
Semaglutide Has Beneficial Effects on Non-Alcoholic Steatohepatitis in Ldlr-/-.Leiden Mice

 , , , , , , ,
, , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
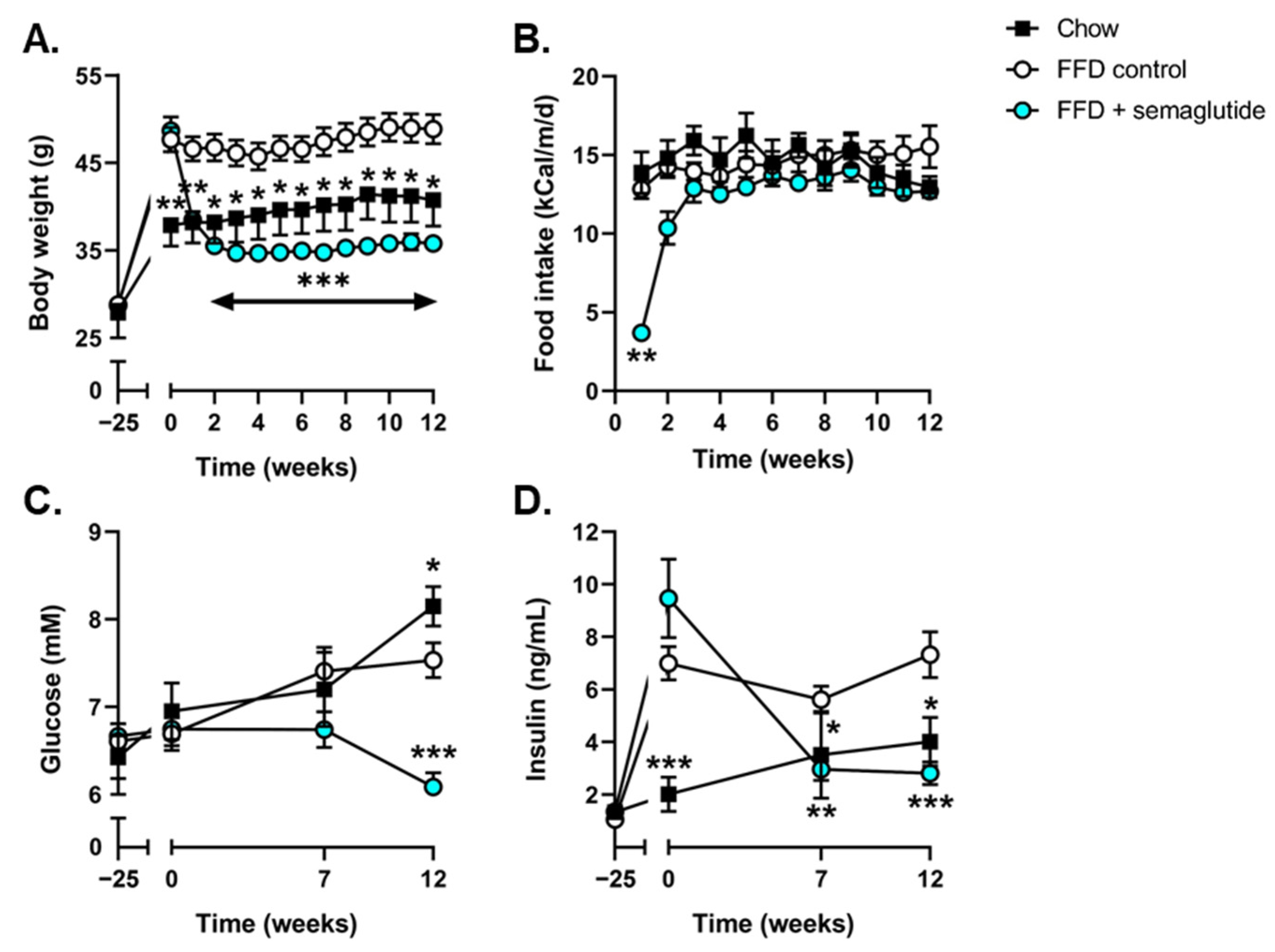
2.1. Semaglutide Improves Metabolic Parameters in Ldlr-/-.Leiden Mice
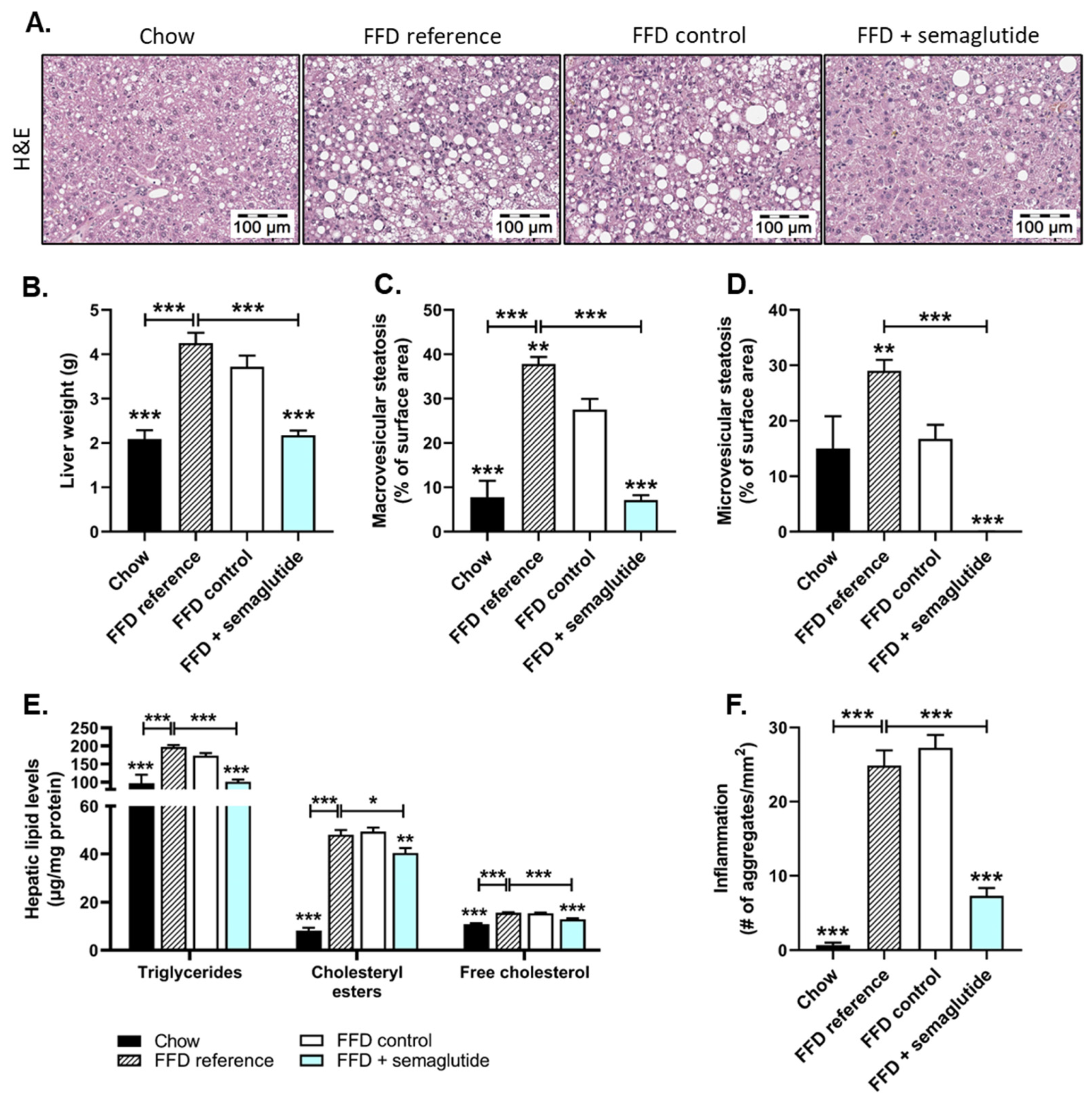
2.2. Treatment with Semaglutide Has Strong Beneficial Effects on Hepatic Steatosis and Inflammation
2.3. Semaglutide Does Not Affect Overall Liver Fibrosis Yet Does Improve Collagen Network Architecture
2.4. Semaglutide Has Minimal Effects on Severe Atherosclerosis
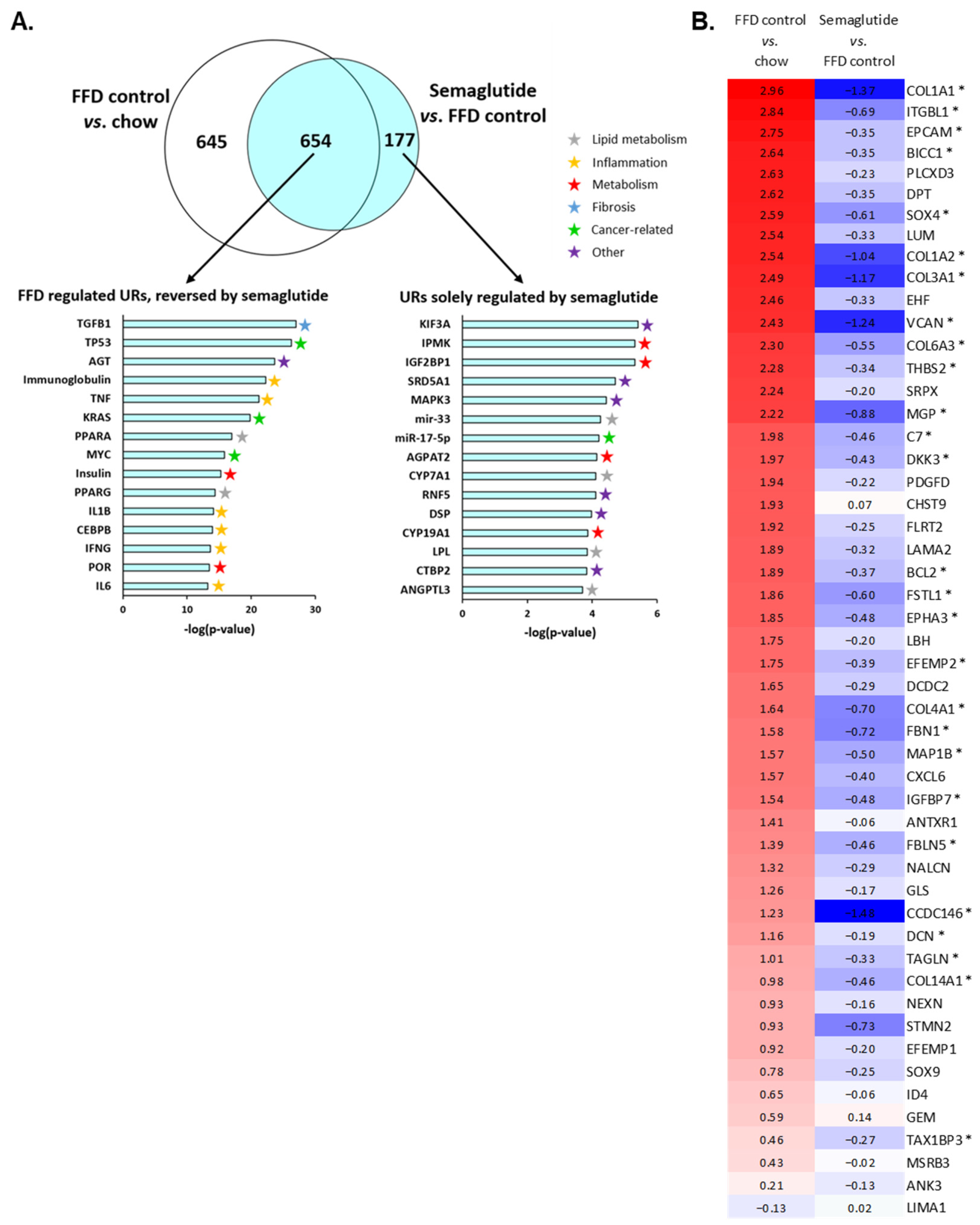
2.5. Ldlr-/-.Leiden Mice on FFD Closely Represent Human NASH and Semaglutide Improves Hepatic Gene Expression
3. Discussion
4. Materials and Methods
4.1. Experimental Design
4.2. Plasma and Liver Biochemical Analyses
4.3. Histological Assessment of NASH
4.4. Histological Assessment of Atherosclerosis
4.5. Transcriptome Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gautier, J.F.; Choukem, S.P.; Girard, J. Physiology of incretins (GIP and GLP-1) and abnormalities in type 2 diabetes. Diabetes Metab. 2008, 34, S65–S72. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef] [PubMed]
- Ahrén, B.; Dimas, A.L.; Miossec, P.; Saubadu, S.; Aronson, R. Efficacy and safety of lixisenatide once-daily morning or evening injections in type 2 diabetes inadequately controlled on metformin (GetGoal-M). Diabetes Care 2013, 36, 2543–2550. [Google Scholar] [CrossRef] [PubMed]
- Wysham, C.; Blevins, T.; Arakaki, R.; Colon, G.; Garcia, P.; Atisso, C.; Kuhstoss, D.; Lakshmanan, M. Efficacy and safety of dulaglutide added onto pioglitazone and metformin versus exenatide in type 2 diabetes in a randomized controlled trial (AWARD-1). Diabetes Care 2014, 37, 2159–2167. [Google Scholar] [CrossRef]
- Knudsen, L.B.; Lau, J. The discovery and development of liraglutide and semaglutide. Front. Endocrinol. 2019, 10, 155. [Google Scholar] [CrossRef]
- Blundell, J.; Finlayson, G.; Axelsen, M.; Flint, A.; Gibbons, C.; Kvist, T.; Hjerpsted, J.B. Effects of once-weekly semaglutide on appetite, energy intake, control of eating, food preference and body weight in subjects with obesity. Diabetes Obes. Metab. 2017, 19, 1242–1251. [Google Scholar] [CrossRef]
- Pratley, R.E.; Aroda, V.R.; Lingvay, I.; Lüdemann, J.; Andreassen, C.; Navarria, A.; Viljoen, A. Semaglutide versus dulaglutide once weekly in patients with type 2 diabetes (SUSTAIN 7): A randomised, open-label, phase 3b trial. Lancet Diabetes Endocrinol. 2018, 6, 275–286. [Google Scholar] [CrossRef]
- Ahmann, A.J.; Capehorn, M.; Charpentier, G.; Dotta, F.; Henkel, E.; Lingvay, I.; Holst, A.G.; Annett, M.P.; Aroda, V.R. Efficacy and Safety of Once-Weekly Semaglutide Versus Exenatide ER in Subjects with Type 2 Diabetes (SUSTAIN 3): A 56-Week, Open-Label, Randomized Clinical Trial. Diabetes Care 2018, 41, 258–266. [Google Scholar] [CrossRef]
- Davies, M.; Færch, L.; Jeppesen, O.K.; Pakseresht, A.; Pedersen, S.D.; Perreault, L.; Rosenstock, J.; Shimomura, I.; Viljoen, A.; Wadden, T.A.; et al. Semaglutide 2.4 mg once a week in adults with overweight or obesity, and type 2 diabetes (STEP 2): A randomised, double-blind, double-dummy, placebo-controlled, phase 3 trial. Lancet 2021, 397, 971–984. [Google Scholar] [CrossRef]
- Wadden, T.A.; Bailey, T.S.; Billings, L.K.; Davies, M.; Frias, J.P.; Koroleva, A.; Lingvay, I.; O’Neil, P.M.; Rubino, D.M.; Skovgaard, D.; et al. Effect of Subcutaneous Semaglutide vs Placebo as an Adjunct to Intensive Behavioral Therapy on Body Weight in Adults with Overweight or Obesity: The STEP 3 Randomized Clinical Trial. JAMA 2021, 325, 1403–1413. [Google Scholar] [CrossRef]
- Rubino, D.M.; Greenway, F.L.; Khalid, U.; O’Neil, P.M.; Rosenstock, J.; Sørrig, R.; Wadden, T.A.; Wizert, A.; Garvey, W.T. Effect of Weekly Subcutaneous Semaglutide vs Daily Liraglutide on Body Weight in Adults with Overweight or Obesity without Diabetes: The STEP 8 Randomized Clinical Trial. JAMA 2022, 327, 138–150. [Google Scholar] [CrossRef]
- Kadowaki, T.; Isendahl, J.; Khalid, U.; Lee, S.Y.; Nishida, T.; Ogawa, W.; Tobe, K.; Yamauchi, T.; Lim, S. Semaglutide once a week in adults with overweight or obesity, with or without type 2 diabetes in an east Asian population (STEP 6): A randomised, double-blind, double-dummy, placebo-controlled, phase 3a trial. Lancet Diabetes Endocrinol. 2022, 10, 193–206. [Google Scholar] [CrossRef]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.S.; Harrison, S.A. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef]
- Flint, A.; Andersen, G.; Hockings, P.; Johansson, L.; Morsing, A.; Sundby Palle, M.; Vogl, T.; Loomba, R.; Plum-Mörschel, L. Randomised clinical trial: Semaglutide versus placebo reduced liver steatosis but not liver stiffness in subjects with non-alcoholic fatty liver disease assessed by magnetic resonance imaging. Aliment. Pharmacol. Ther. 2021, 54, 1150–1161. [Google Scholar] [CrossRef]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; Van den Hoek, A.M. Establishment of a General NAFLD Scoring System for Rodent Models and Comparison to Human Liver Pathology. PLoS ONE 2014, 9, 1072. [Google Scholar] [CrossRef]
- Morrison, M.C.; Mulder, P.; Salic, K.; Verheij, J.; Liang, W.; Van Duyvenvoorde, W.; Menke, A.; Kooistra, T.; Kleemann, R.; Wielinga, P.Y. Intervention with a caspase-1 inhibitor reduces obesity-associated hyperinsulinemia, non-alcoholic steatohepatitis and hepatic fibrosis in LDLR−/−.Leiden mice. Int. J. Obes. 2016, 40, 1416–1423. [Google Scholar] [CrossRef]
- Van Koppen, A.; Verschuren, L.; Van den Hoek, A.M.; Verheij, J.; Morrison, M.C.; Li, K.; Nagabukuro, H.; Costessi, A.; Caspers, M.P.M.; Van den Broek, T.J.; et al. Uncovering a Predictive Molecular Signature for the Onset of NASH-Related Fibrosis in a Translational NASH Mouse Model. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 83–98. [Google Scholar] [CrossRef]
- Morrison, M.C.; Verschuren, L.; Salic, K.; Verheij, J.; Menke, A.; Wielinga, P.Y.; Iruarrizaga-Lejarreta, M.; Gole, L.; Yu, W.M.; Turner, S.; et al. Obeticholic Acid Modulates Serum Metabolites and Gene Signatures Characteristic of Human NASH and Attenuates Inflammation and Fibrosis Progression in Ldlr-/-.Leiden Mice. Hepatol. Commun. 2018, 2, 1513–1532. [Google Scholar] [CrossRef]
- Martínez-Arranz, I.; Bruzzone, C.; Noureddin, M.; Gil-Redondo, R.; Mincholé, I.; Bizkarguenaga, M.; Arretxe, E.; Iruarrizaga-Lejarreta, M.; Fernández-Ramos, D.; Lopitz-Otsoa, F.; et al. Metabolic subtypes of patients with NAFLD exhibit distinctive cardiovascular risk profiles. Hepatology 2022, 76, 1121–1134. [Google Scholar] [CrossRef]
- Van den Hoek, A.M.; Verschuren, L.; Worms, N.; Van Nieuwkoop, A.; De Ruiter, C.; Attema, J.; Menke, A.L.; Caspers, M.P.M.; Radhakrishnan, S.; Salic, K.; et al. A Translational Mouse Model for NASH with Advanced Fibrosis and Atherosclerosis Expressing Key Pathways of Human Pathology. Cells 2020, 9, 2014. [Google Scholar] [CrossRef]
- Gart, E.; Van Duyvenvoorde, W.; Caspers, M.P.M.; Van Trigt, N.; Snabel, J.; Menke, A.; Keijer, J.; Salic, K.; Morrison, M.C.; Kleemann, R. Intervention with isoleucine or valine corrects hyperinsulinemia and reduces intrahepatic diacylglycerols, liver steatosis, and inflammation in Ldlr−/−.Leiden mice with manifest obesity-associated NASH. FASEB J. 2022, 36, e22435. [Google Scholar] [CrossRef] [PubMed]
- Moylan, C.A.; Pang, H.; Dellinger, A.; Suzuki, A.; Garrett, M.E.; Guy, C.D.; Murphy, S.K.; Ashley-Koch, A.E.; Choi, S.S.; Michelotti, G.A.; et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014, 59, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Teufel, A.; Itzel, T.; Erhart, W.; Brosch, M.; Wang, X.Y.; Kim, Y.O.; Von Schönfels, W.; Herrmann, A.; Brückner, S.; Stickel, F.; et al. Comparison of Gene Expression Patterns between Mouse Models of Nonalcoholic Fatty Liver Disease and Liver Tissues from Patients. Gastroenterology 2016, 151, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Charlton, M.; Krishnan, A.; Viker, K.; Sanderson, S.; Cazanave, S.; McConico, A.; Masuoko, H.; Gores, G. Fast food diet mouse: Novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am. J Physiol.-Gastrointest. Liver Physiol. 2011, 301, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef]
- Maeso-Díaz, R.; Boyer-Diaz, Z.; Lozano, J.J.; Ortega-Ribera, M.; Peralta, C.; Bosch, J.; Gracia-Sancho, J. New Rat Model of Advanced NASH Mimicking Pathophysiological Features and Transcriptomic Signature of the Human Disease. Cells 2019, 8, 1062. [Google Scholar] [CrossRef]
- Han, J.E.; Shin, H.B.; Ahn, Y.H.; Cho, H.J.; Cheong, J.Y.; Park, B.; Kim, S.S. Relationship between the dynamics of non-alcoholic fatty liver disease and incident diabetes mellitus. Sci. Rep. 2022, 12, 2538. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Meier, J.J. GLP-1 receptor agonists in the treatment of type 2 diabetes—State-of-the-art. Mol. Metab. 2021, 46, 101102. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef]
- Gabery, S.; Salinas, C.G.; Paulsen, S.J.; Ahnfelt-Rønne, J.; Alanentalo, T.; Baquero, A.F.; Buckley, S.T.; Farkas, E.; Fekete, C.; Frederiksen, K.S.; et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight 2021, 5, e133429. [Google Scholar] [CrossRef]
- Møllerhøj, M.B.; Veidal, S.S.; Thrane, K.T.; Oró, D.; Overgaard, A.; Salinas, C.G.; Madsen, M.R.; Pfisterer, L.; Vyberg, M.; Simon, E.; et al. Hepatoprotective effects of semaglutide, lanifibranor and dietary intervention in the GAN diet-induced obese and biopsy-confirmed mouse model of NASH. Clin. Transl. Sci. 2022, 15, 1167–1186. [Google Scholar] [CrossRef]
- Smits, M.M.; Van Raalte, D.H. Safety of Semaglutide. Front. Endocrinol. 2021, 12, 496. [Google Scholar] [CrossRef]
- Kapitza, C.; Dahl, K.; Jacobsen, J.B.; Axelsen, M.B.; Flint, A. Effects of semaglutide on beta cell function and glycaemic control in participants with type 2 diabetes: A randomised, double-blind, placebo-controlled trial. Diabetologia 2017, 60, 1390. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Hull, D.; Guo, K.; Barton, D.; Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Yu, J.; Gough, S.C.; Newsome, P.N.; et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J. Hepatol. 2016, 64, 399–408. [Google Scholar] [CrossRef]
- Rodbard, H.W.; Rosenstock, J.; Canani, L.H.; Deerochanawong, C.; Gumprecht, J.; Lindberg, S.Ø.; Lingvay, I.; Søndergaard, A.L.; Treppendahl, M.B.; Montanya, E. Oral Semaglutide Versus Empagliflozin in Patients with Type 2 Diabetes Uncontrolled on Metformin: The PIONEER 2 Trial. Diabetes Care 2019, 42, 2272–2281. [Google Scholar] [CrossRef]
- Wang, Y.; Parlevliet, E.T.; Geerling, J.J.; Van der Tuin, S.J.L.; Zhang, H.; Bieghs, V.; Jawad, A.H.M.; Shiri-Sverdlov, R.; Bot, I.; De Jager, S.C.A.; et al. Exendin-4 decreases liver inflammation and atherosclerosis development simultaneously by reducing macrophage infiltration. Br. J. Pharmacol. 2014, 171, 723–734. [Google Scholar] [CrossRef]
- Rakipovski, G.; Rolin, B.; Nøhr, J.; Klewe, I.; Frederiksen, K.S.; Augustin, R.; Hecksher-Sørensen, J.; Ingvorsen, C.; Polex-Wolf, J.; Knudsen, L.B. The GLP-1 Analogs Liraglutide and Semaglutide Reduce Atherosclerosis in ApoE -/- and LDLr -/- Mice by a Mechanism That Includes Inflammatory Pathways. JACC 2018, 3, 844–857. [Google Scholar] [CrossRef]
- Loomba, R.; Abdelmalek, M.F.; Armstrong, M.; Jara, M.; Kjaer, M.; Krarup, N.; Lawitz, E.; Ratziu, V.; Sanyal, A.; Schattenberg, J.; et al. Semaglutide 2.4 mg once weekly improved liver and metabolic parameters, and was well tolerated, in patients with non-alcoholic steatohepatitis-related cirrhosis: A randomised, placebo-controlled phase 2 trial. J. Hepatol. Int. Liver Congr. 2022, 77, S10. [Google Scholar] [CrossRef]
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.; Anania, F.A. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006, 43, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Aviv, V.; Meivar-Levy, I.; Rachmut, I.H.; Rubinek, T.; Mor, E.; Ferber, S. Exendin-4 promotes liver cell proliferation and enhances the PDX-1-induced liver to pancreas transdifferentiation process. J. Biol. Chem. 2009, 284, 33509–33520. [Google Scholar] [CrossRef] [PubMed]
- Tomas, E.; Stanojevic, V.; Habener, J.F. GLP-1 (9-36) amide metabolite suppression of glucose production in isolated mouse hepatocytes. Horm. Metab. Res. 2010, 42, 657–662. [Google Scholar] [CrossRef]
- Panjwani, N.; Mulvihill, E.E.; Longuet, C.; Yusta, B.; Campbell, J.E.; Brown, T.J.; Streutker, C.; Holland, D.; Cao, X.; Baggio, L.L.; et al. GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE(-/-) mice. Endocrinology 2013, 154, 127–139. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378. [Google Scholar] [CrossRef]
- Jacobs, S.A.H.; Gart, E.; Vreeken, D.; Franx, B.A.A.; Wekking, L.; Verweij, V.G.M.; Worms, N.; Schoemaker, M.H.; Gross, G.; Morrison, M.C.; et al. Sex-Specific Differences in Fat Storage, Development of Non-Alcoholic Fatty Liver Disease and Brain Structure in Juvenile HFD-Induced Obese Ldlr-/-.Leiden Mice. Nutrients 2019, 11, 1861. [Google Scholar] [CrossRef]
- Erdfelder, E.; Faul, F.; Buchner, A. GPOWER: A general power analysis program. Behav. Res. Methods Instrum. Comput. 1996, 28, 1–11. [Google Scholar] [CrossRef]
- Post, S.M.; De Crom, R.; Van Haperen, R.; Van Tol, A.; Princen, H.M.G. Increased fecal bile acid excretion in transgenic mice with elevated expression of human phospholipid transfer protein. Arter. Thromb. Vasc. Biol. 2003, 23, 892–897. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu. Rev. Pathol. 2010, 5, 145–171. [Google Scholar] [CrossRef]
- Pouwer, M.G.; Pieterman, E.J.; Worms, N.; Keijzer, N.; Jukema, J.W.; Gromada, J.; Gusarova, V.; Princen, H.M.G. Alirocumab, evinacumab, and atorvastatin triple therapy regresses plaque lesions and improves lesion composition in mice. J. Lipid Res. 2020, 61, 365. [Google Scholar] [CrossRef]
- Kühnast, S.; Van Der Hoorn, J.W.A.; Van Den Hoek, A.M.; Havekes, L.M.; Liau, G.; Jukema, J.W.; Princen, H.M.G. Aliskiren inhibits atherosclerosis development and improves plaque stability in APOE*3Leiden.CETP transgenic mice with or without treatment with atorvastatin. J. Hypertens. 2012, 30, 107–116. [Google Scholar] [CrossRef]
- Kühnast, S.; Van der Tuin, S.J.L.; Van der Hoorn, J.W.A.; Van Klinken, J.B.; Simic, B.; Pieterman, E.J.; Havekes, L.M.; Landmesser, U.; Lüscher, T.F.; Van Dijk, K.W.; et al. Anacetrapib reduces progression of atherosclerosis, mainly by reducing non-HDL-cholesterol, improves lesion stability and adds to the beneficial effects of atorvastatin. Eur. Heart J. 2015, 36, 39–48. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inia, J.A.; Stokman, G.; Morrison, M.C.; Worms, N.; Verschuren, L.; Caspers, M.P.M.; Menke, A.L.; Petitjean, L.; Chen, L.; Petitjean, M.; et al. Semaglutide Has Beneficial Effects on Non-Alcoholic Steatohepatitis in Ldlr-/-.Leiden Mice. Int. J. Mol. Sci. 2023, 24, 8494. https://doi.org/10.3390/ijms24108494
Inia JA, Stokman G, Morrison MC, Worms N, Verschuren L, Caspers MPM, Menke AL, Petitjean L, Chen L, Petitjean M, et al. Semaglutide Has Beneficial Effects on Non-Alcoholic Steatohepatitis in Ldlr-/-.Leiden Mice. International Journal of Molecular Sciences. 2023; 24(10):8494. https://doi.org/10.3390/ijms24108494
Chicago/Turabian StyleInia, José A., Geurt Stokman, Martine C. Morrison, Nicole Worms, Lars Verschuren, Martien P. M. Caspers, Aswin L. Menke, Louis Petitjean, Li Chen, Mathieu Petitjean, and et al. 2023. "Semaglutide Has Beneficial Effects on Non-Alcoholic Steatohepatitis in Ldlr-/-.Leiden Mice" International Journal of Molecular Sciences 24, no. 10: 8494. https://doi.org/10.3390/ijms24108494
APA StyleInia, J. A., Stokman, G., Morrison, M. C., Worms, N., Verschuren, L., Caspers, M. P. M., Menke, A. L., Petitjean, L., Chen, L., Petitjean, M., Jukema, J. W., Princen, H. M. G., & van den Hoek, A. M. (2023). Semaglutide Has Beneficial Effects on Non-Alcoholic Steatohepatitis in Ldlr-/-.Leiden Mice. International Journal of Molecular Sciences, 24(10), 8494. https://doi.org/10.3390/ijms24108494