D. Levels of symmetry adapted perturbation theory (SAPT). I. Efficiency and performance for interaction energies. J. Chem. Phys. 2014, 140, 094106. [Google Scholar] [CrossRef]
- Smith, D.G.A.; Burns, L.A.; Simmonett, A.C.; Parrish, R.M.; Schieber, M.C.; Galvelis, R.; Kraus, P.; Kruse, H.; Di Remigio, R.; Alenaizan, A.; et al. PSI4 1.4: Open-source software for high-throughput quantum chemistry. J. Chem. Phys. 2020, 152, 184108. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Papajak, E.; Truhlar, D.G. Convergent Partially Augmented Basis Sets for Post-Hartree−Fock Calculations of Molecular Properties and Reaction Barrier Heights. J. Chem. Theory Comput. 2011, 7, 10–18. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Average RMSD/Å | RMSE/Å |
|---|---|---|
| CYP19A1-TBTOH complex | 2.072 | 0.136 |
| CYP19A1-TPTOH complex | 2.006 | 0.125 |
| TBTOH | 3.258 | 0.376 |
| TPTOH | 2.045 | 0.278 |
| Ligand | ∆Gbind | ΔEgas | ΔΔGPB | ΔΔGSA | −TΔSgas | |
|---|---|---|---|---|---|---|
| vdW | Coul | |||||
| TBTOH | −78.385 | −184.022 | −18.402 | 115.953 | −21.718 | 29.804 |
| TPTOH | −90.732 | −195.024 | −1.941 | 105.751 | −21.482 | 21.963 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, S.; Yang, J. A Theoretical Study of Organotin Binding in Aromatase. Int. J. Mol. Sci. 2023, 24, 8954. https://doi.org/10.3390/ijms24108954
Cheng S, Yang J. A Theoretical Study of Organotin Binding in Aromatase. International Journal of Molecular Sciences. 2023; 24(10):8954. https://doi.org/10.3390/ijms24108954
Chicago/Turabian StyleCheng, Shuming, and Jing Yang. 2023. "A Theoretical Study of Organotin Binding in Aromatase" International Journal of Molecular Sciences 24, no. 10: 8954. https://doi.org/10.3390/ijms24108954
APA StyleCheng, S., & Yang, J. (2023). A Theoretical Study of Organotin Binding in Aromatase. International Journal of Molecular Sciences, 24(10), 8954. https://doi.org/10.3390/ijms24108954