

The Males Absent on the First (MOF) Mediated Acetylation Alters the Protein Stability and Transcriptional Activity of YY1 in HCT116 Cells
 and
and 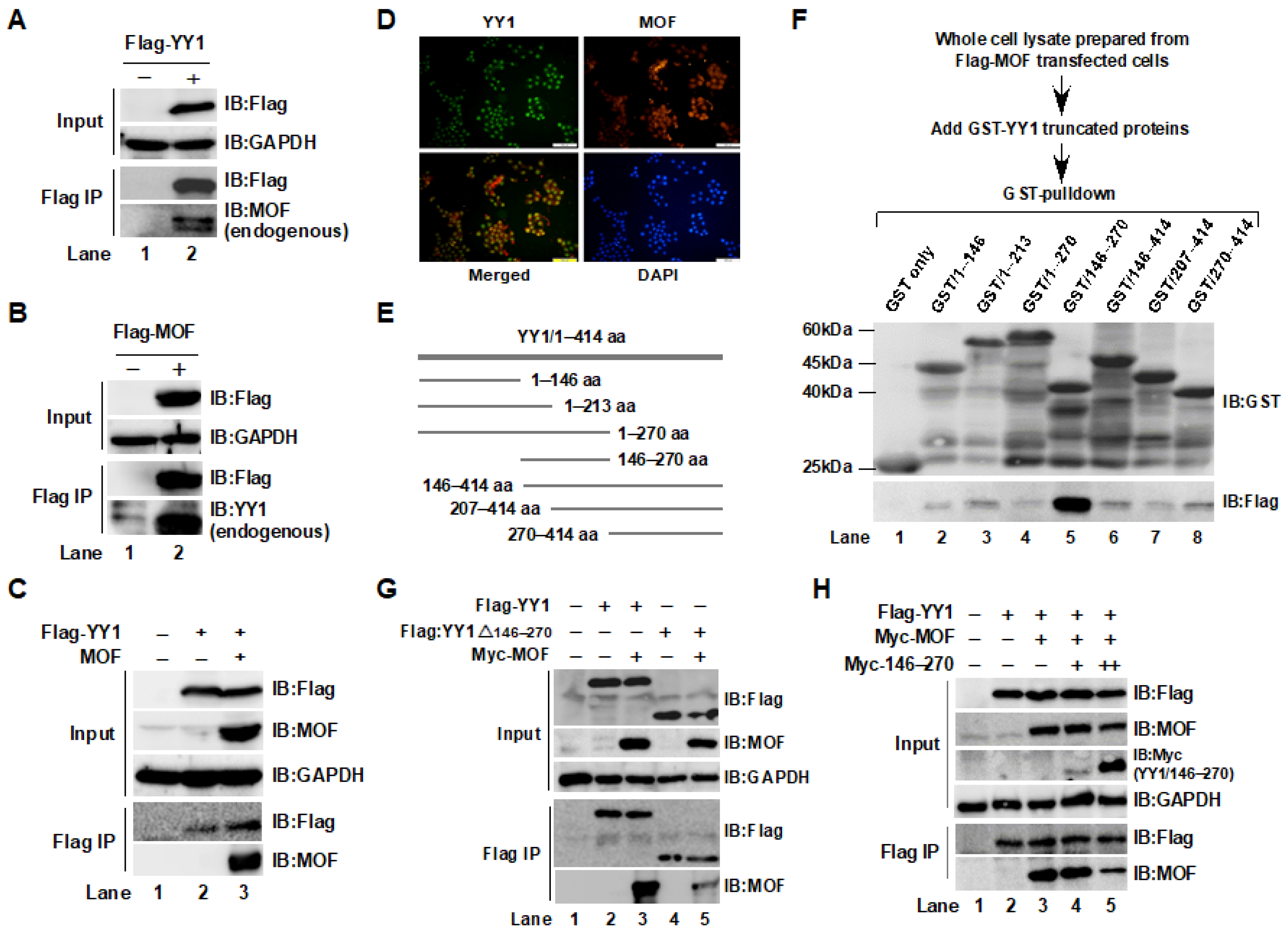{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Interaction between YY1 and MOF Was Confirmed by Immunoprecipitation (IP) and GST-Pulldown Assays
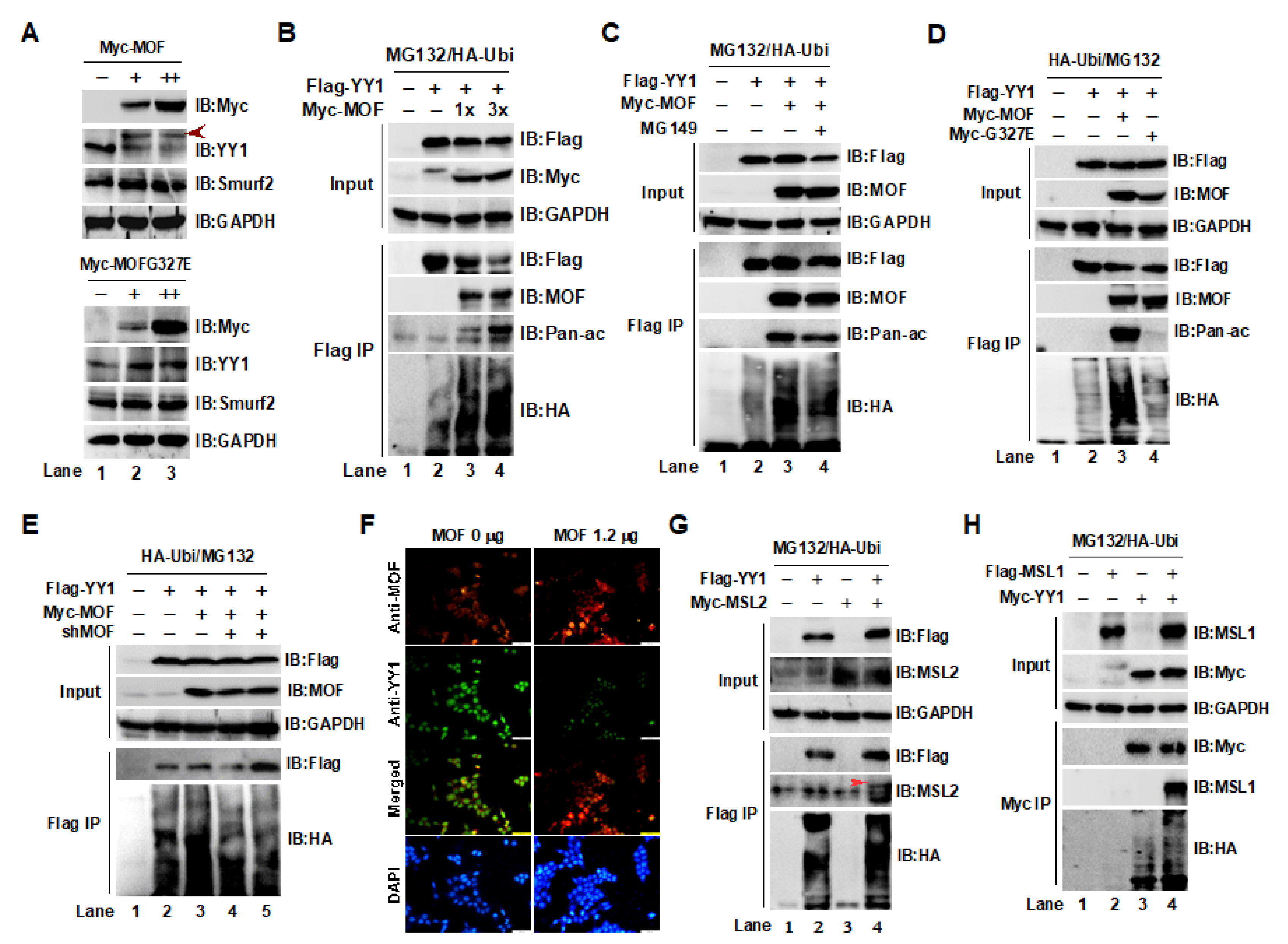
2.2. Acetylation of YY1 by the MOF/MSL Complex Was Closely Associated with YY1 Ubiquitin-Proteasome Degradation in HCT116 Cells
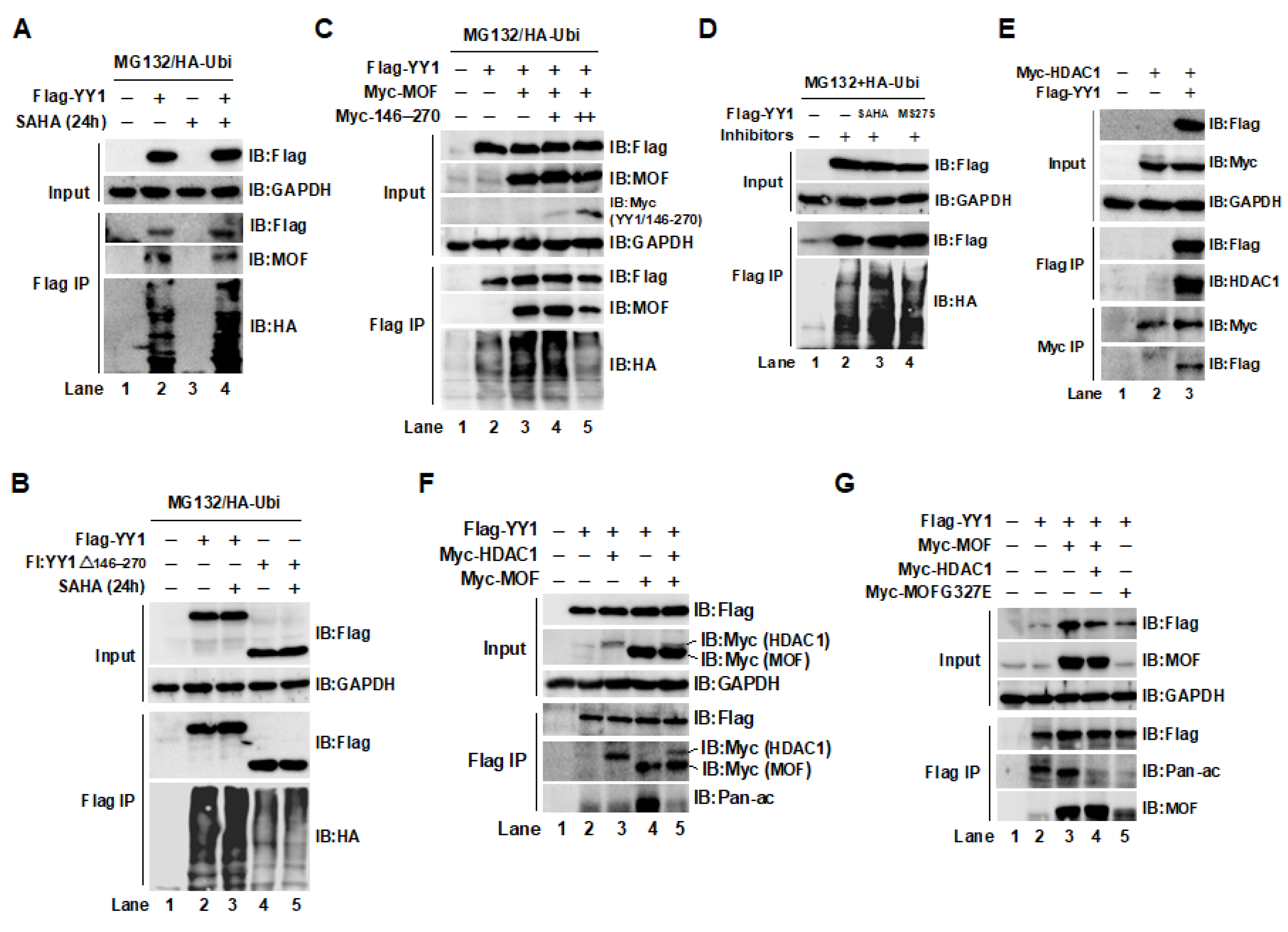
2.3. The Acetylation of YY1 in HCT116 Cells Was Regulated by MOF and HDAC1
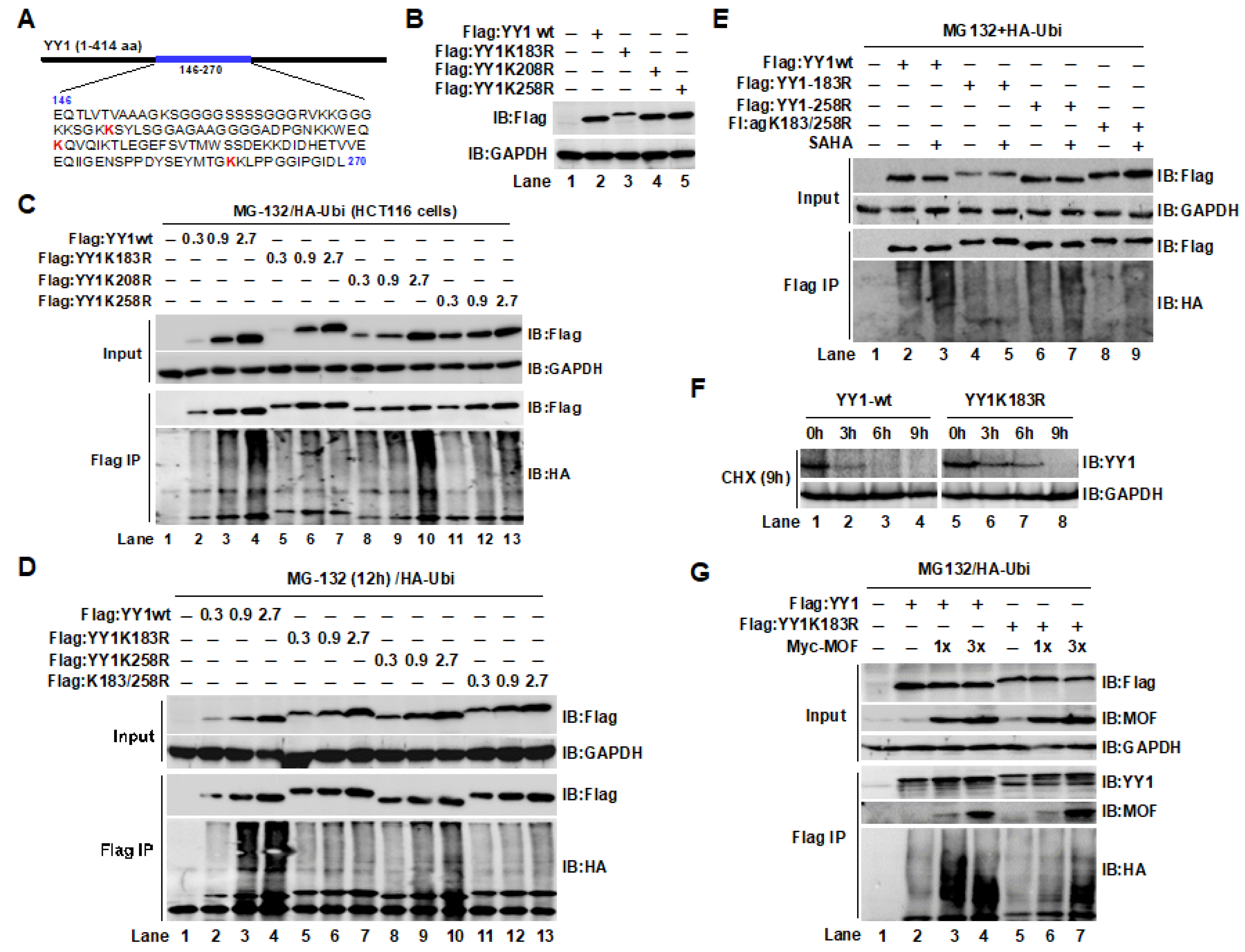
2.4. The YY1K183 Site Is the Main Ubiquitylation Site That Maintains YY1 Stability
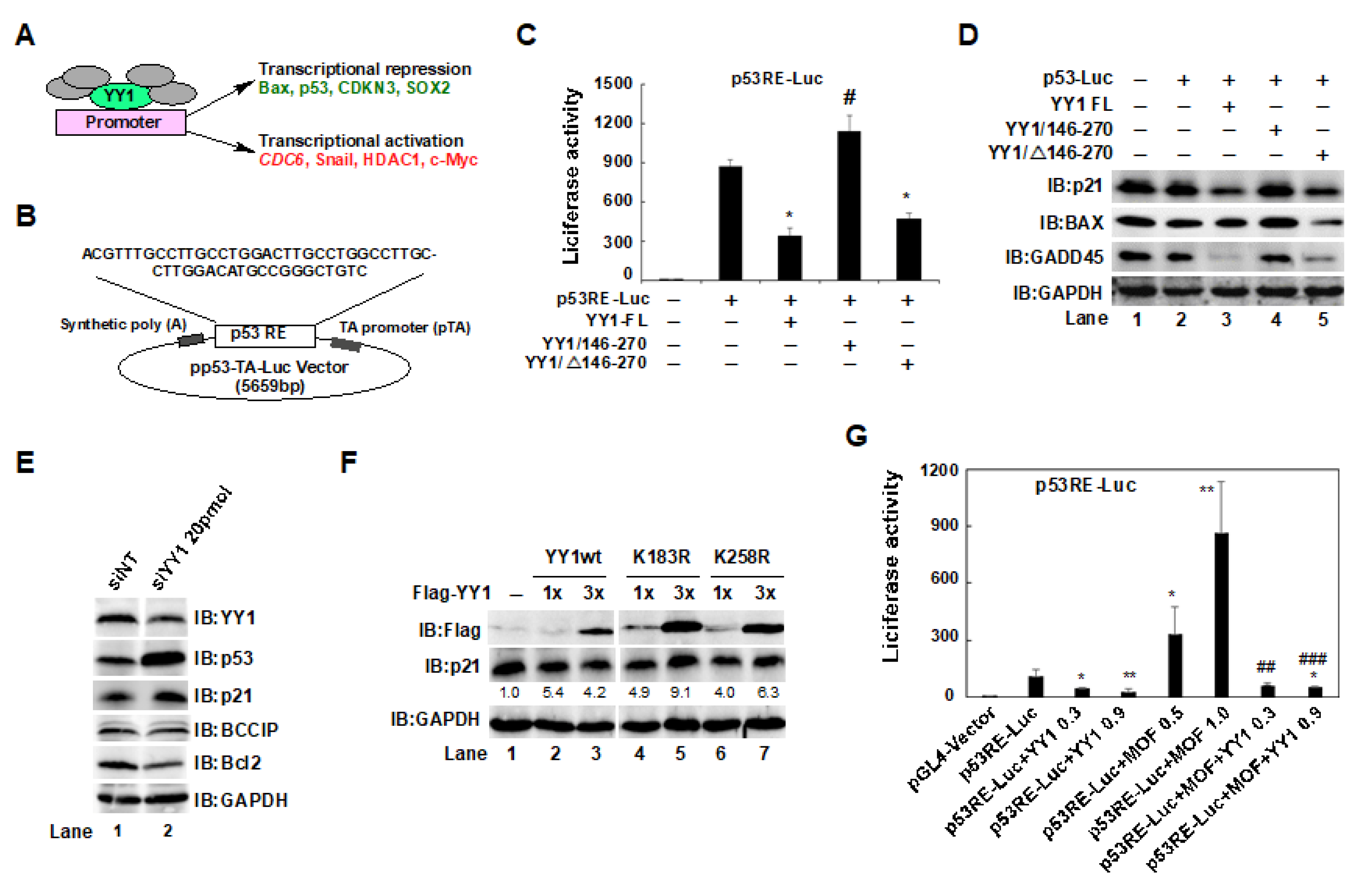
2.5. The YY1/146–270 Region and the YY1K183 Site Interfered with p53-Mediated p21 Expression in HCT116 Cells
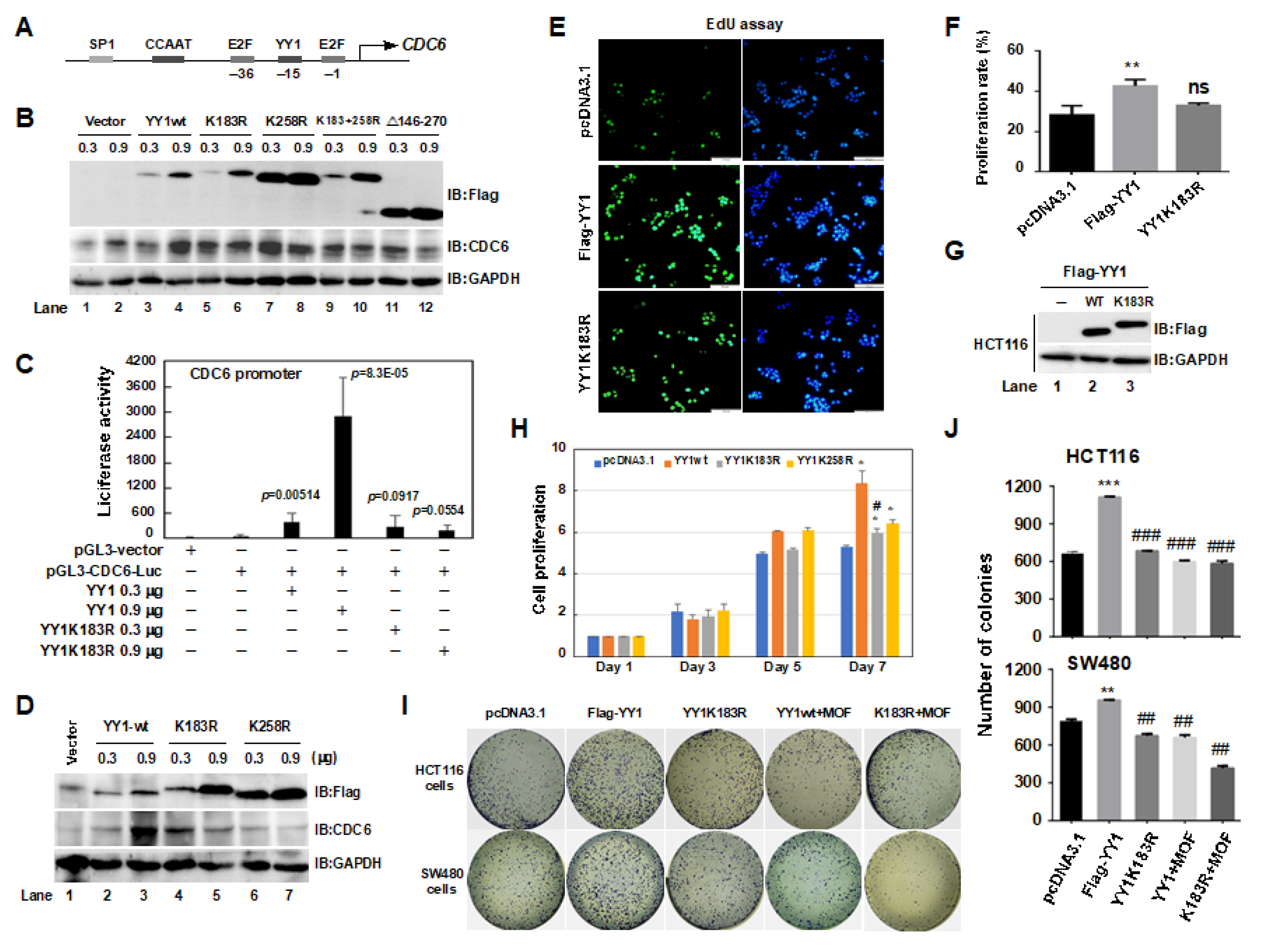
2.6. A YY1K183R Mutant Inhibited CDC6 Transactivation, and Suppressed the Proliferation of HCT116 Cells
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Cell Culture
4.3. Plasmids and Transient Transfection
4.4. Immunoprecipitation (IP)
4.5. Expression of Recombinant Proteins in Escherichia coli
4.6. MTT Assay
4.7. Colony Formation Assay
4.8. Immunofluorescence Staining
4.9. EdU Assay
4.10. Luciferase Reporter Assay
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MOF | Males absent on the first |
| YY1 | Yin-Yang 1 |
| HAT | Histone acetyltransferase |
| HDAC | Histone deacetylase |
| TFs MSL PTM | Transcription factors Male-specific lethal Post-translational modification |
References
- Seto, E.; Shi, Y.; Shenk, T. YY1 is an initiator sequence-binding protein that directs and activates transcription in vitro. Nature 1991, 354, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Seto, E.; Chang, L.S.; Shenk, T. Transcriptional repression by YY1, a human GLI-Krüppel-related protein, and relief of repression by adenovirus E1A protein. Cell 1991, 67, 377–388. [Google Scholar] [CrossRef]
- Galvin, K.M.; Shi, Y. Multiple mechanisms of transcriptional repression by YY1. Mol. Cell. Biol. 1997, 17, 3723–3732. [Google Scholar] [CrossRef]
- Yao, Y.L.; Yang, W.M.; Seto, E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol. Cell. Biol. 2001, 21, 5979–5991. [Google Scholar] [CrossRef] [PubMed]
- Warowicka, A.; Broniarczyk, J.; Węglewska, M.; Kwaśniewski, W.; Goździcka-Józefiak, A. Dual Role of YY1 in HPV Life Cycle and Cervical Cancer Development. Int. J. Mol. Sci. 2022, 23, 3453. [Google Scholar] [CrossRef]
- Lee, J.S.; Galvin, K.M.; See, R.H.; Eckner, R.; Livingston, D.; Moran, E.; Shi, Y. Relief of YY1 transcriptional repression by adenovirus E1A is mediated by E1A-associated protein p300. Genes Dev. 1995, 9, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Rea, S.; Xouri, G.; Akhtar, A. Males absent on the first (MOF): From flies to humans. Oncogene 2007, 26, 5385–5394. [Google Scholar] [CrossRef]
- Keller, C.I.; Akhtar, A. The MSL complex: Juggling RNA-protein interactions for dosage compensation and beyond. Curr. Opin. Genet. Dev. 2015, 31, 1–11. [Google Scholar] [CrossRef]
- Hallacli, E.; Lipp, M.; Georgiev, P.; Spielman, C.; Cusack, S.; Akhtar, A.; Kadlec, J. Msl1-mediated dimerization of the dosage compensation complex is essential for male X-chromosome regulation in Drosophila. Mol. Cell. 2012, 48, 587–600. [Google Scholar] [CrossRef]
- Kadlec, J.; Hallacli, E.; Lipp, M.; Holz, H.; Sanchez-Weatherby, J.; Cusack, S.; Akhtar, A. Structural basis for MOF and MSL3 recruitment into the dosage compensation complex by MSL1. Nat. Struct. Mol. Biol. 2011, 18, 142–149. [Google Scholar] [CrossRef]
- Neal, K.C.; Pannuti, A.; Smith, E.R.; Lucchesi, J.C. A new human member of the MYST family of histone acetyl transferases with high sequence similarity to Drosophila MOF. Biochim. Biophys. Acta 2000, 1490, 170–174. [Google Scholar] [CrossRef]
- Wei, T.; Liu, H.; Zhu, H.; Chen, W.; Wu, T.; Bai, Y.; Zhang, X.; Miao, Y.; Wang, F.; Cai, Y.; et al. Two distinct males absent on the first (MOF)-containing histone acetyltransferases are involved in the epithelial-mesenchymal transition in different ways in human cells. Cell. Mol. Life Sci. 2022, 79, 238. [Google Scholar] [CrossRef]
- Singh, M.; Bacolla, A.; Chaudhary, S.; Hunt, C.R.; Pandita, S.; Chauhan, R.; Gupta, A.; Tainer, J.A.; Pandita, T.K. Histone Acetyltransferase MOF Orchestrates Outcomes at the Crossroad of Oncogenesis, DNA Damage Response, Proliferation, and Stem Cell Development. Mol. Cell. Biol. 2020, 40, e00232-20. [Google Scholar] [CrossRef]
- Mujoo, K.; Pandita, R.K.; Tiwari, A.; Charaka, V.; Chakraborty, S.; Singh, D.K.; Hambarde, S.; Hittelman, W.N.; Horikoshi, N.; Hunt, C.R.; et al. Differentiation of Human Induced Pluripotent or Embryonic Stem Cells Decreases the DNA Damage Repair by Homologous Recombination. Stem Cell Rep. 2017, 9, 1660–1674. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, R.; Wu, D.; Lu, Z.; Sun, W.; Cai, Y.; Wang, C.; Jin, J. Epigenetic change in kidney tumor: Downregulation of histone acetyltransferase MYST1 in human renal cell carcinoma. J. Exp. Clin. Cancer Res. 2013, 32, 8. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Zhu, L.; Yang, J.; Su, J.; Ni, J.; Du, Y.; Liu, D.; Wang, Y.; Wang, F.; Jin, J.; et al. Correlation of low expression of hMOF with clinicopathological features of colorectal carcinoma, gastric cancer and renal cell carcinoma. Int. J. Oncol. 2014, 44, 1207–1214. [Google Scholar] [CrossRef]
- Sykes, S.M.; Mellert, H.S.; Holbert, M.A.; Li, K.; Marmorstein, R.; Lane, W.S.; McMahon, S.B. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell. 2006, 24, 841–851. [Google Scholar] [CrossRef]
- Chen, Z.; Ye, X.; Tang, N.; Shen, S.; Li, Z.; Niu, X.; Lu, S.; Xu, L. The histone acetylranseferase hMOF acetylates Nrf2 and regulates anti-drug responses in human non-small cell lung cancer. Br. J. Pharmacol. 2014, 171, 3196–3211. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Schmitz, K.M.; Mayer, C.; Yuan, X.; Akhtar, A.; Grummt, I. Reversible acetylation of the chromatin remodelling complex NoRC is required for non-coding RNA-dependent silencing. Nat. Cell Biol. 2009, 11, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wei, T.; Sun, L.; Wu, T.; Li, F.; Zhao, J.; Chu, J.; Wang, F.; Cai, Y.; Jin, J. The Non-Specific Lethal (NSL) Histone Acetyltransferase Complex Transcriptionally Regulates Yin Yang 1-Mediated Cell Proliferation in Human Cells. Int. J. Mol. Sci. 2022, 23, 3801. [Google Scholar] [CrossRef]
- Daraiseh, S.I.; Kassardjian, A.; Alexander, K.E.; Rizkallah, R.; Hurt, M.M. c-Abl phosphorylation of Yin Yang 1’s conserved tyrosine 254 in the spacer region modulates its transcriptional activity. Biochim. Biophys. Acta Mol. Cell. Res. 2018, 1865, 1173–1186. [Google Scholar] [CrossRef]
- Jeong, H.M.; Lee, S.H.; Yum, J.; Yeo, C.Y.; Lee, K.Y. Smurf2 regulates the degradation of YY1. Biochim. Biophys. Acta 2014, 1843, 2005–2011. [Google Scholar] [CrossRef]
- Addicks, G.C.; Zhang, H.; Ryu, D.; Vasam, G.; Green, A.E.; Marshall, P.L.; Patel, S.; Kang, B.E.; Kim, D.; Katsyuba, E.; et al. GCN5 maintains muscle integrity by acetylating YY1 to promote dystrophin expression. J. Cell Biol. 2022, 221, e20214022. [Google Scholar] [CrossRef]
- Hiromura, M.; Choi, C.H.; Sabourin, N.A.; Jones, H.; Bachvarov, D.; Usheva, A. YY1 is regulated by O-linked N-acetylglucosaminylation (O-glcNAcylation). J. Biol. Chem. 2003, 278, 14046–14052. [Google Scholar] [CrossRef]
- Hongo, F.; Garban, H.; Huerta-Yepez, S.; Vega, M.; Jazirehi, A.R.; Mizutani, Y.; Miki, T.; Bonavida, B. Inhibition of the transcription factor Yin Yang 1 activity by S-nitrosation. Biochem. Biophys. Res. Commun. 2005, 336, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wan, M.; Sui, G. PIASy-mediated sumoylation of Yin Yang 1 depends on their interaction but not the RING finger. Mol. Cell. Biol. 2007, 27, 3780–3792. [Google Scholar] [CrossRef]
- Griesenbeck, J.; Ziegler, M.; Tomilin, N.; Schweiger, M.; Oei, S.L. Stimulation of the catalytic activity of poly(ADP-ribosyl) transferase by transcription factor Yin Yang 1. FEBS Lett. 1999, 443, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Lee, K.J. Post-translational modifications and their biological functions: Proteomic analysis and systematic approaches. J. Biochem. Mol. Biol. 2004, 37, 35–44. [Google Scholar] [CrossRef]
- Venne, A.S.; Kollipara, L.; Zahedi, R.P. The next level of complexity: Crosstalk of posttranslational modifications. Proteomics 2014, 14, 513–524. [Google Scholar] [CrossRef]
- Caron, C.; Boyault, C.; Khochbin, S. Regulatory cross-talk between lysine acetylation and ubiquitination: Role in the control of protein stability. Bioessays 2005, 27, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yang, Y.; Chen, B.; Guo, X.; Gao, S.; Wang, M.; Duan, M.; Li, X. MOF Regulates TNK2 Transcription Expression to Promote Cell Proliferation in Thyroid Cancer. Front. Pharmacol. 2020, 11, 607605. [Google Scholar] [CrossRef]
- Gaub, A.; Sheikh, B.N.; Basilicata, M.F.; Vincent, M.; Nizon, M.; Colson, C.; Bird, M.J.; Bradner, J.E.; Thevenon, J.; Boutros, M.; et al. Evolutionary conserved NSL complex/BRD4 axis controls transcription activation via histone acetylation. Nat. Commun. 2020, 11, 2243. [Google Scholar] [CrossRef]
- Liu, Y.; Du, J.; Liu, X.; Wang, L.; Han, Y.; Huang, C.; Liang, R.; Zheng, F.; Shi, G.; Li, B. MG149 inhibits histone acetyltransferase KAT8-mediated IL-33 acetylation to alleviate allergic airway inflammation and airway hyperresponsiveness. Signal. Transduct. Target Ther. 2021, 6, 321. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Cheng, Z.; Zhu, J.; Xu, W.; Peng, X.; Chen, C.; Li, W.; Wang, F.; Cao, L.; Yi, X.; et al. Suberoylanilide hydroxamic acid treatment reveals crosstalks among proteome, ubiquitylome and acetylome in non-small cell lung cancer A549 cell line. Sci. Rep. 2015, 5, 9520. [Google Scholar] [CrossRef] [PubMed]
- Sarvagalla, S.; Kolapalli, S.P.; Vallabhapurapu, S. The Two Sides of YY1 in Cancer: A Friend and a Foe. Front. Oncol. 2019, 9, 1230. [Google Scholar] [CrossRef]
- Sui, Y.; Wu, T.; Li, F.; Wang, F.; Cai, Y.; Jin, J. YY1/BCCIP Coordinately Regulates P53-Responsive Element (p53RE)-Mediated Transactivation of p21(Waf1/Cip1). Int. J. Mol. Sci. 2019, 20, 2095. [Google Scholar] [CrossRef]
- Cai, Y.; Jin, J.; Yao, T.; Gottschalk, A.J.; Swanson, S.K.; Wu, S.; Shi, Y.; Washburn, M.P.; Florens, L.; Conaway, R.C.; et al. YY1 functions with INO80 to activate transcription. Nat. Struct. Mol. Biol. 2007, 14, 872–874. [Google Scholar] [CrossRef]
- Borlado, L.R.; Méndez, J. CDC6: From DNA replication to cell cycle checkpoints and oncogenesis. Carcinogenesis 2008, 29, 237–243. [Google Scholar] [CrossRef]
- Zhang, Q.; Stovall, D.B.; Inoue, K.; Sui, G. The oncogenic role of Yin Yang 1. Crit. Rev. Oncog. 2011, 16, 163–197. [Google Scholar] [CrossRef]
- Verheul, T.C.J.; van Hijfte, L.; Perenthaler, E.; Barakat, T.S. The Why of YY1: Mechanisms of Transcriptional Regulation by Yin Yang 1. Front. Cell. Dev. Biol. 2020, 8, 592164. [Google Scholar] [CrossRef]
- Feng, L.; Chen, M.; Li, Y.; Li, M.; Hu, S.; Zhou, B.; Zhu, L.; Yu, L.; Zhou, Q.; Tan, L.; et al. Sirt1 deacetylates and stabilizes p62 to promote hepato-carcinogenesis. Cell Death Dis. 2021, 12, 405. [Google Scholar] [CrossRef]
- Kruse, J.P.; Gu, W. MSL2 promotes Mdm2-independent cytoplasmic localization of p53. J. Biol. Chem. 2009, 284, 3250–3263. [Google Scholar] [CrossRef] [PubMed]
- Sui, G.; Affar el, B.; Shi, Y.; Brignone, C.; Wall, N.R.; Yin, P.; Donohoe, M.; Luke, M.P.; Calvo, D.; Grossman, S.R.; et al. Yin Yang 1 is a negative regulator of p53. Cell 2004, 117, 859–872. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, T.; Zhao, B.; Cai, C.; Chen, Y.; Miao, Y.; Chu, J.; Sui, Y.; Li, F.; Chen, W.; Cai, Y.; et al. The Males Absent on the First (MOF) Mediated Acetylation Alters the Protein Stability and Transcriptional Activity of YY1 in HCT116 Cells. Int. J. Mol. Sci. 2023, 24, 8719. https://doi.org/10.3390/ijms24108719
Wu T, Zhao B, Cai C, Chen Y, Miao Y, Chu J, Sui Y, Li F, Chen W, Cai Y, et al. The Males Absent on the First (MOF) Mediated Acetylation Alters the Protein Stability and Transcriptional Activity of YY1 in HCT116 Cells. International Journal of Molecular Sciences. 2023; 24(10):8719. https://doi.org/10.3390/ijms24108719
Chicago/Turabian StyleWu, Tingting, Bingxin Zhao, Chengyu Cai, Yuyang Chen, Yujuan Miao, Jinmeng Chu, Yi Sui, Fuqiang Li, Wenqi Chen, Yong Cai, and et al. 2023. "The Males Absent on the First (MOF) Mediated Acetylation Alters the Protein Stability and Transcriptional Activity of YY1 in HCT116 Cells" International Journal of Molecular Sciences 24, no. 10: 8719. https://doi.org/10.3390/ijms24108719
APA StyleWu, T., Zhao, B., Cai, C., Chen, Y., Miao, Y., Chu, J., Sui, Y., Li, F., Chen, W., Cai, Y., Wang, F., & Jin, J. (2023). The Males Absent on the First (MOF) Mediated Acetylation Alters the Protein Stability and Transcriptional Activity of YY1 in HCT116 Cells. International Journal of Molecular Sciences, 24(10), 8719. https://doi.org/10.3390/ijms24108719