
The Function of MondoA and ChREBP Nutrient—Sensing Factors in Metabolic Disease


Abstract
1. Introduction
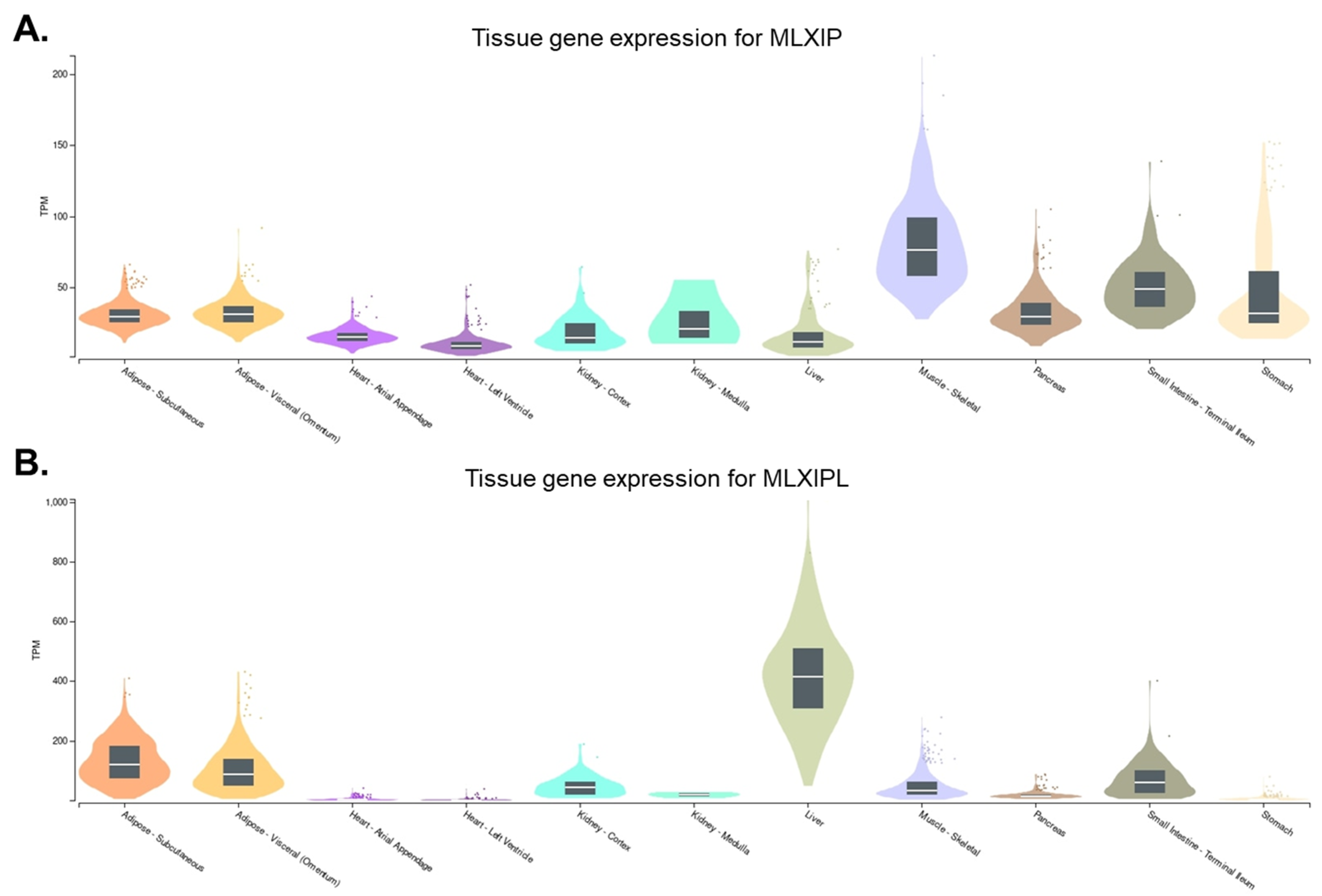
2. Characteristics of MondoA and ChREBP
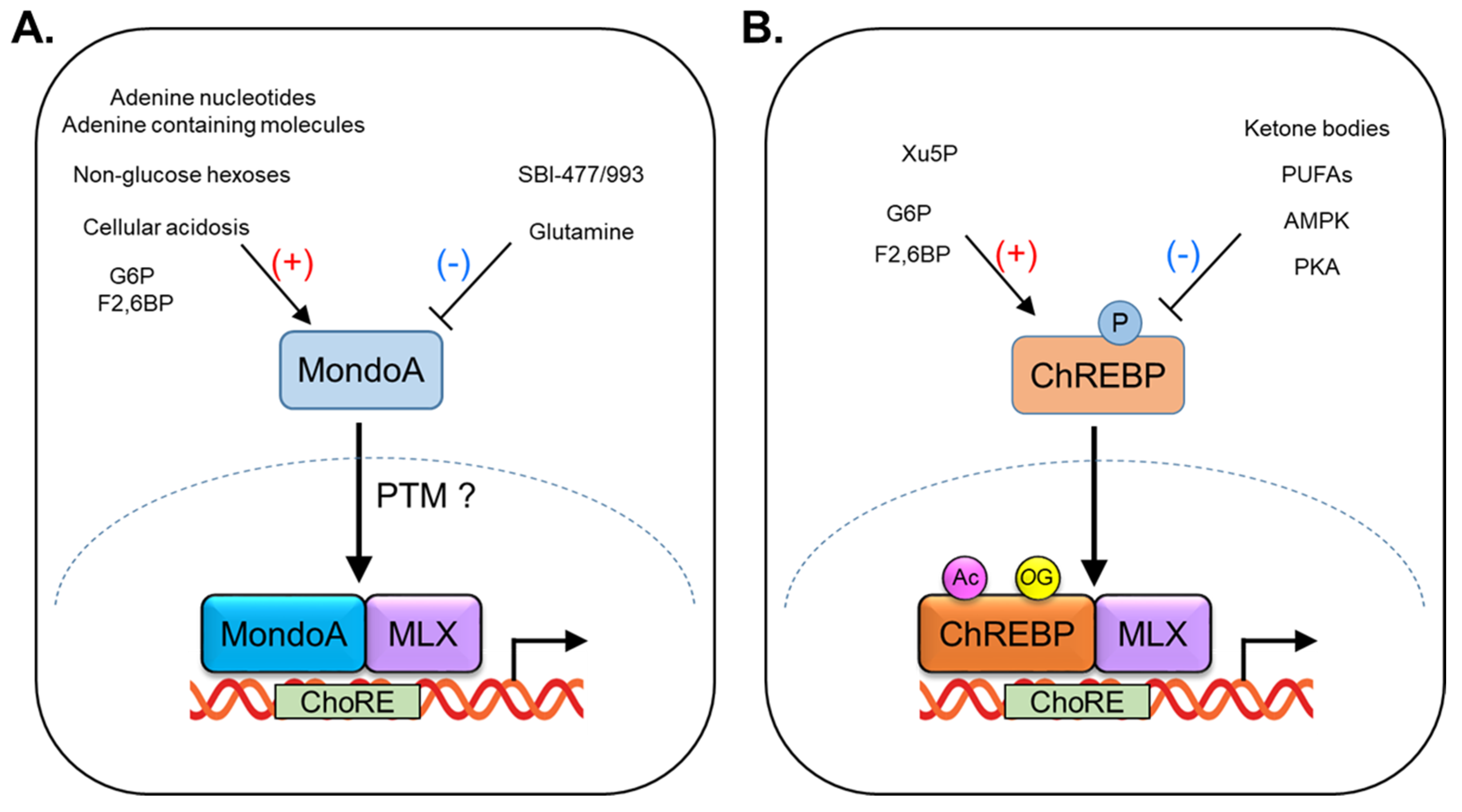
2.1. Regulation of MondoA Activity
2.2. Function of MondoA in Skeletal Muscle
2.3. Regulation of ChREBP Activity
2.4. Function of ChREBP in Liver
2.5. Function of ChREBP in Adipose Tissue
2.6. Function of ChREBP in Pancreas
2.7. Function of TXNIP as a Target of MondoA and ChREBP
2.8. Post-Translational Modification of MondoA and ChREBP

{kind=link}
{kind=link}
| Feature | MondoA | ChREBP | References |
|---|---|---|---|
| Gene symbol and other names | MLXIP (MLX-interacting protein) | MLXIPL (MLX-interacting protein-like) MondoB | [7,8,9] |
| Isoforms and protein size | MondoA (919 AA) | ChREBP-α (852AA) ChREBP-β (675AA) | [7,36,117] |
| Mainly enriched organ | Skeletal muscle | Liver Adipose tissue | [7,36,37,38,39] |
| Major pathway | Insulin signaling Glucose transporter | De novo lipogenesis Glycolysis | [10,11,12,13,14] |
| Post-translational modification | Not available | Phosphorylation Acetylation O-GLcNAcylation | [111,112,113,114,115,116] |
| Inhibitor | SBI-477/993 | Not available | [10] |
| Experimental Models | Context | Diet Duration | Body Weight | Fat Mass | Hepatic Steatosis | Insulin Resistance | References |
|---|---|---|---|---|---|---|---|
| Whole-body MondoA knockout | Chow diet | NC | - | - | NC | [30] | |
| Muscle-specific MondoA knockout | Chow diet | NC | - | NC | NC | [11] | |
| 60% HFD | 16 weeks | NC | - | NC | Improved | [11] | |
| Whole-body ChREBP knockout | Chow diet | NC | NC or down | NC or improved | Moderately induced | [61,62,64] | |
| High-starch diet | 1 week | NC | NC | Improved | Moderately induced | [61] | |
| High-fructose diet (70%) | 3 weeks | Down | - | Improved | NC | [65] | |
| Western diet (41% Fat) | 14 weeks | Down | Down | Improved | - | [64] | |
| Whole-body ChREBP knockout in ob/ob mice | Chow diet | Down | Down | Improved | Moderately improved | [62] | |
| Liver-specific ChREBP knockout | Chow diet | NC | Down | NC | Moderately improved | [67] | |
| 45% HFD | 12 weeks | NC | NC | NC | Improved | [67] | |
| High-carbohydrate diet (70%) | 12 weeks | Down | Down | Improved | Moderately improved | [67] | |
| High-fructose diet (60%) | 9 weeks | Down | Down | NC | Improved | [37] | |
| Liver-specific ChREBP knockdown in ob/ob mice | Chow diet | Down | Down | Improved | Improved | [62] | |
| Liver-specific ChREBP expression | Chow diet | NC | Down | Induced | NC | [68] | |
| 60% HFD | 10 weeks | NC | Down | Induced | Improved | [68] | |
| Adipose-tissue-specific ChREBP knockout | Chow diet | NC | NC | Improved | Induced | [82] | |
| 55% HFD | 16 weeks | NC | NC | NC | Induced | [82] | |
| Adipose-tissue-specific ChREBP expression | Chow diet | NC | NC | NC | NC | [83] | |
| 60% HFD | 10 weeks | Down | Down | Improved | Improved | [83] | |
| Pancreatic b-cell-specific ChREBP expression | Chow diet | Down | - | - | Induced | [63] | |
| SBI-993 (MondoA inhibitor) administration | 60% HFD | 8 weeks | Down | - | Improved | Improved | [10] |
2.9. Development of Therapeutics for MondoA and ChREBP
3. Conclusions and Future Direction
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- The Lancet Gastroenterology Hepatology. Obesity: Another ongoing pandemic. Lancet Gastroenterol. Hepatol. 2021, 6, 411. [Google Scholar] [CrossRef]
- Zimmet, P.; Alberti, K.G.M.M.; Shaw, J. Global and societal implications of the diabetes epidemic. Nature 2001, 414, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-S.; Scherer, P.E. Obesity, Diabetes, and Increased Cancer Progression. Diabetes Metab. J. 2021, 45, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 2003, 46, 3–19. [Google Scholar] [CrossRef]
- Lee, S.-H.; Park, S.-Y.; Choi, C.S. Insulin Resistance: From Mechanisms to Therapeutic Strategies. Diabetes Metab. J. 2022, 46, 15–37. [Google Scholar] [CrossRef]
- Dahlén, A.D.; Dashi, G.; Maslov, I.; Attwood, M.M.; Jonsson, J.; Trukhan, V.; Schiöth, H.B. Trends in Antidiabetic Drug Discovery: FDA Approved Drugs, New Drugs in Clinical Trials and Global Sales. Front. Pharmacol. 2021, 12, 807548. [Google Scholar] [CrossRef]
- Billin, A.; Eilers, A.L.; Coulter, K.L.; Logan, J.S.; Ayer, D.E. MondoA, a novel basic helix-loop-helix-leucine zipper transcriptional activator that constitutes a positive branch of a max-like network. Mol. Cell. Biol. 2000, 20, 8845–8854. [Google Scholar] [CrossRef]
- Yamashita, H.; Takenoshita, M.; Sakurai, M.; Bruick, R.K.; Henzel, W.; Shillinglaw, W.; Arnot, D.; Uyeda, K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. USA 2001, 98, 9116–9121. [Google Scholar] [CrossRef] [PubMed]
- Stoeckman, A.K.; Ma, L.; Towle, H.C. Mlx is the functional heteromeric partner of the carbohydrate response element-binding protein in glucose regulation of lipogenic enzyme genes. J. Biol. Chem. 2004, 279, 15662–15669. [Google Scholar] [CrossRef]
- Ahn, B.; Soundarapandian, M.M.; Sessions, H.; Peddibhotla, S.; Roth, G.P.; Li, J.-L.; Sugarman, E.; Koo, A.; Malany, S.; Wang, M.; et al. MondoA coordinately regulates skeletal myocyte lipid homeostasis and insulin signaling. J. Clin. Investig. 2016, 126, 3567–3579. [Google Scholar] [CrossRef]
- Ahn, B.; Wan, S.; Jaiswal, N.; Vega, R.B.; Ayer, D.E.; Titchenell, P.M.; Han, X.; Won, K.J.; Kelly, D.P. MondoA drives muscle lipid accumulation and insulin resistance. JCI Insight 2019, 5, e129119. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Dentin, R.; Denechaud, P.-D.; Girard, J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu. Rev. Nutr. 2007, 27, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Takao, K.; Kato, T.; Horikawa, Y.; Takeda, J. ChREBP Reciprocally Regulates Liver and Plasma Triacylglycerol Levels in Different Manners. Nutrients 2018, 10, 1699. [Google Scholar] [CrossRef]
- Linden, A.G.; Li, S.; Choi, H.Y.; Fang, F.; Fukasawa, M.; Uyeda, K.; Hammer, R.E.; Horton, J.D.; Engelking, L.J.; Liang, G. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid Res. 2018, 59, 475–487. [Google Scholar] [CrossRef]
- Stoltzman, C.A.; Peterson, C.W.; Breen, K.T.; Muoio, D.M.; Billin, A.N.; Ayer, D.E. Glucose sensing by MondoA:Mlx complexes: A role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6912–6917. [Google Scholar] [CrossRef]
- Petrie, J.L.; Al-Oanzi, Z.H.; Arden, C.; Tudhope, S.J.; Mann, J.; Kieswich, J.; Yaqoob, M.M.; Towle, H.C.; Agius, L. Glucose induces protein targeting to glycogen in hepatocytes by fructose 2,6-bisphosphate-mediated recruitment of MondoA to the promoter. Mol. Cell. Biol. 2013, 33, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Stoltzman, C.A.; Kaadige, M.R.; Peterson, C.W.; Ayer, D. MondoA senses non-glucose sugars: Regulation of thioredoxin-interacting protein (TXNIP) and the hexose transport curb. J. Biol. Chem. 2011, 286, 38027–38034. [Google Scholar] [CrossRef] [PubMed]
- Eilers, A.L.; Sundwall, E.; Lin, M.; Sullivan, A.A.; Ayer, D.E. A novel heterodimerization domain, CRM1, and 14-3-3 control subcellular localization of the MondoA-Mlx heterocomplex. Mol. Cell. Biol. 2002, 22, 8514–8526. [Google Scholar] [CrossRef] [PubMed]
- Li, M.V.; Chang, B.; Imamura, M.; Poungvarin, N.; Chan, L. Glucose-dependent transcriptional regulation by an evolutionarily conserved glucose-sensing module. Diabetes 2006, 55, 1179–1189. [Google Scholar] [CrossRef]
- Sans, C.L.; Satterwhite, D.J.; Stoltzman, C.A.; Breen, K.T.; Ayer, D.E. MondoA-Mlx heterodimers are candidate sensors of cellular energy status: Mitochondrial localization and direct regulation of glycolysis. Mol. Cell. Biol. 2006, 26, 4863–4871. [Google Scholar] [CrossRef]
- Peterson, C.W.; Stoltzman, C.A.; Sighinolfi, M.P.; Han, K.S.; Ayer, D.E. Glucose controls nuclear accumulation, promoter binding, and transcriptional activity of the MondoA-Mlx heterodimer. Mol. Cell. Biol. 2010, 30, 2887–2895. [Google Scholar] [CrossRef] [PubMed]
- Kaadige, M.R.; Looper, R.E.; Kamalanaadhan, S.; Ayer, D.E. Glutamine-dependent anapleurosis dictates glucose uptake and cell growth by regulating MondoA transcriptional activity. Proc. Natl. Acad. Sci. USA 2009, 106, 14878–14883. [Google Scholar] [CrossRef] [PubMed]
- Wilde, B.R.; Ye, Z.; Lim, T.Y.; Ayer, D.E. Cellular acidosis triggers human MondoA transcriptional activity by driving mitochondrial ATP production. Elife 2019, 8, e40199. [Google Scholar] [CrossRef]
- Han, K.S.; Ayer, D.E. MondoA senses adenine nucleotides: Transcriptional induction of thioredoxin-interacting protein. Biochem. J. 2013, 453, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-X.; Goh, S.-R.; Dai, R.-P.; Luo, Y. Adenosine-containing molecules amplify glucose signaling and enhance txnip expression. Mol. Endocrinol. 2009, 23, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Kaadige, M.R.; Yang, J.; Wilde, B.R.; Ayer, D.E. MondoA-Mlx transcriptional activity is limited by mTOR-MondoA interaction. Mol. Cell. Biol. 2015, 35, 101–110. [Google Scholar] [CrossRef]
- Amoasii, L.; Holland, W.; Sanchez-Ortiz, E.; Baskin, K.K.; Pearson, M.; Burgess, S.C.; Nelson, B.R.; Bassel-Duby, R.; Olson, E.N. A MED13-dependent skeletal muscle gene program controls systemic glucose homeostasis and hepatic metabolism. Genes. Dev. 2016, 30, 434–446. [Google Scholar] [CrossRef]
- Thyfault, J.P.; Bergouignan, A. Exercise and metabolic health: Beyond skeletal muscle. Diabetologia 2020, 63, 1464–1474. [Google Scholar] [CrossRef]
- Aune, D.; Norat, T.; Leitzmann, M.; Tonstad, S.; Vatten, L.J. Physical activity and the risk of type 2 diabetes: A systematic review and dose-response meta-analysis. Eur. J. Epidemiol. 2015, 30, 529–542. [Google Scholar] [CrossRef]
- Imamura, M.; Chang, B.H.-J.; Kohjima, M.; Li, M.; Hwang, B.; Taegtmeyer, H.; Harris, R.A.; Chan, L. MondoA deficiency enhances sprint performance in mice. Biochem. J. 2014, 464, 35–48. [Google Scholar] [CrossRef]
- Parikh, H.; Carlsson, E.; Chutkow, W.A.; Johansson, L.E.; Storgaard, H.; Poulsen, P.; Saxena, R.; Ladd, C.; Schulze, P.C.; Mazzini, M.J.; et al. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 2007, 4, e158. [Google Scholar] [CrossRef] [PubMed]
- Dotimas, J.R.; Lee, A.W.; Schmider, A.B.; Carroll, S.H.; Shah, A.; Bilen, J.; Elliott, K.R.; Myers, R.B.; Soberman, R.J.; Yoshioka, J.; et al. Diabetes regulates fructose absorption through thioredoxin-interacting protein. Elife 2016, 5, e18313. [Google Scholar] [CrossRef] [PubMed]
- Waldhart, A.N.; Dykstra, H.; Peck, A.S.; Boguslawski, E.A.; Madaj, Z.B.; Wen, J.; Veldkamp, K.; Hollowell, M.; Zheng, B.; Cantley, L.C.; et al. Phosphorylation of TXNIP by AKT Mediates Acute Influx of Glucose in Response to Insulin. Cell Rep. 2017, 19, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- Ran, H.; Lu, Y.; Zhang, Q.; Hu, Q.; Zhao, J.; Wang, K.; Tong, X.; Su, Q. MondoA Is Required for Normal Myogenesis and Regulation of the Skeletal Muscle Glycogen Content in Mice. Diabetes Metab. J. 2021, 45, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Delibegovic, M.; Armstrong, C.G.; Dobbie, L.; Watt, P.W.; Smith, A.J.; Cohen, P.T. Disruption of the striated muscle glycogen targeting subunit PPP1R3A of protein phosphatase 1 leads to increased weight gain, fat deposition, and development of insulin resistance. Diabetes 2003, 52, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schön, M.R.; Abumrad, N.A.; Blüher, M.; Klein, S.; Kahn, B.B. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012, 484, 333–338. [Google Scholar] [CrossRef]
- Kim, M.; Astapova, I.I.; Flier, S.N.; Hannou, S.A.; Doridot, L.; Sargsyan, A.; Kou, H.H.; Fowler, A.J.; Liang, G.; Herman, M.A. Intestinal, but not hepatic, ChREBP is required for fructose tolerance. JCI Insight 2017, 2, e96703. [Google Scholar] [CrossRef]
- Lee, H.-J.; Cha, J.-Y. Recent insights into the role of ChREBP in intestinal fructose absorption and metabolism. BMB Rep. 2018, 51, 429–436. [Google Scholar] [CrossRef]
- Sanchez-Gurmaches, J.; Tang, Y.; Jespersen, N.Z.; Wallace, M.; Calejman, C.M.; Gujja, S.; Li, H.; Edwards, Y.J.K.; Wolfrum, C.; Metallo, C.M.; et al. Brown Fat AKT2 Is a Cold-Induced Kinase that Stimulates ChREBP-Mediated De Novo Lipogenesis to Optimize Fuel Storage and Thermogenesis. Cell Metab. 2018, 27, 195–209.e6. [Google Scholar] [CrossRef]
- Kabashima, T.; Kawaguchi, T.; Wadzinski, B.E.; Uyeda, K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc. Natl. Acad. Sci. USA 2003, 100, 5107–5112. [Google Scholar] [CrossRef]
- Arden, C.; Tudhope, S.J.; Petrie, J.L.; Al-Oanzi, Z.H.; Cullen, K.S.; Lange, A.J.; Towle, H.C.; Agius, L. Fructose 2,6-bisphosphate is essential for glucose-regulated gene transcription of glucose-6-phosphatase and other ChREBP target genes in hepatocytes. Biochem. J. 2012, 443, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Dentin, R.; Tomas-Cobos, L.; Foufelle, F.; Leopold, J.; Girard, J.; Postic, C.; Ferré, P. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. J. Hepatol. 2012, 56, 199–209. [Google Scholar] [CrossRef]
- Li, M.V.; Chen, W.; Harmancey, R.N.; Nuotio-Antar, A.M.; Imamura, M.; Saha, P.; Taegtmeyer, H.; Chan, L. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochem. Biophys. Res. Commun. 2010, 395, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; Nakagawa, T.; Wynn, R.; Chook, Y.M.; Miller, B.C.; Uyeda, K. Importin-alpha protein binding to a nuclear localization signal of carbohydrate response element-binding protein (ChREBP). J. Biol. Chem. 2011, 286, 28119–28127. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Takeshima, T.; Nakagawa, T.; MacMillan, K.S.; Wynn, R.M.; Wang, H.; Sakiyama, H.; Wei, S.; Li, Y.; Bruick, R.K.; et al. The structure of importin alpha and the nuclear localization peptide of ChREBP, and small compound inhibitors of ChREBP-importin alpha interactions. Biochem. J. 2020, 477, 3253–3269. [Google Scholar] [CrossRef]
- Merla, G.; Howald, C.; Antonarakis, S.E.; Reymond, A. The subcellular localization of the ChoRE-binding protein, encoded by the Williams-Beuren syndrome critical region gene 14, is regulated by 14-3-3. Hum. Mol. Genet. 2004, 13, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
- Sakiyama, H.; Wynn, R.M.; Lee, W.R.; Fukasawa, M.; Mizuguchi, H.; Gardner, K.H.; Repa, J.J.; Uyeda, K. Regulation of nuclear import/export of carbohydrate response element-binding protein (ChREBP): Interaction of an alpha-helix of ChREBP with the 14-3-3 proteins and regulation by phosphorylation. J. Biol. Chem. 2008, 283, 24899–24908. [Google Scholar] [CrossRef]
- Li, M.V.; Chen, W.; Poungvarin, N.; Imamura, M.; Chan, L. Glucose-mediated transactivation of carbohydrate response element-binding protein requires cooperative actions from Mondo conserved regions and essential trans-acting factor 14-3-3. Mol. Endocrinol. 2008, 22, 1658–1672. [Google Scholar] [CrossRef]
- Nakagawa, T.; Ge, Q.; Pawlosky, R.; Wynn, R.M.; Veech, R.L.; Uyeda, K. Metabolite regulation of nucleo-cytosolic trafficking of carbohydrate response element-binding protein (ChREBP): Role of ketone bodies. J. Biol. Chem. 2013, 288, 28358–28367. [Google Scholar] [CrossRef]
- Sato, S.; Jung, H.; Nakagawa, T.; Pawlosky, R.; Takeshima, T.; Lee, W.R.; Sakiyama, H.; Laxman, S.; Wynn, R.M.; Tu, B.P.; et al. Metabolite Regulation of Nuclear Localization of Carbohydrate-response Element-binding Protein (ChREBP): Role of AMP as an Allosteric Inhibitor. J. Biol. Chem. 2016, 291, 10515–10527. [Google Scholar] [CrossRef]
- Dentin, R.; Benhamed, F.; Pégorier, J.-P.; Foufelle, F.; Viollet, B.; Vaulont, S.; Girard, J.; Postic, C. Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. J. Clin. Investig. 2005, 115, 2843–2854. [Google Scholar] [CrossRef]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar]
- Nordlie, R.C.; Foster, J.D.; Lange, A.J. Regulation of glucose production by the liver. Annu. Rev. Nutr. 1999, 19, 379–406. [Google Scholar] [CrossRef]
- Titchenell, P.M.; Lazar, M.A.; Birnbaum, M.J. Unraveling the Regulation of Hepatic Metabolism by Insulin. Trends Endocrinol. Metab. 2017, 28, 497–505. [Google Scholar] [CrossRef]
- del Pozo, C.H.; Vesperinas-García, G.; Rubio, M.Á.; Corripio-Sánchez, R.; Torres-García, A.J.; Obregon, M.J.; Calvo, R.M. ChREBP expression in the liver, adipose tissue and differentiated preadipocytes in human obesity. Biochim. Biophys. Acta 2011, 1811, 1194–1200. [Google Scholar] [CrossRef]
- Eissing, L.; Scherer, T.; Tödter, K.; Knippschild, U.; Greve, J.W.; Buurman, W.A.; Pinnschmidt, H.O.; Rensen, S.S.; Wolf, A.M.; Bartelt, A.; et al. De novo lipogenesis in human fat and liver is linked to ChREBP-beta and metabolic health. Nat. Commun. 2013, 4, 1528. [Google Scholar] [CrossRef]
- Stamatikos, A.D.; da Silva, R.P.; Lewis, J.T.; Douglas, D.N.; Kneteman, N.M.; Jacobs, R.L.; Paton, C.M. Tissue Specific Effects of Dietary Carbohydrates and Obesity on ChREBPalpha and ChREBPbeta Expression. Lipids 2016, 51, 95–104. [Google Scholar] [CrossRef]
- Ishii, S.; Iizuka, K.; Miller, B.C.; Uyeda, K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 15597–15602. [Google Scholar] [CrossRef]
- Ma, L.; Robinson, L.N.; Towle, H.C. ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver. J. Biol. Chem. 2006, 281, 28721–28730. [Google Scholar] [CrossRef]
- Iizuka, K.; Takao, K.; Yabe, D. ChREBP-Mediated Regulation of Lipid Metabolism: Involvement of the Gut Microbiota, Liver, and Adipose Tissue. Front. Endocrinol. 2020, 11, 587189. [Google Scholar] [CrossRef]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef]
- Iizuka, K.; Miller, B.; Uyeda, K. Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E358–E364. [Google Scholar] [CrossRef]
- Poungvarin, N.; Lee, J.K.; Yechoor, V.K.; Li, M.V.; Assavapokee, T.; Suksaranjit, P.; Thepsongwajja, J.J.; Saha, P.K.; Oka, K.; Chan, L. Carbohydrate response element-binding protein (ChREBP) plays a pivotal role in beta cell glucotoxicity. Diabetologia 2012, 55, 1783–1796. [Google Scholar] [CrossRef]
- Wu, W.; Tsuchida, H.; Kato, T.; Niwa, H.; Horikawa, Y.; Takeda, J.; Iizuka, K. Fat and carbohydrate in western diet contribute differently to hepatic lipid accumulation. Biochem. Biophys. Res. Commun. 2015, 461, 681–686. [Google Scholar] [CrossRef]
- Zhang, D.; Tong, X.; VanDommelen, K.; Gupta, N.; Stamper, K.; Brady, G.F.; Meng, Z.; Lin, J.; Rui, L.; Omary, M.B.; et al. Lipogenic transcription factor ChREBP mediates fructose-induced metabolic adaptations to prevent hepatotoxicity. J. Clin. Investig. 2017, 127, 2855–2867. [Google Scholar] [CrossRef]
- Dentin, R.; Benhamed, F.; Hainault, I.; Fauveau, V.; Foufelle, F.; Dyck, J.R.; Girard, J.; Postic, C. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 2006, 55, 2159–2170. [Google Scholar]
- Jois, T.; Chen, W.; Howard, V.; Harvey, R.; Youngs, K.; Thalmann, C.; Saha, P.; Chan, L.; Cowley, M.A.; Sleeman, M.W. Deletion of hepatic carbohydrate response element binding protein (ChREBP) impairs glucose homeostasis and hepatic insulin sensitivity in mice. Mol. Metab. 2017, 6, 1381–1394. [Google Scholar] [CrossRef]
- Benhamed, F.; Denechaud, P.-D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J.; et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Investig. 2012, 122, 2176–2194. [Google Scholar] [CrossRef]
- Iizuka, K.; Takeda, J.; Horikawa, Y. Glucose induces FGF21 mRNA expression through ChREBP activation in rat hepatocytes. FEBS Lett. 2009, 583, 2882–2886. [Google Scholar] [CrossRef]
- Fisher, F.M.; Kim, M.; Doridot, L.; Cunniff, J.C.; Parker, T.S.; Levine, D.M.; Hellerstein, M.K.; Hudgins, L.C.; Maratos-Flier, E.; Herman, M.A. A critical role for ChREBP-mediated FGF21 secretion in hepatic fructose metabolism. Mol. Metab. 2017, 6, 14–21. [Google Scholar] [CrossRef]
- Iroz, A.; Montagner, A.; Benhamed, F.; Levavasseur, F.; Polizzi, A.; Anthony, E.; Régnier, M.; Fouché, E.; Lukowicz, C.; Cauzac, M.; et al. A Specific ChREBP and PPARalpha Cross-Talk Is Required for the Glucose-Mediated FGF21 Response. Cell Rep. 2017, 21, 403–416. [Google Scholar] [CrossRef]
- Lewis, J.E.; Ebling, F.J.; Samms, R.J.; Tsintzas, K. Going Back to the Biology of FGF21: New Insights. Trends Endocrinol. Metab. 2019, 30, 491–504. [Google Scholar] [CrossRef]
- Tezze, C.; Romanello, V.; Sandri, M. FGF21 as Modulator of Metabolism in Health and Disease. Front. Physiol. 2019, 10, 419. [Google Scholar] [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef]
- Carobbio, S.; Pellegrinelli, V.; Vidal-Puig, A. Adipose Tissue Function and Expandability as Determinants of Lipotoxicity and the Metabolic Syndrome. Adv. Exp. Med. Biol. 2017, 960, 161–196. [Google Scholar]
- Booth, A.; Magnuson, A.; Fouts, J.; Foster, M.T. Adipose tissue: An endocrine organ playing a role in metabolic regulation. Horm. Mol. Biol. Clin. Investig. 2016, 26, 25–42. [Google Scholar] [CrossRef]
- Lynes, M.D.; Tseng, Y.H. Deciphering adipose tissue heterogeneity. Ann. N. Y. Acad. Sci. 2018, 1411, 5–20. [Google Scholar] [CrossRef]
- Luong, Q.; Huang, J.; Lee, K.Y. Deciphering White Adipose Tissue Heterogeneity. Biology 2019, 8, 23. [Google Scholar] [CrossRef]
- Townsend, K.L.; Tseng, Y.H. Brown fat fuel utilization and thermogenesis. Trends Endocrinol. Metab. 2014, 25, 168–177. [Google Scholar] [CrossRef]
- Ricquier, D. UCP1, the mitochondrial uncoupling protein of brown adipocyte: A personal contribution and a historical perspective. Biochimie 2017, 134, 3–8. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Kajimura, S. Metabolic adaptation and maladaptation in adipose tissue. Nat. Metab. 2019, 1, 189–200. [Google Scholar] [CrossRef]
- Vijayakumar, A.; Aryal, P.; Wen, J.; Syed, I.; Vazirani, R.P.; Moraes-Vieira, P.M.; Camporez, J.P.; Gallop, M.R.; Perry, R.J.; Peroni, O.D.; et al. Absence of Carbohydrate Response Element Binding Protein in Adipocytes Causes Systemic Insulin Resistance and Impairs Glucose Transport. Cell Rep. 2017, 21, 1021–1035. [Google Scholar] [CrossRef]
- Nuotio-Antar, A.M.; Poungvarin, N.; Li, M.; Schupp, M.; Mohammad, M.; Gerard, S.; Zou, F.; Chan, L. FABP4-Cre Mediated Expression of Constitutively Active ChREBP Protects Against Obesity, Fatty Liver, and Insulin Resistance. Endocrinology 2015, 156, 4020–4032. [Google Scholar] [CrossRef]
- Fernandez, S.; Viola, J.M.; Torres, A.; Wallace, M.; Trefely, S.; Zhao, S.; Affronti, H.C.; Gengatharan, J.M.; Guertin, D.A.; Snyder, N.W.; et al. Adipocyte ACLY Facilitates Dietary Carbohydrate Handling to Maintain Metabolic Homeostasis in Females. Cell Rep. 2019, 27, 2772–2784.e6. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wallace, M.; Sanchez-Gurmaches, J.; Hsiao, W.Y.; Li, H.; Lee, P.L.; Vernia, S.; Metallo, C.M.; Guertin, D.A. Adipose tissue mTORC2 regulates ChREBP-driven de novo lipogenesis and hepatic glucose metabolism. Nat. Commun. 2016, 7, 11365. [Google Scholar] [CrossRef]
- Witte, N.; Muenzner, M.; Rietscher, J.; Knauer, M.; Heidenreich, S.; Nuotio-Antar, A.M.; Graef, F.A.; Fedders, R.; Tolkachov, A.; Goehring, I.; et al. The Glucose Sensor ChREBP Links De Novo Lipogenesis to PPARgamma Activity and Adipocyte Differentiation. Endocrinology 2015, 156, 4008–4019. [Google Scholar] [CrossRef]
- Katz, L.S.; Xu, S.; Ge, K.; Scott, D.K.; Gershengorn, M.C. T3 and Glucose Coordinately Stimulate ChREBP-Mediated Ucp1 Expression in Brown Adipocytes from Male Mice. Endocrinology 2018, 159, 557–569. [Google Scholar] [CrossRef]
- Wei, C.; Ma, X.; Su, K.; Qi, S.; Zhu, Y.; Lin, J.; Wang, C.; Yang, R.; Chen, X.; Wang, W.; et al. ChREBP-beta regulates thermogenesis in brown adipose tissue. J. Endocrinol. 2020, 245, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Röder, P.V.; Wu, B.; Liu, Y.; Han, W. Pancreatic regulation of glucose homeostasis. Exp. Mol. Med. 2016, 48, e219. [Google Scholar] [CrossRef] [PubMed]
- Honka, H.; Hannukainen, J.C.; Tarkia, M.; Karlsson, H.; Saunavaara, V.; Salminen, P.; Soinio, M.; Mikkola, K.; Kudomi, N.; Oikonen, V.; et al. Pancreatic metabolism, blood flow, and β-cell function in obese humans. J. Clin. Endocrinol. Metab. 2014, 99, E981–E990. [Google Scholar] [CrossRef]
- Metukuri, M.R.; Zhang, P.; Stewart, A.F.; Vasavada, R.C.; Garcia-Ocaña, A.; Scott, D.K.; Basantani, M.K.; Chin, C.; Stamateris, R.E.; Alonso, L.C.; et al. ChREBP Mediates Glucose-Stimulated Pancreatic β-Cell Proliferation. Diabetes 2012, 61, 2004–2015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Kumar, A.; Katz, L.S.; Li, L.; Paulynice, M.; Herman, M.A.; Scott, D.K. Induction of the ChREBPβ Isoform Is Essential for Glucose-Stimulated β-Cell Proliferation. Diabetes 2015, 64, 4158–4170. [Google Scholar] [CrossRef]
- Schmidt, S.F.; Madsen, J.G.; Frafjord, K.Ø.; Poulsen, L.L.; Salö, S.; Boergesen, M.; Loft, A.; Larsen, B.D.; Madsen, M.S.; Holst, J.J.; et al. Integrative Genomics Outlines a Biphasic Glucose Response and a ChREBP-RORγ Axis Regulating Proliferation in β Cells. Cell Rep. 2016, 16, 2359–2372. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Katz, L.S.; Schulz, A.M.; Kim, M.; Honig, L.B.; Li, L.; Davenport, B.; Homann, D.; Garcia-Ocaña, A.; Herman, M.A.; et al. Activation of Nrf2 Is Required for Normal and ChREBPα-Augmented Glucose-Stimulated β-Cell Proliferation. Diabetes 2018, 67, 1561–1575. [Google Scholar] [CrossRef]
- Jing, G.; Chen, J.; Xu, G.; Shalev, A. Islet ChREBP-β is increased in diabetes and controls ChREBP-α and glucose-induced gene expression via a negative feedback loop. Mol. Metab. 2016, 5, 1208–1215. [Google Scholar] [CrossRef]
- Minn, A.H.; Hafele, C.; Shalev, A. Thioredoxin-Interacting Protein Is Stimulated by Glucose through a Carbohydrate Response Element and Induces β-Cell Apoptosis. Endocrinology 2005, 146, 2397–2405. [Google Scholar] [CrossRef]
- Chen, J.; Saxena, G.; Mungrue, I.N.; Lusis, A.J.; Shalev, A. Thioredoxin-Interacting Protein: A Critical Link Between Glucose Toxicity and β-Cell Apoptosis. Diabetes 2008, 57, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Spindel, O.N.; World, C.; Berk, B.C. Thioredoxin interacting protein: Redox dependent and independent regulatory mechanisms. Antioxid. Redox Signal. 2012, 16, 587–596. [Google Scholar] [CrossRef]
- Alhawiti, N.M.; Al Mahri, S.; Aziz, M.A.; Malik, S.S.; Mohammad, S. TXNIP in Metabolic Regulation: Physiological Role and Therapeutic Outlook. Curr. Drug. Targets 2017, 18, 1095–1103. [Google Scholar]
- Chen, Y.; Ning, J.; Cao, W.; Wang, S.; Du, T.; Jiang, J.; Feng, X.; Zhang, B. Research Progress of TXNIP as a Tumor Suppressor Gene Participating in the Metabolic Reprogramming and Oxidative Stress of Cancer Cells in Various Cancers. Front. Oncol. 2020, 10, 568574. [Google Scholar] [CrossRef] [PubMed]
- Thielen, L.; Shalev, A. Diabetes pathogenic mechanisms and potential new therapies based upon a novel target called TXNIP. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 75–80. [Google Scholar] [CrossRef]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.-H.; Wen, J.; Asara, J.; McGraw, T.E.; et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 2013, 49, 1167–1175. [Google Scholar] [CrossRef]
- Yoshihara, E.; Fujimoto, S.; Inagaki, N.; Okawa, K.; Masaki, S.; Yodoi, J.; Masutani, H. Disruption of TBP-2 ameliorates insulin sensitivity and secretion without affecting obesity. Nat. Commun. 2010, 1, 127. [Google Scholar] [CrossRef]
- Yoshioka, J.; Imahashi, K.; Gabel, S.A.; Chutkow, W.A.; Burds, A.A.; Gannon, J.; Schulze, P.C.; MacGillivray, C.; London, R.E.; Murphy, E.; et al. Targeted Deletion of Thioredoxin-Interacting Protein Regulates Cardiac Dysfunction in Response to Pressure Overload. Circ. Res. 2007, 101, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, J.; Chutkow, W.A.; Lee, S.; Kim, J.B.; Yan, J.; Tian, R.; Lindsey, M.L.; Feener, E.P.; Seidman, C.E.; Seidman, J.G.; et al. Deletion of thioredoxin-interacting protein in mice impairs mitochondrial function but protects the myocardium from ischemia-reperfusion injury. J. Clin. Investig. 2012, 122, 267–279. [Google Scholar] [CrossRef]
- Johnson, L.N.; Noble, M.; Owen, D.J. Active and inactive protein kinases: Structural basis for regulation. Cell 1996, 85, 149–158. [Google Scholar] [CrossRef]
- Walma, D.A.C.; Chen, Z.; Bullock, A.N.; Yamada, K.M. Ubiquitin ligases: Guardians of mammalian development. Nat. Rev. Mol. Cell Biol. 2022, 23, 350–367. [Google Scholar] [CrossRef] [PubMed]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Takenoshita, M.; Kabashima, T.; Uyeda, K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. USA 2001, 98, 13710–13715. [Google Scholar] [CrossRef] [PubMed]
- Denechaud, P.-D.; Bossard, P.; Lobaccaro, J.-M.A.; Millatt, L.; Staels, B.; Girard, J.; Postic, C. ChREBP, but not LXRs, is required for the induction of glucose-regulated genes in mouse liver. J. Clin. Investig. 2008, 118, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Bricambert, J.; Miranda, J.; Benhamed, F.; Girard, J.; Postic, C.; Dentin, R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J. Clin. Investig. 2010, 120, 4316–4331. [Google Scholar] [CrossRef]
- Ortega-Prieto, P.; Postic, C. Carbohydrate Sensing Through the Transcription Factor ChREBP. Front. Genet. 2019, 10, 472. [Google Scholar] [CrossRef]
- Guinez, C.; Filhoulaud, G.; Rayah-Benhamed, F.; Marmier, S.; Dubuquoy, C.; Dentin, R.; Moldes, M.; Burnol, A.F.; Yang, X.; Lefebvre, T.; et al. O-GlcNAcylation Increases ChREBP Protein Content and Transcriptional Activity in the Liver. Diabetes 2011, 60, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Lane, E.A.; Choi, D.W.; Garcia-Haro, L.; Levine, Z.G.; Tedoldi, M.; Walker, S.; Danial, N.N. HCF-1 Regulates De Novo Lipogenesis through a Nutrient-Sensitive Complex with ChREBP. Mol. Cell. 2019, 75, 357–371.e7. [Google Scholar] [CrossRef]
- Yu, Y.; Maguire, T.G.; Alwine, J.C. ChREBP, a glucose-responsive transcriptional factor, enhances glucose metabolism to support biosynthesis in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. USA 2014, 111, 1951–1956. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, B. The Function of MondoA and ChREBP Nutrient—Sensing Factors in Metabolic Disease. Int. J. Mol. Sci. 2023, 24, 8811. https://doi.org/10.3390/ijms24108811
Ahn B. The Function of MondoA and ChREBP Nutrient—Sensing Factors in Metabolic Disease. International Journal of Molecular Sciences. 2023; 24(10):8811. https://doi.org/10.3390/ijms24108811
Chicago/Turabian StyleAhn, Byungyong. 2023. "The Function of MondoA and ChREBP Nutrient—Sensing Factors in Metabolic Disease" International Journal of Molecular Sciences 24, no. 10: 8811. https://doi.org/10.3390/ijms24108811
APA StyleAhn, B. (2023). The Function of MondoA and ChREBP Nutrient—Sensing Factors in Metabolic Disease. International Journal of Molecular Sciences, 24(10), 8811. https://doi.org/10.3390/ijms24108811