

Phytochemical Constituents and Derivatives of Cannabis sativa; Bridging the Gap in Melanoma Treatment



Abstract
1. Introduction
2. Factors Effecting Melanoma Classification, Onset, Progression, and Risk
2.1. Subtypes of Melanoma
2.2. Risk Factors of Melanoma
2.2.1. Reactive Oxygen Species
2.2.2. Chronic Inflammation
2.2.3. Reducing Oxidative Stress Risk
2.3. Genetic Influences of Melanoma
3. Treatment Outlooks for Melanoma
3.1. Surgical Implications
3.2. Pharmacological Treatments of Melanoma
3.2.1. Targeted Therapy
3.2.2. Immune Therapy
3.2.3. Multi-Drug Administration Effectiveness

{kind=link}
| Treatment | Patient Population | Effect on Survival | Side Effects | Reference |
|---|---|---|---|---|
| Dabrafenib + Trametinib | Unresectable or metastatic melanoma with BRAFV600E/K | ↑ Treatment response rate ↑ PFS in patients with advanced melanoma | Pyrexia, nausea, arthralgia, fatigue, diarrhea, chills, vomiting, headache | [83] |
| Nivolumab + Ipilimumab | Previously untreated advanced melanoma | ↑ OS ↑ PFS | High occurrence of gastrointestinal (diarrhea, colitis), skin-related (pruritus, rash) and pyrexia events | [84] |
| Vemurafenib + Cobimetinib | Previously untreated, unresectable locally advanced or metastatic BRAFV600 | ↑ PFS with BRAFV600 metastatic melanoma. ↓ Incidence of cutaneous secondary cancers | Rash, diarrhoea, photosensitivity, hepatic-enzyme abnormalities | [82] |
| Encorafenib + Binimetinib | Unresectable or metastatic melanoma with BRAF mutations | ↑ OS ↑ PFS | Nausea, diarrhoea, fatigue, arthralgia, omitting, pyrexia, and increased aspartate aminotransferase (AST) | [81] |
| Nivolumab + Relatimab | Previously untreated advanced melanoma | ↑ PFS | Well tolerated with a manageable safety profile | [110] |
3.3. Limitations of Existing Treatment Options
3.3.1. Treatment Resistance
3.3.2. Intolerance and Toxicity
3.3.3. Low Long-Term Survival Outlooks
3.3.4. Treatment Cost
3.4. Bridging the Gap in Melanoma Treatment
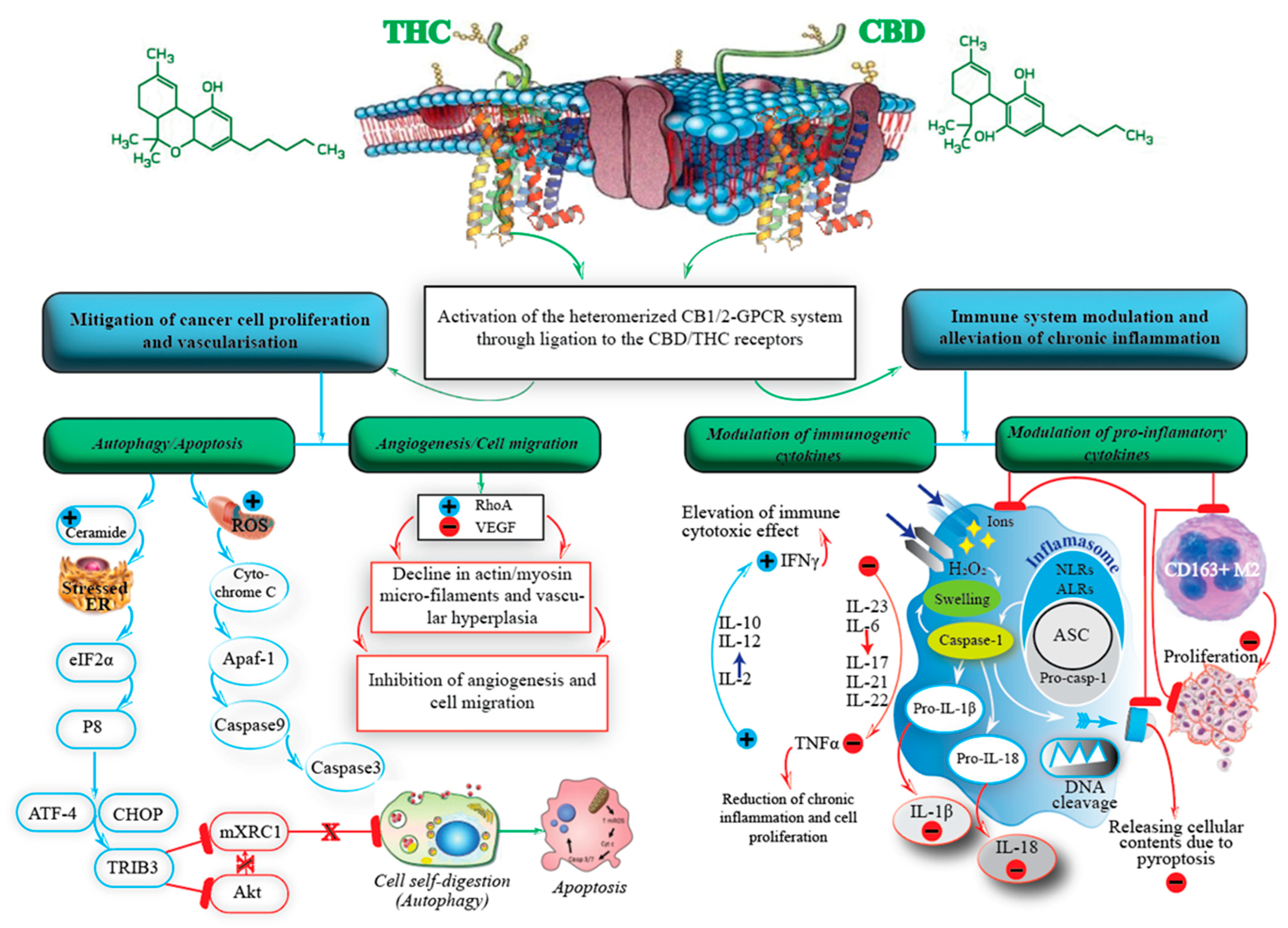
4. The Endocannabinoid System
5. Cannabis and Its Constituents
5.1. Classification of Cannabis
5.2. Classification of Cannabinoids
5.3. Endogenous Cannabinoids
5.4. Phytocannabinoids
5.5. Synthocannabinoids
| Cannabinoid | Cell Type | Mutation | In Vitro/ In Vivo | Dose | Effect | Receptor | Reference |
|---|---|---|---|---|---|---|---|
| Exogenous | |||||||
| THC | |||||||
| Synthetic | A375, SK-MEL-28, CHL-1 | BRAF, CDKN2A and TERT (A375) and BRAF, CDK4, CCLE, EGFR, PTEN, TERT, TP53 (SK-MEL-28) | In vitro | 1–5 μM | Primary melanocytes unaffected up to 6 μM THC ↓ Cell viability ↑ Apoptosis ↑ Beclin1 and Ambra1-independent autophagyRegardless of BRAF mutational status | N/A | [141] |
| CHL-1 injected athymic mice | CDKN2A, MAPK3, TERT, TP53 | In vivo | 15 mg kg−1 | ↓ Cell viability ↓ Tumour growth ↑ Apoptosis ↑ Antitumor response | N/A | [141] | |
| HCmel12 and B16 | DMBA-induced HGF-CDK4R24C melanoma (HCmel12) and spontaneous mouse mutant (B16) | In vitro | 5 and 10 μM | No effect | CB1, CB2 Low expression | [142] | |
| HCmel12 injected HGF-CDK4R24C and Cnr1/2−/− mice and B16 injected HGF-CDK4R24C mice | DMBA-induced HGF-CDK4R24C Cnr1/2−/− | In vivo | 5 mg/kg body weight | ↓ Tumour growth ↓ Inflammatory response ↓ Infiltration of CD45+ immune cells Tumour angiogenesis unaffected B16 and HCmel12 Cnr1/2−/− mice not significantly affected | CB receptor-dependent affect for HCmel12 injected mice, CB1, CB2 | [142] | |
| Plant based | B16, A375, MelJuso | Spontaneous mouse mutant (B16), BRAF, CDKN2A and TERT (A375), HRAS and NRAS (MelJuso) | In vitro | 1, 2, 2.5 and 3 μM | ↓ Viability ↓ Proliferation ↑ Apoptosis No effect on mouse melan-c and human Hermes 2b healthy melanocytes | CB1, CB2, Comparable CB1 expression in mouse melan-c and human Hermes 2b healthy melanocytes | [143] |
| CBD | B16 | Spontaneous mouse mutant | In vitro | 0.0016–0.2 mg/mL | ↓ Cell growth | N/A | [144] |
| B16F1, A375 | Spontaneous mouse mutant (B16), BRAF, CDKN2A and TERT (A375), HRAS and NRAS (MelJuso) | In vivo | 5 mg/kg twice per week | ↑ Survival duration ↑ Quality of life ↑ Movement ↓ Tumour growth | N/A | [121] | |
| CBG | Mouse skin melanoma cells | Unknown | In vitro | 31.31 µg/mL | ↓ Proliferation | N/A | [145] |
| WIN 55,212–2 (CB1/CB2 agonist) | COLO38, SK-MEL-28, OCM-1 | MPG antigen (COLO38), HLA-allotyped, BRAF, CDK4, EGFR, PTEN, TERT, TP53 (SK-MEL-28) | In vitro | 500 nM, 2 µM, 5 µM. | ↓ Cell growth ↑ Apoptosis | CB1 independent affect, CB2 independent affect, VR1 independent affect | [146] |
| B16, A375, MelJuso | Spontaneous mouse mutant (B16), BRAF, CDKN2A and TERT (A375), HRAS and NRAS (MelJuso) | In vitro | 100 nM | ↓ Cell viability ↓ Cell proliferation No effect on mouse melan-c and human Hermes 2b healthy melanocytes | CB receptor-dependent affect for B16 and A375, CB1, CB2, Comparable CB1 expression in mouse melan-c and human Hermes 2b healthy melanocytes | [144] | |
| B16 injected immune-deficient nude mice and C57BL/6 mice | Spontaneous mouse mutant | In vivo | 50 µg/day | ↓ Metastasis ↓ Tumour vascularisation ↓ Metastatic nodules in liver and lungs ↑ Apoptosis ↓ Tumour volume Independent of immune response Inhibition of cell cycle at G1-S transition ↓ Tumour proliferation Akt dependent inhibition | CB1, CB2, Comparable CB1 expression in mouse melan-c and human Hermes 2b healthy melanocytes | [144] | |
| JWH-133 (CB2-selective agonist) | A2058 | BRAF, TERT, TP53, TP63 | In vitro | 10 µM | ↓ Transendothelial migration ↓ Adhesion to brain endothelial cells Involvement of Gi/Goα subunits Downregulation of ICAM, VCAM and MMP | CB2 GPR55 and GPR119 independent | [147] |
| B16 injected Immune-deficient nude mice and C57BL/6 mice | Spontaneous mouse mutant | In vivo | 50 µg/day | ↓ Tumour volume ↓ Tumour vascularisation ↑ Apoptosis ↓ Tumour proliferation ↓ Cell cycle at G1-S transition | CB1, CB2, Comparable CB1 expression in mouse melan-c and human Hermes 2b healthy melanocytes | [143] | |
| OCM-1A, COLO38 | BRAF, CDKN2A and TP53 heterozygous (OCM-1A) MPG antigen (COLO38) | In vitro | 500, 2 and 5 µM | No effect | CB1, CB2 | [143] | |
| AM251 (CB1 receptor antagonist) | HT168-M1 | HLA-DR antigen | In vitro | 1–10 µM | ↑ Apoptosis ↓ Cell cycle at G2/M | CB1 | [148] |
| A375 | BRAF, CDKN2A and TERT | In vitro | 6 µM | No effect | CB1 | [149] | |
| OCM-1A, COLO38 | BRAF, CDKN2A, TP53 heterozygous (OCM-1A), MPG antigen (COLO38) | In vitro | 500, 2 and 5 µM | No effect | CB1 | [146] | |
| AM630 (CB2 receptor antagonist) | OCM-1A, COLO38 | BRAF, CDKN2A, TP53 heterozygous (OCM-1A), MPG antigen (COLO38) | In vitro | 1 µM | No effect | CB2 | [146] |
| Endocannabinoids | |||||||
| PEA | B16 in C57BL/6 mice | Spontaneous mouse mutant | In vitro | 1, 10 and 20 μM | ↑ Apoptosis ↓ Cell viability ↑ Cytotoxicity | CB1 | [150] |
| AEA | A375 | BRAF, CDKN2A and TERT (A375) | In vitro | 0.1–100 mM | ↓ Cell growth ↓ Cell viability ↑ Cytotoxicity ↑ Caspase-dependent apoptosis Via FAAH inhibition Mitigated by COX-2 and LOX inhibition Possible role of lipid raft and GPR55 | CB1, GPR55 | [149] |
| HT168-M1 | HLA-DR antigen | In vitro | 1–10 μM | ↑ Apoptosis ↓ Cell growth ↓ Metastasis ↓ Migration ↑ Necrosis Cell-cycle Arrest at G2/M | CB1 | [148] | |
| HT168-M1 in SCID mice | HLA-DR antigen | In vivo | 10–30 μM | ↓ Migration | CB1 | [148] | |
| Met-F-AEA (stable AEA analogue) | HT-168-M1, WM35, HT199 | HLA-DR antigen (HT-168-M1), BRAF (WM35), BRAF, TP53 (WM983B) | In vitro | 1–10 µM | ↓ Proliferation | CB1 | [148] |
| HT168-M1 in SCID mice | HLA-DR antigen | In vivo | 0.24 or 1.2 mg/kg | ↓ Cell growth ↓ Colonization ↓ Liver colonization ↓ Metastasis ↓ Migration No effect on tumour growth | CB1 | [148] | |
| Combined | |||||||
| THC + CBD (Sativex) | A375, SK-MEL-28, CHL-1 | BRAF, CDKN2A and TERT (A375) and BRAF, CDK4, EGFR, PTEN, TERT, TP53 (SK-MEL-28), CDKN2A, MAPK3, TERT, TP53 (CHL-1) | In vitro | 1:1 ratio of THC:CBD from 0.5–2.5 μM ideal dosage ratio selected at 1 μM THC+1 μM CBD | ↑ Apoptosis ↑ Beclin1 and Ambra1-independent autophagy Independent of BRAF mutational status ↓ Cell viability | Not assessed | [132] |
| CHL-1 in athymic nude mice | CDKN2A, MAPK3, TERT, TP53 | In vivo | 7.5 mg kg−1 of 1 μM THC + 1 μM CBD | ↑ Apoptosis ↑ Autophagy ↓ Tumour growth | N/A | [141] | |
| THC + Trametinib | WM35, A375, SK-MEL-28 | CD271 exogenously overexpressed (WM35) BRAF, CDKN2A and TERT (A375) and BRAF, CDK4, EGFR, PTEN, TERT, TP53 (SK-MEL-28) | In vitro | 5 µmol L−1 THC + 16 nmol L−1 Trametinib | ↓ Invasion ↓ Metastasis ↓ Viability | N/A | [151] |
| A375 in Zebrafish | BRAF, CDKN2A and TERT (A375) | In vivo | 5 µmol L−1 THC + 16 nmol L−1 trametinib | ↓Autophagy ↓ Invasion ↓ Metastasis ↓ Viability | N/A | [151] | |
| OCM-1A, COLO38 | BRAF, CDKN2A, TP53 heterozygous (OCM-1A), MPG antigen (COLO38) | In vitro | 500 nM, 2 µM, 5 µM | ↑ Apoptosis ↓ Cell viability Via lipid raft machinery Involves cleavage of caspases 9 and 7, ERK phosphorylation | CB1 independent affect, CB2 independent affect, VR1 independent affect | [146] | |
| WIN 55,212–2 + AM251 (CB1 receptor antagonist) + AM630 (CB2 receptor antagonist) | OCM-1A, COLO38 | BRAF, CDKN2A, TP53 heterozygous (OCM-1A), COLO38 | In vitro | WIN 500nM + AM251 1 µM + AM630 1 µM WIN 5 µM + AM251 1 µM + AM630 1 µM | ↓ Cell viability | CB1, CB2 | [146] |
| WIN 55,212–2 + AM251 (CB1 receptor antagonist) | OCM-1A, COLO38 | BRAF, CDKN2A, TP53 heterozygous (OCM-1A), COLO38 | In vitro | WIN 500nM + AM251 1 µM + AM630 1 µM WIN 5 µM + AM251 1 µM + AM630 1 µM | ↓ Cell viability | CB1, CB2 | [146] |
| WIN 55,212–2 + AM630 (CB2 receptor antagonist) | OCM-1A, COLO38 | BRAF, CDKN2A, TP53 heterozygous (OCM-1A), COLO38 | In vitro | WIN 500nM + AM630 1 µM WIN 5 µM + AM630 1 µM | ↓ Cell viability | CB1, CB2 | [146] |
| WIN 55,212–2 + SB36679 (VR1 receptor antagonist) | OCM-1A, COLO38 | BRAF, CDKN2A, TP53 heterozygous (OCM-1A), COLO38 | In vitro | 500 nM, 5 µM (WIN 55,212-2) + 20 nM (SB36679) | ↓ Cell viability | VR1 | [146] |
| AM251 (CB1 receptor antagonist) + AM630 (CB2 receptor antagonist) | OCM-1A, COLO38 | BRAF, CDKN2A, TP53 heterozygous (OCM-1A), COLO38 | In vitro | 1 µM of each | No effect | CB1, CB2 | [146] |
| JWH-133 + ACEA | HCmel12 and B1 | DMBA-induced HGF-CDK4R24C (HCmel12) and spontaneous mouse mutant (B16) | In vitro | 500 nM, 2 µM, 5 µM of each | No effect | CB1, CB2 | [142] |
| AM251 (CB1 receptor antagonist) + Celecoxib | A375 | BRAF, CDKN2A and TERT (A375) | In vitro | 0.1–10 µM (AM251) + 0.1–100 µM (Celecoxib) | ↑ Apoptosis ↓ Cell growth ↑ Cell-cycle arrest ↓ Expression of antiapoptotic BCL2 and surviving protein expression ↑ Expression of proapoptotic BAX gene ↓ Proliferative at the G2/M transition | CB1, GPR55, TRPA1, and COX-2 in the AM251 | [152] |
| PEA + URB597 (FAAH inhibitor) | B16 and MZ2-MEL.43 | Spontaneous mouse mutant (B16), NRAS, RAF1 heterozygous (MZ2-MEL.43) | In vitro | 10 μM (PEA) + 10 μM (URB597) | ↑ Apoptosis ↓ Cell growth ↑ Necrosis ↓Viability Cytotoxicity may involve PEA regulatory affects | Potential weak TRPV1 activation CB1, PPARα, PPARγ and GPR55 independent affect | [150] |
| B16 in C57BL/6 mice | Spontaneous mouse mutant | In vivo | Both 10 mg/kg/day | ↑ Apoptosis ↑ Necrosis ↓ Tumor volume ↓ Tumor progression Angiogenesis unaffected | Potential weak TRPV1 activation CB1, PPARα, PPARγ and GPR55 independent affect | [150] | |
| Met-F-AEA (stable AEA analogue) + AM251 (CB1 receptor antagonist) | HT168-M1 | HLA-DR antigen (HT-168-M1) | In vitro | 5 µM Met-F-AEA + 1–10 µM AM251 | ↑ Apoptosis ↓ Cell growth ↓ Cell migration ↓ Metastasis ↑ Necrosis Cell-cycle arrest at the G2/M transition | CB1 | [148] |
| AEA + URB597 (FAAH inhibitor) | A375 | BRAF, CDKN2A and TERT (A375) | In vitro | 0.1–100 mM (AEA) + 1 µM (URB597) | ↓ Cell viability | CB1, GPR55, TRPV1 independent | [149] |
5.6. Whole Cannabis Considerations
5.7. The Entourage Effect
5.8. Terpenoids
5.9. Other Polyphenols of Cannabis
6. Application of Cannabis Flavonoids in Melanoma
6.1. Cannflavins
6.2. Apigenin
6.3. Chrysin
6.4. Ferulic Acid
6.5. Genistein
6.6. Luteolin
6.7. P-Coumaric Acid
6.8. Quercetin
6.9. Rutin
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AJCC | American Joint Committee on Cancer |
| AEA-N | Arachidonoylethanolamine |
| 2-AG | 2-Arachidonoglycerol |
| CB1 | Cannabinoid receptor 1 |
| CB2 | Cannabinoid receptor 2 |
| CFL-A | Cannflavin A |
| CFL-B | Cannflavin B |
| CFL-C | Cannflavin C |
| CBD | Cannabidiol |
| CBG | Cannabigerol |
| CBN | Cannabinol |
| CBC | Cannabichromene |
| CBV | Cannabidivarin |
| CBL | Cannabicyclol |
| CBE | Cannabielsoin |
| CBDA | Cannabidiolic acid |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| CDK4 | Cyclin-dependent kinase 4 |
| CDK6 | Cyclin-dependent kinase 6 |
| CK2 | Casein kinase 2 |
| CMM | Cutaneous malignant melanoma |
| Cyto c | Cytochrome c |
| CTLA-4 | Cytotoxic T lymphocyte-associated protein 4 |
| ECS | Endocannabinoid system |
| ER | Endoplasmic reticulum |
| EE | The entourage effect |
| EGCG | Epigallocatechin gallate |
| FAAH | Fatty acid amide hydrolase |
| FABPs | Fatty acid-binding proteins |
| FAF | Fas-associated Factors |
| HT | Hypertrophic |
| HO2• | Hydroperoxyl |
| ITM | In-transit melanoma metastasis |
| IsoB | Isocannflavin B |
| LDL | Lactate dehydrogenase level |
| MEK | Mitogen-activated protein kinase |
| MiRNAs | MicroRNAs |
| MDM2 | Mouse double minute 2 homolog |
| MITF | Microphthalmia transcription factor |
| NF1 | Neurofibromatosis type 1 |
| NEAs | N-acyleethanolamines |
| OEA | Oleoylethanolamide |
| OS | Overall survival |
| OH | Hydroxyl free radicals |
| O2¯ | Anion superoxides |
| p-CA | p-Coumaric acid |
| PDI | Protein disulfide isomerase |
| PD-1 | Programmed cell death protein 1 |
| PEA | Palmitoyethanolamide |
| PGE2 | Prostaglandin E2 |
| ROS | Reactive oxygen species |
| RO2• | Peroxyl |
| SCC | Squamous cell carcinoma |
| STAT3 | Transcription 3 |
| THC | Δ9-tetrahydrocannabinol |
| TRPV1 | TRP vanilloid type 1 |
| TRPV2 | TRP vanilloid type 2 |
| TRPV3 | TRP vanilloid type 3 |
| TRPV4 | TRP vanilloid type 4 |
| TRPVA1 | TRP ankyrin 1 |
| TRPM8 | TRP melastatin 8 |
| UV | Ultraviolet |
| UVR | Ultraviolet radiation |
| VEGF | Vascular endothelial growth factor |
References
- Cust, A.; Mishra, K.; Berwick, M. Melanoma–role of the environment and genetics. Photochem. Photobiol. Sci. 2018, 17, 1853–1860. [Google Scholar] [CrossRef] [PubMed]
- Langley, A.; Levesque, L.; Baetz, T.; Asai, Y. Brief Report: Increase in Melanoma Incidence in Ontario. J. Cutan. Med. Surg. 2018, 22, 476–478. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.; Geller, A.C.; Tucker, M.A.; Guy, G.P., Jr.; Weinstock, M.A. Melanoma burden and recent trends among non-Hispanic whites aged 15–49 years, United States. Prev. Med. 2016, 91, 294–298. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weir, H.K.; Bs, T.D.T.; Soman, A.; Møller, B.; Ms, S.L. The past, present, and future of cancer incidence in the United States: 1975 through 2020. Cancer 2015, 121, 1827–1837. [Google Scholar] [CrossRef]
- Rebecca, V.W.; Somasundaram, R.; Herlyn, M. Pre-clinical modeling of cutaneous melanoma. Nat. Commun. 2020, 11, 2858. [Google Scholar] [CrossRef] [PubMed]
- Soura, E.; Eliades, P.J.; Shannon, K.; Stratigos, A.J.; Tsao, H. Hereditary melanoma: Update on syndromes and management. J. Am. Acad. Dermatol. 2016, 74, 395–407. [Google Scholar] [CrossRef]
- Whiteman, D.C.; Green, A.C.; Olsen, C.M. The Growing Burden of Invasive Melanoma: Projections of Incidence Rates and Numbers of New Cases in Six Susceptible Populations through 2031. J. Investig. Dermatol. 2016, 136, 1161–1171. [Google Scholar] [CrossRef]
- Bandarchi, B.; Ma, L.; Navab, R.; Seth, A.; Rasty, G. From Melanocyte to Metastatic Malignant Melanoma. Dermatol. Res. Pract. 2010, 2010, 583748. [Google Scholar] [CrossRef]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.-J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Prim. 2015, 1, 15003. [Google Scholar] [CrossRef]
- Sarna, M.; Krzykawska-Serda, M.; Jakubowska, M.; Zadlo, A.; Urbanska, K.; Sarna, M.; Krzykawska-Serda, M.; Jakubowska, M.; Zadlo, A.; Urbanska, K. Melanin presence inhibits melanoma cell spread in mice in a unique mechanical fashion. Sci. Rep. 2019, 9, 9280. [Google Scholar] [CrossRef]
- Hamza, H.S.; Elhusseiny, A.M. Choroidal melanoma resection. Middle East Afr. J. Ophthalmol. 2018, 25, 65–70. [Google Scholar] [CrossRef]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef] [PubMed]
- Keilholz, U.; Ascierto, P.; Dummer, R.; Robert, C.; Lorigan, P.; van Akkooi, A.; Arance, A.; Blank, C.; Sileni, V.C.; Donia, M.; et al. ESMO consensus conference recommendations on the management of metastatic melanoma: Under the auspices of the ESMO Guidelines Committee. Ann. Oncol. 2020, 31, 1435–1448. [Google Scholar] [CrossRef] [PubMed]
- Madunić, J.; Madunić, I.V.; Gajski, G.; Popić, J.; Garaj-Vrhovac, V. Apigenin: A dietary flavonoid with diverse anticancer properties. Cancer Lett. 2018, 413, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Davoodvandi, A.; Darvish, M.; Borran, S.; Nejati, M.; Mazaheri, S.; Tamtaji, O.R.; Hamblin, M.R.; Masoudian, N.; Mirzaei, H. The therapeutic potential of resveratrol in a mouse model of melanoma lung metastasis. Int. Immunopharmacol. 2020, 88, 106905. [Google Scholar] [CrossRef]
- Louis, B.W.-S. Cannabinoids and the entourage effect. In Cannabis; Chapman and Hall/CRC: Boca Raton, FL, USA, 2018; pp. 87–96. [Google Scholar] [CrossRef]
- Muller, C.; Morales, P.; Reggio, P.H. Cannabinoid Ligands Targeting TRP Channels. Front. Mol. Neurosci. 2019, 11, 487. [Google Scholar] [CrossRef]
- Maccarrone, M.; Maldonado, R.; Casas, M.; Henze, T.; Centonze, D. Cannabinoids therapeutic use: What is our current understanding following the introduction of THC, THC: CBD oromucosal spray and others? Expert Rev. Clin. Pharmacol. 2017, 10, 443–455. [Google Scholar] [CrossRef]
- Brand, E.J.; Zhao, Z.J.F.i.p. Cannabis in Chinese medicine: Are some traditional indications referenced in ancient literature related to cannabinoids? Front. Pharmacol. 2017, 8, 108. [Google Scholar] [CrossRef]
- Clarke, R.; Merlin, M. Ethnobotanical History and Contemporary Context of Medicinal Cannabis. In Cannabis; University of California Press: Berkeley, CA, USA, 2013; pp. 241–256. [Google Scholar] [CrossRef]
- Mechoulam, R. Cannabinoids as Therapeutic Agents; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Fraguas-Sánchez, A.I.; Fernández-Carballido, A.; Torres-Suárez, A.I. Phyto-, endo- and synthetic cannabinoids: Promising chemotherapeutic agents in the treatment of breast and prostate carcinomas. Expert Opin. Investig. Drugs 2016, 25, 1311–1323. [Google Scholar] [CrossRef]
- Velasco, G.; Sánchez, C.; Guzmán, M. Towards the use of cannabinoids as antitumour agents. Nat. Rev. Cancer 2012, 12, 436–444. [Google Scholar] [CrossRef]
- Velasco, G.; Sánchez, C.; Guzmán, M. Anticancer Mechanisms of Cannabinoids. Curr. Oncol. 2016, 23, 23–32. [Google Scholar] [CrossRef]
- Darmani, N.A.; Belkacemi, L.; Zhong, W. Δ9-THC and related cannabinoids suppress substance P- induced neurokinin NK1-receptor-mediated vomiting via activation of cannabinoid CB1 receptor. Eur. J. Pharmacol. 2019, 865, 172806. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, L.; Balestrino, M.; Mori, L.; Puce, L.; Rosa, G.M.; Giorello, L.; Currà, A.; Fattapposta, F.; Serrati, C.; Gandolfo, C.; et al. A randomised controlled cross-over double-blind pilot study protocol on THC:CBD oromucosal spray efficacy as an add-on therapy for post-stroke spasticity. BMJ Open 2017, 7, e016843. [Google Scholar] [CrossRef]
- Badowski, M.E.; Yanful, P.K. Dronabinol oral solution in the management of anorexia and weight loss in AIDS and cancer. Ther. Clin. Risk Manag. 2018, 14, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Rock, E.; Bolognini, D.; Limebeer, C.; Cascio, M.; Anavi-Goffer, S.; Fletcher, P.; Mechoulam, R.; Pertwee, R.; Parker, L. Cannabidiol, a non-psychotropic component of cannabis, attenuates vomiting and nausea-like behaviour via indirect agonism of 5-HT1A somatodendritic autoreceptors in the dorsal raphe nucleus. Br. J. Pharmacol. 2011, 165, 2620–2634. [Google Scholar] [CrossRef]
- Silvestro, S.; Mammana, S.; Cavalli, E.; Bramanti, P.; Mazzon, E. Use of Cannabidiol in the Treatment of Epilepsy: Efficacy and Security in Clinical Trials. Molecules 2019, 24, 1459. [Google Scholar] [CrossRef] [PubMed]
- Huestis, M.A.; Solimini, R.; Pichini, S.; Pacifici, R.; Carlier, J.; Busardò, F.P. Cannabidiol Adverse Effects and Toxicity. Curr. Neuropharmacol. 2019, 17, 974–989. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Wolf, G.; Cremer-Schaeffer, P. Three years of cannabis as medicine-preliminary results of the survey accompanying the prescription of medical cannabis in Germany. Bundesgesundheitsblatt Gesundh. Gesundh. 2021, 64, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Markovic, S.N.; Erickson, L.A.; Rao, R.D.; McWilliams, R.R.; Kottschade, L.A.; Creagan, E.T.; Weenig, R.H.; Hand, J.L.; Pittelkow, M.R.; Pockaj, B.A. Malignant melanoma in the 21st century, part 1: Epidemiology, risk factors, screening, prevention, and diagnosis. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2007; pp. 364–380. [Google Scholar]
- Huang, R.; Rofstad, E.K. Integrins as therapeutic targets in the organ-specific metastasis of human malignant melanoma. J. Exp. Clin. Cancer Res. 2018, 37, 92. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Zhu, X.; Liu, Z.; Sun, N.; Gu, L.; Wei, Y.; Cheng, X.; Zhang, Z.; Xie, B.; Yang, S.; et al. Clinical characteristics of malignant melanoma in central China and predictors of metastasis. Oncol. Lett. 2019, 19, 1452–1464. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Scolyer, R.A.; Thompson, J.F.; Shaw, H.M.; McCarthy, S.W. The importance of mitotic rate as a prognostic factor for localized primary cutaneous melanoma. J. Cutan. Pathol. 2006, 33, 395–396. [Google Scholar] [CrossRef] [PubMed]
- Visconti, A.; Ribero, S.; Sanna, M.; Spector, T.D.; Bataille, V.; Falchi, M. Body site-specific genetic effects influence naevus count distribution in women. Pigment Cell Melanoma Res. 2019, 33, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Xu, W.-H.; Wang, J.-X.; Hou, J.-M.; Zhang, H.-L.; Zhao, X.-Y.; Shen, G.-L. Identification, Validation, and Functional Annotations of Genome-Wide Profile Variation between Melanocytic Nevus and Malignant Melanoma. BioMed Res. Int. 2020, 2020, 1840415. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.D.; Chinta, S.; Yeh, C.; Shah, V.P.; Shah, R.; Paskhover, B.; Schwartz, R.A. An analysis of lactate dehydrogenase (LDH) levels in advanced stage IV melanoma of the skin: Prognostic capabilities and demographic variability. Arch. Dermatol. Res. 2022, 1–8. [Google Scholar] [CrossRef]
- McMeniman, E.; Duffy, D.; Jagirdar, K.; Lee, K.; Peach, E.; McInerney-Leo, A.; De’Ambrosis, B.; Rayner, J.; Smithers, B.; Soyer, H.; et al. The interplay of sun damage and genetic risk in Australian multiple and single primary melanoma cases and controls. Br. J. Dermatol. 2020, 183, 357–366. [Google Scholar] [CrossRef]
- Carr, S.; Smith, C.; Wernberg, J. Epidemiology and Risk Factors of Melanoma. Surg. Clin. 2019, 100, 1–12. [Google Scholar] [CrossRef]
- Oliveria, S.A.; Saraiya, M.; Geller, A.C.; Heneghan, M.K.; Jorgensen, C. Sun exposure and risk of melanoma. Arch. Dis. Child. 2005, 91, 131–138. [Google Scholar] [CrossRef]
- Duffy, D.; Lee, K.; Jagirdar, K.; Pflugfelder, A.; Stark, M.; McMeniman, E.; Soyer, H.; Sturm, R.A. High naevus count and MC 1R red hair alleles contribute synergistically to increased melanoma risk. Br. J. Dermatol. 2019, 181, 1009–1016. [Google Scholar] [CrossRef]
- Espinoza, C.D.R.; Roberts, N.; Chen, S.; Leacy, F.P.; Alexandrov, L.B.; Pornputtapong, N.; Halaban, R.; Krauthammer, M.; Cui, R.; Bishop, D.T.; et al. Germline MC1R status influences somatic mutation burden in melanoma. Nat. Commun. 2016, 7, 12064. [Google Scholar] [CrossRef]
- Aitken, J.F.; Elwood, M.; Baade, P.D.; Youl, P.; English, D. Clinical whole-body skin examination reduces the incidence of thick melanomas. Int. J. Cancer 2009, 126, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M. NADPH oxidase. Curr. Opin. Immunol. 2004, 16, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Yepes, J.; Zavala-Flores, L.; Anandhan, A.; Wang, F.; Skotak, M.; Chandra, N.; Li, M.; Pappa, A.; Martinez-Fong, D.; Del Razo, L.M.; et al. Antioxidant gene therapy against neuronal cell death. Pharmacol. Ther. 2013, 142, 206–230. [Google Scholar] [CrossRef] [PubMed]
- Kurian, N.K.; Nair, H.P.; Bhat, S.G. Evaluation of anti-inflammatory property of melanin from marine Bacillus spp. BTCZ31. Asian J. Pharm. Clin. Res. 2015, 8, 251–255. [Google Scholar]
- Maric, H.; Supic, G.; Kandolf-Sekulovic, L.; Maric, V.; Mijuskovic, Z.; Radevic, T.; Rajovic, M.; Magic, Z. DNMT1 and DNMT3B genetic polymorphisms affect the clinical course and outcome of melanoma patients. Melanoma Res. 2019, 29, 596–602. [Google Scholar] [CrossRef]
- Talantov, D.; Mazumder, A.; Yu, J.X.; Briggs, T.; Jiang, Y.; Backus, J.; Atkins, D.; Wang, Y. Novel Genes Associated with Malignant Melanoma but not Benign Melanocytic Lesions. Clin. Cancer Res. 2005, 11, 7234–7242. [Google Scholar] [CrossRef]
- Tas, F.; Erturk, K.J. BRAF V600E mutation as a prognostic factor in cutaneous melanoma patients. Dermatol. Ther. 2020, 33, e13270. [Google Scholar] [CrossRef]
- Amaral, T.; Sinnberg, T.; Meier, F.; Krepler, C.; Levesque, M.; Niessner, H.; Garbe, C. MAPK pathway in melanoma part II—Secondary and adaptive resistance mechanisms to BRAF inhibition. Eur. J. Cancer 2017, 73, 93–101. [Google Scholar] [CrossRef]
- Karachaliou, N.; Pilotto, S.; Teixidó, C.; Viteri, S.; González-Cao, M.; Riso, A.; Morales-Espinosa, D.; Molina, M.A.; Chaib, I.; Santarpia, M.; et al. Melanoma: Oncogenic drivers and the immune system. Ann. Transl. Med. 2015, 3, 265. [Google Scholar] [CrossRef]
- Boniol, M.; Autier, P.; Boyle, P.; Gandini, S. Cutaneous melanoma attributable to sunbed use: Systematic review and meta-analysis. BMJ 2012, 345, e4757. [Google Scholar] [CrossRef] [PubMed]
- Reichrath, J.; Lindqvist, P.G.; Pilz, S.; März, W.; Grant, W.B.; Holick, M.F.; De Gruijl, F.R. Sunbeds and Melanoma Risk: Many Open Questions, Not Yet Time to Close the Debate. Anticancer Res. 2019, 40, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Roh, M.R.; Eliades, P.; Gupta, S.; Grant-Kels, J.M.; Tsao, H. Cutaneous melanoma in women. Int. J. Women’s Dermatol. 2017, 3, S11–S15. [Google Scholar] [CrossRef] [PubMed]
- Hutchenreuther, J.; Leask, A. Why target the tumor stroma in melanoma? J. Cell Commun. Signal. 2017, 12, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Fecher, L.A.; Amaravadi, R.K.; Flaherty, K.T. The MAPK pathway in melanoma. Curr. Opin. Oncol. 2008, 20, 183–189. [Google Scholar] [CrossRef]
- Leung, G.P.; Feng, T.; Sigoillot, F.D.; Geyer, F.C.; Shirley, M.D.; Ruddy, D.A.; Rakiec, D.P.; Freeman, A.K.; Engelman, J.A.; Jaskelioff, M.; et al. Hyperactivation of MAPK Signaling Is Deleterious to RAS/RAF-mutant Melanoma. Mol. Cancer Res. 2019, 17, 199–211. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genom. 2017, 11, 1–19. [Google Scholar] [CrossRef]
- Hansson, J. Familial Cutaneous Melanoma. In Diseases of DNA Repair; Springer: Berlin/Heidelberg, Germany, 2010; Volume 685, pp. 134–145. [Google Scholar] [CrossRef]
- Hogg, D.; Brill, H.; Liu, L.; Monzon, J.; Summers, A.; From, L.; Lassam, N.J. Role of the Cyclin-Dependent Kinase Inhibitor CDKN2A in Familial Melanoma. J. Cutan. Med. Surg. 1998, 2, 172–179. [Google Scholar] [CrossRef]
- Testori, A.; Ribero, S.; Bataille, V. Diagnosis and treatment of in-transit melanoma metastases. Eur. J. Surg. Oncol. 2017, 43, 544–560. [Google Scholar] [CrossRef]
- Turner, N.; Ware, O.; Bosenberg, M. Genetics of metastasis: Melanoma and other cancers. Clin. Exp. Metastasis 2018, 35, 379–391. [Google Scholar] [CrossRef]
- Frank, C.; Sundquist, J.; Hemminki, A.; Hemminki, K. Risk of other Cancers in Families with Melanoma: Novel Familial Links. Sci. Rep. 2017, 7, 42601. [Google Scholar] [CrossRef] [PubMed]
- Domingues, B.; Lopes, J.M.; Soares, P.; Populo, H. Melanoma treatment in review. Immuno Targets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Lin, C.C.; Mariotto, A.B.; Siegel, R.L.; Stein, K.D.; Kramer, J.L.; Alteri, R.; Robbins, A.S.; Jemal, A. Cancer treatment and survivorship statistics, 2014. CA Cancer J. Clin. 2014, 64, 252–271. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Li, S.-Y.; Zhang, P.; Yu, B. Clinical characteristics, treatment, and prognosis of squamous cell carcinoma arising from extremity chronic osteomyelitis: A synthesis analysis of one hundred and seventy six reported cases. Int. Orthop. 2020, 44, 2457–2471. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Sawada, S.; Yoshioka, I.; Ohashi, Y.; Matsuo, M.; Harimaya, Y.; Tsukada, K.; Saiki, I. Increased surgical stress promotes tumor metastasis. Surgery 2003, 133, 547–555. [Google Scholar] [CrossRef]
- Tai, L.-H.; De Souza, C.T.; Bélanger, S.; Ly, L.; Alkayyal, A.A.; Zhang, J.; Rintoul, J.L.; Ananth, A.A.; Lam, T.; Breitbach, C.J.; et al. Preventing postoperative metastatic disease by inhibiting surgery-induced dysfunction in natural killer cells. Cancer Res. 2013, 73, 97–107. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Inflammation. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Kienzl, M.; Kargl, J.; Schicho, R. The Immune Endocannabinoid System of the Tumor Microenvironment. Int. J. Mol. Sci. 2020, 21, 8929. [Google Scholar] [CrossRef]
- Kishimoto, S.; Muramatsu, M.; Gokoh, M.; Oka, S.; Waku, K.; Sugiura, T. Endogenous Cannabinoid Receptor Ligand Induces the Migration of Human Natural Killer Cells. J. Biochem. 2005, 137, 217–223. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Lee, H.; Kim, S.-H.; Jin, H.; Bae, J.; Choi, H.-K. Discovery of potential biomarkers in human melanoma cells with different metastatic potential by metabolic and lipidomic profiling. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Peris, K.; Hauschild, A.; Saiag, P.; Middleton, M.; Spatz, A.; Grob, J.-J.; Malvehy, J.; Newton-Bishop, J.; Stratigos, A.; et al. Diagnosis and treatment of melanoma: European consensus-based interdisciplinary guideline. Eur. J. Cancer 2010, 46, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller Jr, W.H.; Kaempgen, E.J.T.L. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Blankenstein, S.A.; Aarts, M.J.B.; van den Berkmortel, F.W.P.J.; Boers-Sonderen, M.J.; van den Eertwegh, A.J.M.; Franken, M.G.; de Groot, J.W.B.; Haanen, J.B.A.G.; Hospers, G.A.P.; Kapiteijn, E.; et al. Surgery for Unresectable Stage IIIC and IV Melanoma in the Era of New Systemic Therapy. Cancers 2020, 12, 1176. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef]
- Long, G.V.; Flaherty, K.T.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: Long-term survival and safety analysis of a phase 3 study. Ann. Oncol. 2017, 28, 1631–1639. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Eggermont, A.M.; Spatz, A.; Robert, C. Cutaneous melanoma. Lancet 2014, 383, 816–827. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. New Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Bradish, J.R.; Montironi, R.; Lopez-Beltran, A.; Post, K.M.; MacLennan, G.T.; Cheng, L. Towards personalized therapy for patients with malignant melanoma: Molecular insights into the biology of BRAF mutations. Future Oncol. 2013, 9, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Champiat, S.; Dercle, L.; Ammari, S.; Massard, C.; Hollebecque, A.; Postel-Vinay, S.; Chaput, N.; Eggermont, A.; Marabelle, A.; Soria, J.-C.; et al. Hyperprogressive Disease Is a New Pattern of Progression in Cancer Patients Treated by Anti-PD-1/PD-L1Hyperprogressive Disease with Anti-PD-1/PD-L1 Therapy. Clin. Cancer Res. 2017, 23, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.; Chiarion-Sileni, V.; Grob, J.-J.; Dummer, R.; Wolchok, J.D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Adjuvant ipilimumab versus placebo after complete resection of stage III melanoma: Long-term follow-up results of the European Organisation for Research and Treatment of Cancer 18071 double-blind phase 3 randomised trial. Eur. J. Cancer 2019, 119, 1–10. [Google Scholar] [CrossRef]
- Heinzerling, L.; Eigentler, T.K.; Fluck, M.; Hassel, J.C.; Heller-Schenck, D.; Leipe, J.; Pauschinger, M.; Vogel, A.; Zimmer, L.; Gutzmer, R. Tolerability of BRAF/MEK inhibitor combinations: Adverse event evaluation and management. ESMO Open 2019, 4, e000491. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.C.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef]
- Del Curatolo, A.; Conciatori, F.; Incani, U.C.; Bazzichetto, C.; Falcone, I.; Corbo, V.; D’Agosto, S.; Eramo, A.; Sette, G.; Sperduti, I.; et al. Therapeutic potential of combined BRAF/MEK blockade in BRAF-wild type preclinical tumor models. J. Exp. Clin. Cancer Res. 2018, 37, 1–14. [Google Scholar] [CrossRef]
- Park, Y.-J.; Kuen, D.-S.; Chung, Y. Future prospects of immune checkpoint blockade in cancer: From response prediction to overcoming resistance. Exp. Mol. Med. 2018, 50, 1–13. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Elliott, T.M.; Whiteman, D.C.; Olsen, C.M.; Gordon, L.G. Estimated Healthcare Costs of Melanoma in Australia Over 3 Years Post-Diagnosis. Appl. Health Econ. Health Policy 2017, 15, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Eroglu, Z.; Infante, J.; Patel, S.; Daud, A.; Johnson, D.B.; Gonzalez, R.; Kefford, R.; Hamid, O.; Schuchter, L.; et al. Long-Term Outcomes in Patients With BRAF V600–Mutant Metastatic Melanoma Who Received Dabrafenib Combined With Trametinib. J. Clin. Oncol. 2018, 36, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Liu, X.; Yang, J.; Zhang, M.; Jin, H.; Ma, X.; Shi, H. Combination of Immunotherapy With Targeted Therapy: Theory and Practice in Metastatic Melanoma. Front. Immunol. 2019, 10, 990. [Google Scholar] [CrossRef] [PubMed]
- Kandel, M.; Allayous, C.; Dalle, S.; Mortier, L.; Dalac, S.; Dutriaux, C.; Leccia, M.; Guillot, B.; Saiag, P.; Lacour, J.; et al. Update of survival and cost of metastatic melanoma with new drugs: Estimations from the MelBase cohort. Eur. J. Cancer 2018, 105, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- Achkar, T.; Tarhini, A.A. The use of immunotherapy in the treatment of melanoma. J. Hematol. Oncol. 2017, 10, 1–9. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Hamid, O.; Gonzalez, R.; Infante, J.R.; Patel, M.R.; Hodi, F.S.; Lewis, K.D.; Tawbi, H.A.; Hernandez, G.; Wongchenko, M.J.; et al. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat. Med. 2019, 25, 929–935. [Google Scholar] [CrossRef]
- Dummer, R.; Fernández, A.M.A.; Hansson, J.; Larkin, J.M.G.; Long, G.V.; Gasal, E.; Kaper, M.; Upalawanna, A.; Mookerjee, B.; Atkinson, V. Preliminary findings from part 1 of COMBI-i: A phase III study of anti–PD-1 antibody PDR001 combined with dabrafenib (D) and trametinib (T) in previously untreated patients (pts) with advanced BRAF V600-mutant melanoma. J. Clin. Oncol. 2018, 36, 189. [Google Scholar] [CrossRef]
- Minor, D.R.; Puzanov, I.; Callahan, M.K.; Hug, B.A.; Hoos, A. Severe gastrointestinal toxicity with administration of trametinib in combination with dabrafenib and ipilimumab. Pigment. Cell Melanoma Res. 2015, 28, 611–612. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.J.; Corrie, P.G. Management of BRAF and MEK inhibitor toxicities in patients with metastatic melanoma. Ther. Adv. Med. Oncol. 2015, 7, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Boada, A.; Carrera, C.; Segura, S.; Collgros, H.; Pasquali, P.; Bodet, D.; Puig, S.; Malvehy, J. Cutaneous toxicities of new treatments for melanoma. Clin. Transl. Oncol. 2018, 20, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Gutiérrez, E.C.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Kähler, K.C.; Hauschild, A. Treatment and side effect management of CTLA-4 antibody therapy in metastatic melanoma. JDDG J. Dtsch. Dermatol. Ges. 2010, 9, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Reddy, H.G.; Schneider, B.J.; Tai, A.W. Immune Checkpoint Inhibitor-Associated Colitis and Hepatitis. Clin. Transl. Gastroenterol. 2018, 9, e180. [Google Scholar] [CrossRef]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.-J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A. Melanoma. Lancet 2015, 1, 1–20. [Google Scholar] [CrossRef]
- Lu, H.-C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2015, 79, 516–525. [Google Scholar] [CrossRef]
- Mouslech, Z.; Valla, V. Endocannabinoid system: An overview of its potential in current medical practice. Neuro Endocrinol. Lett. 2009, 30, 153–179. [Google Scholar]
- Silver, R.J. The Endocannabinoid System of Animals. Animals 2019, 9, 686. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.; Onaivi, E.S. Endocannabinoid System Components: Overview and Tissue Distribution. Adv. Exp. Med. Biol. 2019, 1162, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Devane, W.A.; Dysarz, F.A., 3rd; Johnson, M.R.; Melvin, L.S.; Howlett, A.C. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988, 34, 605–613. [Google Scholar] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Kendall, D.A.; Yudowski, G.A. Cannabinoid Receptors in the Central Nervous System: Their Signaling and Roles in Disease. Front. Cell. Neurosci. 2017, 10, 294. [Google Scholar] [CrossRef]
- Simmerman, E.; Qin, X.; Yu, J.C.; Baban, B. Cannabinoids as a Potential New and Novel Treatment for Melanoma: A Pilot Study in a Murine Model. J. Surg. Res. 2019, 235, 210–215. [Google Scholar] [CrossRef]
- Howlett, A.C.; Reggio, P.H.; Childers, S.R.; Hampson, R.E.; Ulloa, N.M.; Deutsch, D.G. Endocannabinoid tone versus constitutive activity of cannabinoid receptors. Br. J. Pharmacol. 2011, 163, 1329–1343. [Google Scholar] [CrossRef]
- McPartland, J.M. Cannabis systematics at the levels of family, genus, and species. Cannabis Cannabinoid Res. 2018, 3, 203–212. [Google Scholar] [CrossRef]
- Small, E.; Cronquist, A. A Practical and Natural Taxonomy for Cannabis. Taxon 1976, 25, 405–435. [Google Scholar] [CrossRef]
- Reimann-Philipp, U.; Speck, M.; Orser, C.; Johnson, S.; Hilyard, A.; Turner, H.; Stokes, A.J.; Small-Howard, A.L. Cannabis Chemovar Nomenclature Misrepresents Chemical and Genetic Diversity; Survey of Variations in Chemical Profiles and Genetic Markers in Nevada Medical Cannabis Samples. Cannabis Cannabinoid Res. 2020, 5, 215–230. [Google Scholar] [CrossRef]
- Tsuboi, K.; Uyama, T.; Okamoto, Y.; Ueda, N. Endocannabinoids and related N-acylethanolamines: Biological activities and metabolism. Inflamm. Regen. 2018, 38, 28. [Google Scholar] [CrossRef] [PubMed]
- Correia-Sá, I.B.; Carvalho, C.M.; Serrão, P.V.; Loureiro, A.I.; Fernandes-Lopes, C.; Marques, M.; Vieira-Coelho, M.A. A new role for anandamide: Defective link between the systemic and skin endocannabinoid systems in hypertrophic human wound healing. Sci. Rep. 2020, 10, 11134. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Quang, D.N.; Nukada, M.; Asakawa, Y. Isolation, synthesis and biological activity of grifolic acid dervivatives from the inedible mushroom Albatrellus dispansus. Heterocycles 2005, 65, 2431. [Google Scholar]
- Gülck, T.; Møller, B.L. Phytocannabinoids: Origins and Biosynthesis. Trends Plant Sci. 2020, 25, 985–1004. [Google Scholar] [CrossRef] [PubMed]
- Happyana, N.; Kayser, O. Monitoring Metabolite Profiles of Cannabis sativa L. Trichomes during Flowering Period Using 1H NMR-Based Metabolomics and Real-Time PCR. Planta Med. 2016, 82, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Roberto, D.; Klotz, L.H.; Venkateswaran, V.J. Cannabinoid WIN 55,212-2 induces cell cycle arrest and apoptosis, and inhibits proliferation, migration, invasion, and tumor growth in prostate cancer in a cannabinoid-receptor 2 dependent manner. Prostate 2019, 79, 151–159. [Google Scholar] [CrossRef]
- Nallathambi, R.; Mazuz, M.; Namdar, D.; Shik, M.; Namintzer, D.; Vinayaka, A.C.; Ion, A.; Faigenboim, A.; Nasser, A.; Laish, I.; et al. Identification of Synergistic Interaction Between Cannabis-Derived Compounds for Cytotoxic Activity in Colorectal Cancer Cell Lines and Colon Polyps That Induces Apoptosis-Related Cell Death and Distinct Gene Expression. Cannabis Cannabinoid Res. 2018, 3, 120–135. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Ligresti, A.; Moriello, A.S.; Iappelli, M.; Verde, R.; Stott, C.G.; Cristino, L.; Orlando, P.; Di Marzo, V. Non-THC cannabinoids inhibit prostate carcinoma growthin vitroandin vivo: Pro-apoptotic effects and underlying mechanisms. Br. J. Pharmacol. 2012, 168, 79–102. [Google Scholar] [CrossRef]
- Gaoni, Y.; Mechoulam, R. Isolation, Structure, and Partial Synthesis of an Active Constituent of Hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. [Google Scholar] [CrossRef]
- Dalzell, A.M.; Bartlett, H.; Lilleyman, J.S. Nabilone: An alternative antiemetic for cancer chemotherapy.. Arch. Dis. Child. 1986, 61, 502–505. [Google Scholar] [CrossRef]
- Sun, N.; Cunha, N.; Amar, S.; Brown, S. Synthetic cannabinoid for the treatment of severe chronic noncancer pain in children and adolescents. Can. J. Pain 2022, 6, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; MacDougall, D. CADTH Rapid Response Reports. In Nabilone for the Treatment of Nausea and Vomiting or Anorexia: A Review of Clinical Effectiveness and Guidelines; Canadian Agency for Drugs and Technologies in Health Copyright: Ottawa, ON, Canada, 2019. [Google Scholar]
- N.S.W. Health. Cannabis Medicines Registered in Australia. Available online: https://www.medicinalcannabis.nsw.gov.au/health-professionals/product (accessed on 30 November 2022).
- Karschner, E.L.; Darwin, W.D.; Goodwin, R.S.; Wright, S.; Huestis, M.A. Plasma Cannabinoid Pharmacokinetics following Controlled Oral Δ9-Tetrahydrocannabinol and Oromucosal Cannabis Extract Administration. Clin. Chem. 2011, 57, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Benito, S.B.; Seijo-Vila, M.; Caro-Villalobos, M.; Tundidor, I.; Andradas, C.; García-Taboada, E.; Wade, J.; Smith, S.; Guzmán, M.; Pérez-Gómez, E.; et al. Appraising the “entourage effect”: Antitumor action of a pure cannabinoid versus a botanical drug preparation in preclinical models of breast cancer. Biochem. Pharmacol. 2018, 157, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.L.; Hill, D.S.; McKee, C.S.; Hernandez-Tiedra, S.; Lorente, M.; Lopez-Valero, I.; Anagnostou, M.E.; Babatunde, F.; Corazzari, M.; Redfern, C.P.F.; et al. Exploiting Cannabinoid-Induced Cytotoxic Autophagy to Drive Melanoma Cell Death. J. Investig. Dermatol. 2015, 135, 1629–1637. [Google Scholar] [CrossRef]
- Glodde, N.; Jakobs, M.; Bald, T.; Tüting, T.; Gaffal, E. Differential role of cannabinoids in the pathogenesis of skin cancer. Life Sci. 2015, 138, 35–40. [Google Scholar] [CrossRef]
- Blázquez, C.; Carracedo, A.; Barrado, L.; Real, P.J.; Fernández-Luna, J.L.; Velasco, G.; Malumbres, M.; Guzmán, M. Cannabinoid receptors as novel targets for the treatment of melanoma. FASEB J. 2006, 20, 2633–2635. [Google Scholar] [CrossRef]
- Burch, R.; Mortuza, A.; Blumenthal, E.; Mustafa, A. Effects of cannabidiol (CBD) on the inhibition of melanoma cells in vitro. J. Immunoass. Immunochem. 2021, 42, 285–291. [Google Scholar] [CrossRef]
- Baek, S.-H.; Du Han, S.; Yook, C.N.; Kim, Y.C.; Kwak, J.S. Synthesis and antitumor activity of cannabigerol. Arch. Pharmacal Res. 1996, 19, 228–230. [Google Scholar] [CrossRef]
- Scuderi, M.R.; Cantarella, G.; Scollo, M.; Lempereur, L.; Palumbo, M.; Saccani-Jotti, G.; Bernardini, R. The antimitogenic effect of the cannabinoid receptor agonist WIN55212-2 on human melanoma cells is mediated by the membrane lipid raft. Cancer Lett. 2011, 310, 240–249. [Google Scholar] [CrossRef]
- Haskó, J.; Fazakas, C.; Molnar, J.; Nyúl-Tóth, Á; Herman, H.; Hermenean, A.; Wilhelm, I.; Persidsky, Y.; Krizbai, I.A. CB2 Receptor Activation Inhibits Melanoma Cell Transmigration through the Blood-Brain Barrier. Int. J. Mol. Sci. 2014, 15, 8063–8074. [Google Scholar] [CrossRef]
- Kenessey, I.; Bánki, B.; Márk, Á.; Varga, N.; Tóvári, J.; Ladányi, A.; Rásó, E.; Tímár, J. Revisiting CB1 Receptor as Drug Target in Human Melanoma. Pathol. Oncol. Res. 2012, 18, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, B.; Romanini, A.; Vanni, A.; Martinotti, E.; Chicca, A.; Fogli, S.; Nieri, P. Anticancer activity of anandamide in human cutaneous melanoma cells. Eur. J. Pharmacol. 2013, 718, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Hamtiaux, L.; Masquelier, J.; Muccioli, G.G.; Bouzin, C.; Feron, O.; Gallez, B.; Lambert, D.M. The association of N-palmitoylethanolamine with the FAAH inhibitor URB597 impairs melanoma growth through a supra-additive action. BMC Cancer 2012, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Verykiou, S.; Alexander, M.; Edwards, N.; Plummer, R.; Chaudhry, B.; Lovat, P.; Hill, D.S. Harnessing autophagy to overcome mitogen-activated protein kinase kinase inhibitor-induced resistance in metastatic melanoma. Br. J. Dermatol. 2018, 180, 346–356. [Google Scholar] [CrossRef]
- Carpi, S.; Fogli, S.; Romanini, A.; Pellegrino, M.; Adinolfi, B.; Podestà, A.; Costa, B.; Da Pozzo, E.; Martini, C.; Breschi, M.C.; et al. AM251 induces apoptosis and G2/M cell cycle arrest in A375 human melanoma cells. Anti-Cancer Drugs 2015, 26, 754–762. [Google Scholar] [CrossRef]
- Matsuo, A.L.; Figueiredo, C.R.; Arruda, D.C.; Pereira, F.V.; Scutti, J.A.B.; Massaoka, M.H.; Travassos, L.R.; Sartorelli, P.; Lago, J.H.G. α-Pinene isolated from Schinus terebinthifolius Raddi (Anacardiaceae) induces apoptosis and confers antimetastatic protection in a melanoma model. Biochem. Biophys. Res. Commun. 2011, 411, 449–454. [Google Scholar] [CrossRef]
- Schomberg, J.; Wang, Z.; Farhat, A.; Guo, K.L.; Xie, J.; Zhou, Z.; Liu, J.; Kovacs, B.; Liu-Smith, F. Luteolin inhibits melanoma growth in vitro and in vivo via regulating ECM and oncogenic pathways but not ROS. Biochem. Pharmacol. 2020, 177, 114025. [Google Scholar] [CrossRef]
- Woo, J.S.; Choo, G.S.; Yoo, E.S.; Kim, S.H.; Lee, J.H.; Han, S.H.; Kim, H.J.; Jung, S.H.; Park, Y.S.; Kim, B.S.; et al. Apigenin induces apoptosis by regulating Akt and MAPK pathways in human melanoma cell A375SM. Mol. Med. Rep. 2020, 22, 4877–4889. [Google Scholar] [CrossRef]
- Yang, G.-W.; Jiang, J.-S.; Lu, W.-Q. Ferulic Acid Exerts Anti-Angiogenic and Anti-Tumor Activity by Targeting Fibroblast Growth Factor Receptor 1-Mediated Angiogenesis. Int. J. Mol. Sci. 2015, 16, 24011–24031. [Google Scholar] [CrossRef]
- Peng, D.; Chen, L.; Sun, Y.; Sun, L.; Yin, Q.; Deng, S.; Niu, L.; Lou, F.; Wang, Z.; Xu, Z.; et al. Melanoma suppression by quercein is correlated with RIG-I and type I interferon signaling. Biomed. Pharmacother. 2020, 125, 109984. [Google Scholar] [CrossRef]
- Sturza, A.; Pavel, I.; Ancușa, S.; Danciu, C.; Dehelean, C.; Duicu, O.; Muntean, D. Quercetin exerts an inhibitory effect on cellular bioenergetics of the B164A5 murine melanoma cell line. Mol. Cell. Biochem. 2018, 447, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Thangasamy, T.; Sittadjody, S.; Lanza-Jacoby, S.; Wachsberger, P.R.; Limesand, K.H.; Burd, R. Quercetin Selectively Inhibits Bioreduction and Enhances Apoptosis in Melanoma Cells That Overexpress Tyrosinase. Nutr. Cancer 2007, 59, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Kubo, I.; Nitoda, T.; Nihei, K.-I. Effects of Quercetin on Mushroom Tyrosinase and B16-F10 Melanoma Cells. Molecules 2007, 12, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.-H.; Chu, J.-H.; Kwan, H.Y.; Su, T.; Yu, H.; Cheng, B.C.-Y.; Fu, X.-Q.; Guo, H.; Li, T.; Tse, A.K.-W.; et al. Inhibition of the STAT3 signaling pathway contributes to apigenin-mediated anti-metastatic effect in melanoma. Sci. Rep. 2016, 6, 21731. [Google Scholar] [CrossRef]
- Kim, S.-H.; Yoo, E.-S.; Woo, J.-S.; Han, S.-H.; Lee, J.-H.; Jung, S.-H.; Kim, H.-J.; Jung, J.-Y. Antitumor and apoptotic effects of quercetin on human melanoma cells involving JNK/P38 MAPK signaling activation. Eur. J. Pharmacol. 2019, 860, 172568. [Google Scholar] [CrossRef] [PubMed]
- Soll, F.; Ternent, C.; Berry, I.M.; Kumari, D.; Moore, T.C. Quercetin Inhibits Proliferation and Induces Apoptosis of B16 Melanoma CellsIn Vitro. ASSAY Drug Dev. Technol. 2020, 18, 261–268. [Google Scholar] [CrossRef]
- Cao, H.-H.; Cheng, C.-Y.; Su, T.; Fu, X.-Q.; Guo, H.; Li, T.; Tse, A.K.-W.; Kwan, H.-Y.; Yu, H.; Yu, Z.-L. Quercetin inhibits HGF/c-Met signaling and HGF-stimulated melanoma cell migration and invasion. Mol. Cancer 2015, 14, 103. [Google Scholar] [CrossRef]
- Baram, L.; Peled, E.; Berman, P.; Yellin, B.; Besser, E.; Benami, M.; Louria-Hayon, I.; Lewitus, G.M.; Meiri, D. The heterogeneity and complexity of Cannabis extracts as antitumor agents. Oncotarget 2019, 10, 4091–4106. [Google Scholar] [CrossRef]
- Russo, E.B. Beyond cannabis: Plants and the endocannabinoid system. Trends Pharmacol. Sci. 2016, 37, 594–605. [Google Scholar] [CrossRef]
- Lukhele, S.T.; Motadi, L.R. Cannabidiol rather than Cannabis sativa extracts inhibit cell growth and induce apoptosis in cervical cancer cells. BMC Complement. Altern. Med. 2016, 16, 335. [Google Scholar] [CrossRef]
- Finlay, D.B.; Sircombe, K.J.; Nimick, M.; Jones, C.; Glass, M. Terpenoids From Cannabis Do Not Mediate an Entourage Effect by Acting at Cannabinoid Receptors. Front. Pharmacol. 2020, 11, 359. [Google Scholar] [CrossRef] [PubMed]
- Tomko, A.M.; Whynot, E.G.; Ellis, L.D.; Dupré, D.J. Anti-Cancer Potential of Cannabinoids, Terpenes, and Flavonoids Present in Cannabis. Cancers 2020, 12, 1985. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Ligresti, A.; Moriello, A.S.; Allarà, M.; Bisogno, T.; Petrosino, S.; Stott, C.G.; Di Marzo, V. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br. J. Pharmacol. 2011, 163, 1479–1494. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.B. Taming THC: Potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br. J. Pharmacol. 2011, 163, 1344–1364. [Google Scholar] [CrossRef]
- Heblinski, M.; Santiago, M.; Fletcher, C.; Stuart, J.; Connor, M.; McGregor, I.S.; Arnold, J.C. Terpenoids Commonly Found in Cannabis sativa Do Not Modulate the Actions of Phytocannabinoids or Endocannabinoids on TRPA1 and TRPV1 Channels. Cannabis Cannabinoid Res. 2020, 5, 305–317. [Google Scholar] [CrossRef]
- Di Giacomo, S.; Mariano, A.; Gullì, M.; Fraschetti, C.; Vitalone, A.; Filippi, A.; Mannina, L.; D’Abusco, A.S.; Di Sotto, A. Role of Caryophyllane Sesquiterpenes in the Entourage Effect of Felina 32 Hemp Inflorescence Phytocomplex in Triple Negative MDA-MB-468 Breast Cancer Cells. Molecules 2021, 26, 6688. [Google Scholar] [CrossRef]
- Russo, E.B. The Case for the Entourage Effect and Conventional Breeding of Clinical Cannabis: No “Strain,” No Gain. Front. Plant Sci. 2019, 9, 1969. [Google Scholar] [CrossRef]
- Ramer, R.; Schwarz, R.; Hinz, B. Modulation of the Endocannabinoid System as a Potential Anticancer Strategy. Front. Pharmacol. 2019, 10, 430. [Google Scholar] [CrossRef] [PubMed]
- Pyszniak, M.; Tabarkiewicz, J.; Łuszczki, J.J. Endocannabinoid system as a regulator of tumor cell malignancy—biological pathways and clinical significance. OncoTargets Ther. 2016, 9, 4323–4336. [Google Scholar] [CrossRef]
- Perveen, S.; Al-Taweel, A. Terpenes and Terpenoids; Books on Demand: Norderstedt, Germany, 2018. [Google Scholar]
- Jin, D.; Dai, K.; Xie, Z.; Chen, J. Secondary Metabolites Profiled in Cannabis Inflorescences, Leaves, Stem Barks, and Roots for Medicinal Purposes. Sci. Rep. 2020, 10, 3309–3314. [Google Scholar] [CrossRef]
- Lewis, M.A.; Russo, E.B.; Smith, K.M. Pharmacological Foundations of Cannabis Chemovars. Planta Med. 2018, 84, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Abbas, M.; Saeed, F.; Anjum, F.M.; Afzaal, M.; Tufail, T.; Bashir, M.S.; Ishtiaq, A.; Hussain, S.; Suleria, H.A.R. Natural polyphenols: An overview. Int. J. Food Prop. 2017, 20, 1689–1699. [Google Scholar] [CrossRef]
- Watson, R.R.; Preedy, V.R.; Zibadi, S. Polyphenols: Prevention and Treatment of Human Disease; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Deborah, J.K.; Kuhn, D.; Lam, W.H.; Kazi, A.; Daniel, K.G.; Song, S.; Chow, L.M.C.; Chan, T.H.; Dou, Q.P. Synthetic peracetate tea polyphenols as potent proteasome inhibitors and apoptosis inducers in human cancer cells. Front. Biosci. 2005, 10, 1010–1023. [Google Scholar] [CrossRef]
- Bishop, K.; Ferguson, L.; Braakhuis, A. Polyphenols for Cancer Treatment or Prevention; MDPI AG-Multidisciplinary Digital Publishing Institute: Basel, Switzerland, 2018. [Google Scholar]
- Cheng, Y.-C.; Sheen, J.-M.; Hu, W.L.; Hung, Y.-C. Polyphenols and Oxidative Stress in Atherosclerosis-Related Ischemic Heart Disease and Stroke. Oxidative Med. Cell. Longev. 2017, 2017, 8526438. [Google Scholar] [CrossRef] [PubMed]
- Murillo, A.G.; Fernandez, M.L. The relevance of dietary polyphenols in cardiovascular protection. Curr. Pharm. Des. 2017, 23, 2444–2452. [Google Scholar] [CrossRef] [PubMed]
- Naseri, R.; Farzaei, F.; Fakhri, S.; El-Senduny, F.F.; Altouhamy, M.; Bahramsoltani, R.; Ebrahimi, F.; Rahimi, R.; Farzaei, M.H. Polyphenols for diabetes associated neuropathy: Pharmacological targets and clinical perspective. DARU J. Pharm. Sci. 2019, 27, 781–798. [Google Scholar] [CrossRef]
- Sassi, A.; Maatouk, M.; El Gueder, D.; Bzéouich, I.M.; Hatira, S.A.-B.; Jemni-Yacoub, S.; Ghedira, K.; Chekir-Ghedira, L. Chrysin, a natural and biologically active flavonoid suppresses tumor growth of mouse B16F10 melanoma cells: In vitro and In vivo study. Chem.-Biol. Interact. 2018, 283, 10–19. [Google Scholar] [CrossRef]
- Caltagirone, S.; Rossi, C.; Poggi, A.; Ranelletti, F.O.; Natali, P.G.; Brunetti, M.; Aiello, F.B.; Piantelli, M. Flavonoids apigenin and quercetin inhibit melanoma growth and metastatic potential. Int. J. Cancer 2000, 87, 595–600. [Google Scholar] [CrossRef]
- George, V.C.; Dellaire, G.; Rupasinghe, H.V. Plant flavonoids in cancer chemoprevention: Role in genome stability. J. Nutr. Biochem. 2017, 45, 1–14. [Google Scholar] [CrossRef]
- Wu, J.-C.; Lai, C.-S.; Lee, P.-S.; Ho, C.-T.; Liou, W.-S.; Wang, Y.-J.; Pan, M.-H. Anti-cancer efficacy of dietary polyphenols is mediated through epigenetic modifications. Curr. Opin. Food Sci. 2016, 8, 1–7. [Google Scholar] [CrossRef]
- Amiot, M.J.; Riva, C.; Vinet, A. Effects of dietary polyphenols on metabolic syndrome features in humans: A systematic review. Obes. Rev. 2016, 17, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Santhakumar, A.B.; Battino, M.; Alvarez-Suarez, J.M. Dietary polyphenols: Structures, bioavailability and protective effects against atherosclerosis. Food Chem. Toxicol. 2018, 113, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Shahzad, B.; Rehman, A.; Bhardwaj, R.; Landi, M.; Zheng, B. Response of Phenylpropanoid Pathway and the Role of Polyphenols in Plants under Abiotic Stress. Molecules 2019, 24, 2452. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, H.; Kucinska, M.; Murias, M. Biological activity of piceatannol: Leaving the shadow of resveratrol. Mutat. Res. Mol. Mech. Mutagen. 2012, 750, 60–82. [Google Scholar] [CrossRef]
- Chen, M.-K.; Liu, Y.-T.; Lin, J.-T.; Lin, C.-C.; Chuang, Y.-C.; Lo, Y.-S.; Hsi, Y.-T.; Hsieh, M.-J. Pinosylvin reduced migration and invasion of oral cancer carcinoma by regulating matrix metalloproteinase-2 expression and extracellular signal-regulated kinase pathway. Biomed. Pharmacother. 2019, 117, 109160. [Google Scholar] [CrossRef]
- Martínez Conesa, C.; Vicente Ortega, V.; Yáñez Gascón, M.J.; Alcaraz Baños, M.; Canteras Jordana, M.; Benavente-García, O.; Castillo, J. Treatment of metastatic melanoma B16F10 by the flavonoids tangeretin, rutin, and diosmin. J. Agric. Food Chem. 2005, 53, 6791–6797. [Google Scholar] [CrossRef]
- García-Lafuente, A.; Guillamón, E.; Villares, A.; Rostagno, M.A.; Martínez, J.A. Flavonoids as anti-inflammatory agents: Implications in cancer and cardiovascular disease. Inflamm. Res. 2009, 58, 537–552. [Google Scholar] [CrossRef]
- Liskova, A.; Koklesova, L.; Samec, M.; Smejkal, K.; Samuel, S.M.; Varghese, E.; Abotaleb, M.; Biringer, K.; Kudela, E.; Danko, J.; et al. Flavonoids in Cancer Metastasis. Cancers 2020, 12, 1498. [Google Scholar] [CrossRef]
- Njenga, P.K.; Mugo, S.M.; Zhou, T.J. Characterization of Polyphenols, Flavonoids and Their Anti-microbial Activity in the Fruits of Vangueria madagascariensis JF Gmel. Eur. J. Med. Plants 2020, 31, 24–37. [Google Scholar] [CrossRef]
- Pedrosa, M.O.; Da Cruz, R.D.; Viana, J.O.; De Moura, R.; Ishiki, H.; Filho, J.B.; Diniz, M.; Scotti, M.; Scotti, L.; Mendonca, F.B. Hybrid Compounds as Direct Multitarget Ligands: A Review. Curr. Top. Med. Chem. 2017, 17, 1044–1079. [Google Scholar] [CrossRef]
- Wu, F.; Cui, L.J. Resveratrol suppresses melanoma by inhibiting NF-κB/miR-221 and inducing TFG expression. Arch. Dermatol. Res. 2017, 309, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Abotaleb, M.; Samuel, S.M.; Varghese, E.; Varghese, S.; Kubatka, P.; Liskova, A.; Büsselberg, D. Flavonoids in Cancer and Apoptosis. Cancers 2018, 11, 28. [Google Scholar] [CrossRef]
- Menezes, J.C.J.M.D.S.; Orlikova, B.; Morceau, F.; Diederich, M. Natural and Synthetic Flavonoids: Structure–Activity Relationship and Chemotherapeutic Potential for the Treatment of Leukemia. Crit. Rev. Food Sci. Nutr. 2015, 56, S4–S28. [Google Scholar] [CrossRef] [PubMed]
- Sirerol, J.A.; Rodríguez, M.L.; Mena, S.; Asensi, M.A.; Estrela, J.M.; Ortega, A.L. Role of Natural Stilbenes in the Prevention of Cancer. Oxidative Med. Cell. Longev. 2016, 2016, 3128951. [Google Scholar] [CrossRef] [PubMed]
- Spoerlein, C.; Mahal, K.; Schmidt, H.; Schobert, R. Effects of chrysin, apigenin, genistein and their homoleptic copper (II) complexes on the growth and metastatic potential of cancer cells. J. Inorg. Biochem. 2013, 127, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Kasala, E.R.; Bodduluru, L.N.; Madana, R.M.; Gogoi, R.; Barua, C.C. Chemopreventive and therapeutic potential of chrysin in cancer: Mechanistic perspectives. Toxicol. Lett. 2015, 233, 214–225. [Google Scholar] [CrossRef]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef]
- Estrela, J.M.; Mena, S.; Obrador, E.; Benlloch, M.; Castellano, G.; Salvador, R.; Dellinger, R.W. Polyphenolic Phytochemicals in Cancer Prevention and Therapy: Bioavailability versus Bioefficacy. J. Med. Chem. 2017, 60, 9413–9436. [Google Scholar] [CrossRef]
- Senggunprai, L.; Kukongviriyapan, V.; Prawan, A.; Kukongviriyapan, U. Quercetin and EGCG Exhibit Chemopreventive Effects in Cholangiocarcinoma Cells via Suppression of JAK/STAT Signaling Pathway. Phyther. Res. 2014, 28, 841–848. [Google Scholar] [CrossRef]
- Arias, N.; Macarulla, M.T.; Aguirre, L.; Milton, I.; Portillo, M.P. The combination of resveratrol and quercetin enhances the individual effects of these molecules on triacylglycerol metabolism in white adipose tissue. Eur. J. Nutr. 2015, 55, 341–348. [Google Scholar] [CrossRef]
- Brents, L.K.; Medina-Bolivar, F.; Seely, K.A.; Nair, V.; Bratton, S.M.; Ñopo-Olazabal, L.; Patel, R.Y.; Liu, H.; Doerksen, R.J.; Prather, P.L.; et al. Natural prenylated resveratrol analogs arachidin-1 and -3 demonstrate improved glucuronidation profiles and have affinity for cannabinoid receptors. Xenobiotica 2011, 42, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.-C.; Wu, J.M. Targeting CWR22Rv1 prostate cancer cell proliferation and gene expression by combinations of the phytochemicals EGCG, genistein and quercetin. Anticancer Res. 2009, 29, 4025–4032. [Google Scholar] [PubMed]
- Erridge, S.; Mangal, N.; Salazar, O.; Pacchetti, B.; Sodergren, M.H. Cannflavins–From plant to patient: A scoping review. Fitoterapia 2020, 146, 104712. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.; Scutt, A.; Evans, F.J. Cannflavin A and B, prenylated flavones from Cannabis sativa L. Experientia 1986, 42, 452–453. [Google Scholar] [CrossRef]
- Barrett, M.; Gordon, D.; Evans, F.J. Isolation from Cannabis sativa L. of cannflavin—A novel inhibitor of prostaglandin production. Biochem Pharmacol. 1985, 34, 2019–2024. [Google Scholar] [CrossRef]
- Seegers, J. Identification and Characterization of Natural Products as Dual Inhibitors of Microsomal Prostaglandin E2 Synthase-1 and 5-Lipoxygenase. Ph.D. Thesis, Universität Tübingen, Tübingen, Germany, 2014. [Google Scholar]
- Werz, O.; Seegers, J.; Schaible, A.M.; Weinigel, C.; Barz, D.; Koeberle, A.; Allegrone, G.; Pollastro, F.; Zampieri, L.; Grassi, G.; et al. Cannflavins from hemp sprouts, a novel cannabinoid-free hemp food product, target microsomal prostaglandin E2 synthase-1 and 5-lipoxygenase. PharmaNutrition 2014, 2, 53–60. [Google Scholar] [CrossRef]
- Kim, S.-H.; Hashimoto, Y.; Cho, S.-N.; Roszik, J.; Milton, D.R.; Dal, F.; Kim, S.F.; Menter, D.G.; Yang, P.; Ekmekcioglu, S.; et al. Microsomal PGE2 synthase-1 regulates melanoma cell survival and associates with melanoma disease progression. Pigment Cell Melanoma Res. 2016, 29, 297–308. [Google Scholar] [CrossRef]
- Choi, E.J.; Kim, G.-H. Apigenin causes G2/M arrest associated with the modulation of p21Cip1 and Cdc2 and activates p53-dependent apoptosis pathway in human breast cancer SK-BR-3 cells. J. Nutr. Biochem. 2009, 20, 285–290. [Google Scholar] [CrossRef]
- Seo, H.-S.; Choi, H.-S.; Kim, S.-R.; Choi, Y.K.; Woo, S.-M.; Shin, I.; Woo, J.-K.; Park, S.-Y.; Shin, Y.C.; Ko, S.-K.J.M.; et al. Apigenin induces apoptosis via extrinsic pathway, inducing p53 and inhibiting STAT3 and NFκB signaling in HER2-overexpressing breast cancer cells. Mol. Cell. Biochem. 2012, 366, 319–334. [Google Scholar] [CrossRef]
- Shukla, S.; Gupta, S. Apigenin-induced prostate cancer cell death is initiated by reactive oxygen species and p53 activation. Free. Radic. Biol. Med. 2008, 44, 1833–1845. [Google Scholar] [CrossRef]
- Mirzoeva, S.; Franzen, C.A.; Pelling, J.C. Apigenin inhibits TGF-β-induced VEGF expression in human prostate carcinoma cells via a Smad2/3-and Src-dependent mechanism. Mol. Carcinog. 2014, 53, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Chao, S.-C.; Huang, S.-C.; Hu, D.-N.; Lin, H.-Y. Subtoxic levels of apigenin inhibit expression and secretion of VEGF by uveal melanoma cells via suppression of ERK1/2 and PI3K/Akt pathways. Evid.-Based Complement. Altern. Med. 2013, 2013, 817674. [Google Scholar] [CrossRef] [PubMed]
- Kiraly, A.J.; Soliman, E.; Jenkins, A.; Van Dross, R.T. Apigenin inhibits COX-2, PGE2, and EP1 and also initiates terminal differentiation in the epidermis of tumor bearing mice. Prostaglandins Leukot. Essent. Fat. Acids 2015, 104, 44–53. [Google Scholar] [CrossRef]
- Yan, X.; Qi, M.; Li, P.; Zhan, Y.; Shao, H. Apigenin in cancer therapy: Anti-cancer effects and mechanisms of action. Cell Biosci. 2017, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yufei, Z.; Yuqi, W.; Binyue, H.; Lingchen, T.; Xi, C.; Hoffelt, D.; Fuliang, H. Chrysin Inhibits Melanoma Tumor Metastasis via Interfering with the FOXM1/β-Catenin Signaling. J. Agric. Food Chem. 2020, 68, 9358–9367. [Google Scholar] [CrossRef]
- Maruyama, H.; Kawakami, F.; Lwin, T.-T.; Imai, M.; Shamsa, F. Biochemical Characterization of Ferulic Acid and Caffeic Acid Which Effectively Inhibit Melanin Synthesis via Different Mechanisms in B16 Melanoma Cells. Biol. Pharm. Bull. 2018, 41, 806–810. [Google Scholar] [CrossRef]
- Park, H.J.; Cho, J.H.; Hong, S.H.; Kim, D.H.; Jung, H.Y.; Kang, I.K.; Cho, Y.J. Whitening and anti-wrinkle activities of ferulic acid isolated from Tetragonia tetragonioides in B16F10 melanoma and CCD-986sk fibroblast cells. J. Nat. Med. 2018, 72, 127–135. [Google Scholar] [CrossRef]
- Kim, J.K.; Park, S.U. A recent overview on the biological and pharmacological activities of ferulic acid. Excli J. 2019, 18, 132–138. [Google Scholar]
- Kamm, A.; Przychodzeń, P.; Kuban-Jankowska, A.; Marino Gammazza, A.; Cappello, F.; Daca, A.; Żmijewski, M.A.; Woźniak, M.; Górska-Ponikowska, M. 2-Methoxyestradiol and its combination with a natural compound, ferulic acid, induces melanoma cell death via downregulation of Hsp60 and Hsp90. J. Oncol. 2019, 2019, 9293416. [Google Scholar] [CrossRef]
- Kumar, N.; Pruthi, V. Potential applications of ferulic acid from natural sources. Biotechnol. Rep. 2014, 4, 86–93. [Google Scholar] [CrossRef]
- Borgo, C.; Ruzzene, M.J. Role of protein kinase CK2 in antitumor drug resistance. J. Exp. Clin. Cancer Res. 2019, 38, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.S.; Lee, N.-H.; Hyun, C.-G.; Shin, D.-B. Differential effects of methoxylated p-coumaric acids on melanoma in B16/F10 cells. Prev. Nutr. Food Sci. 2015, 20, 73. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Wang, J.; Wu, Q.; Qian, J.; Yang, C.; Bo, P. Genistein inhibits the growth and regulates the migration and invasion abilities of melanoma cells via the FAK/paxillin and MAPK pathways. Oncotarget 2017, 8, 21674–21691. [Google Scholar] [CrossRef] [PubMed]
- Venza, I.; Visalli, M.; Oteri, R.; Beninati, C.; Teti, D.; Venza, M. Genistein reduces proliferation of EP3-expressing melanoma cells through inhibition of PGE2-induced IL-8 expression. Int. Immunopharmacol. 2018, 62, 86–95. [Google Scholar] [CrossRef]
- Yao, X.; Jiang, W.; Yu, D.; Yan, Z. Luteolin inhibits proliferation and induces apoptosis of human melanoma cells in vivo and in vitro by suppressing MMP-2 and MMP-9 through the PI3K/AKT pathway. Food Funct. 2019, 10, 703–712. [Google Scholar] [CrossRef]
- Kubo, I.; Nihei, K.-I.; Tsujimoto, K. Methyl p-coumarate, a melanin formation inhibitor in B16 mouse melanoma cells. Bioorganic Med. Chem. 2004, 12, 5349–5354. [Google Scholar] [CrossRef]
- Shen, Y.; Song, X.; Li, L.; Sun, J.; Jaiswal, Y.; Huang, J.; Liu, C.; Yang, W.; Williams, L.; Zhang, H.; et al. Protective effects of p-coumaric acid against oxidant and hyperlipidemia-an in vitro and in vivo evaluation. Biomed. Pharmacother. 2018, 110, 579–587. [Google Scholar] [CrossRef]
- Sharma, S.H.; Kumar, J.S.; Chellappan, D.R.; Nagarajan, S. Molecular chemoprevention by morin—A plant flavonoid that targets nuclear factor kappa B in experimental colon cancer. Biomed. Pharmacother. 2018, 100, 367–373. [Google Scholar] [CrossRef]
- Arruda, C.; Ribeiro, V.P.; Almeida, M.O.; Mejía, J.A.A.; Casoti, R.; Bastos, J.K. Effect of light, oxygen and temperature on the stability of artepillin C and p-coumaric acid from Brazilian green propolis. J. Pharm. Biomed. Anal. 2020, 178, 112922. [Google Scholar] [CrossRef]
- Ferry, D.R.; Smith, A.; Malkhandi, J.; Fyfe, D.W.; Detakats, P.G.; Anderson, D.; Baker, J.; Kerr, D.J. Phase I clinical trial of the flavonoid quercetin: Pharmacokinetics and evidence for in vivo tyrosine kinase inhibition.. Clin. Cancer Res. 1996, 2, 659–668. [Google Scholar]
- Sharmila, G.; Bhat, F.; Arunkumar, R.; Elumalai, P.; Singh, P.R.; Senthilkumar, K.; Arunakaran, J. Chemopreventive effect of quercetin, a natural dietary flavonoid on prostate cancer in in vivo model. Clin. Nutr. 2013, 33, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Ezzati, M.; Yousefi, B.; Velaei, K.; Safa, A. A review on anti-cancer properties of Quercetin in breast cancer. Life Sci. 2020, 248, 117463. [Google Scholar] [CrossRef] [PubMed]
- Hundsberger, H.; Stierschneider, A.; Sarne, V.; Ripper, D.; Schimon, J.; Weitzenböck, H.; Schild, D.; Jacobi, N.; Eger, A.; Atzler, J.; et al. Concentration-Dependent Pro- and Antitumor Activities of Quercetin in Human Melanoma Spheroids: Comparative Analysis of 2D and 3D Cell Culture Models. Molecules 2021, 26, 717. [Google Scholar] [CrossRef] [PubMed]
- Guruvayoorappan, C.; Kuttan, G.J. Antiangiogenic effect of rutin and its regulatory effect on the production of VEGF, IL-1β and TNF-α in tumor associated macrophages. J. Biol. Sci. 2007, 7, 1511–1519. [Google Scholar] [CrossRef][Green Version]
- Menon, L.G.; Kuttan, R.; Kuttan, G. Inhibition of lung metastasis in mice induced by B16F10 melanoma cells by polyphenolic compounds. Cancer Lett. 1995, 95, 221–225. [Google Scholar] [CrossRef]
- Corina, D.; Florina, B.; Iulia, P.; Cristina, D.; Rita, A.; Alexandra, P.; Virgil, P.; Hancianu, M.; Daliana, M.; Codruta, S. Rutin and Its Cyclodextrin Inclusion Complexes: Physico-chemical Evaluation and in vitro Activity on B164A5 Murine Melanoma Cell Line. Curr. Pharm. Biotechnol. 2018, 18, 1067–1077. [Google Scholar] [CrossRef]
- Beyerstedt, S.; Franco, M.; Oliveira, T.; Mendonça, G.; Alves-Fernandes, D.; Maria-Engler, S.; Machado-Neto, J.; Lopes, L. Targeting protein disulfide isomerase to overcome resistance to BRAF inhibitors in melanoma. Free Radic. Biol. Med. 2018, 128, S62. [Google Scholar] [CrossRef]
- Nobili, S.; Lippi, D.; Witort, E.; Donnini, M.; Bausi, L.; Mini, E.; Capaccioli, S. Natural compounds for cancer treatment and prevention. Pharmacol. Res. 2009, 59, 365–378. [Google Scholar] [CrossRef]
- Otvos, R.A.; Still, K.B.; Somsen, G.W.; Smit, A.B.; Kool, J. Drug Discovery on Natural Products: From Ion Channels to nAChRs, from Nature to Libraries, from Analytics to Assays. SLAS Discov. Adv. Sci. Drug Discov. 2019, 24, 362–385. [Google Scholar] [CrossRef]

| Terpenoid | Cell Type | Effect | In Vitro/ In Vivo | Reference |
|---|---|---|---|---|
| α-Pinene | B16F10 murine melanoma cell line syngeneic in C57Bl/6 mice | ↓ Lung tumor nodules ↓ Metastatic activity ↑ ROS production ↑ Early apoptotic features such as: DNA fragmentation Phosphatidylserine on the cell surface Disruption of mitochondrial membrane potential | In vitro/In vivo | [153] |
| Luteolin | SK-Mel2, A375 and SK-Mel28, WM3211 Athymic nude mice (Strain 490) | ↓ Cell growth via: Extracellular matrix, oncogenic signaling, and immune response pathways Not through ROS induction | In vitro/In vivo | [154] |
| Phenol | Cell Lines | Anticancer Effect | Mechanism of Action | In Vivo/In Vitro | References |
|---|---|---|---|---|---|
| Apigenin | A375SM human melanoma cells | ↓ Cell viability ↑ Apoptosis ↓ Tumour growth | ↑ Apoptosis via regulating the Akt and mitogen-activated protein kinase signalling pathway | In vitro/In vivo | [155] |
| Ferulic Acid | Melanoma A375, CHL-1, SK-MEL-2, B16F10 cells B16F10 cells in female C57BL mice | ↓ Proliferation ↓ Angiogenesis In vivo | FGFR1-mediated PI3K-Akt signalling pathway Blocking of the PI3K-Akt pathway | In vitro/In vivo | [156] |
| Quercetin | B16 and A375 murine model | ↓ Tumour growth ↓ Proliferation ↑ Apoptosis | ↑ IFN-α and IFN-β expression through activation of RIG-I promoter in B16 cells | In vivo | [157] |
| B164A5 murine melanoma cell line | ↓ Mitochondrial respiration ↑ Apoptotic ↓ Proliferative | ↓ OCR ↓ ECAR Modulated glycolytic and mitochondrial pathways for ATP production | In vitro | [158] | |
| DB-1 | Targetted Tyr+ expressing melanoma cells ↑ Apoptosis | ↓GSH ↓ Bio reduction capacity ↑ ROS | In vitro | [159] | |
| Murine B16-F10 melanoma cells | ↑ Melanin production ↓ Cell viability | ↑ Activity and synthesis of tyrosinase | In vitro | [160] | |
| Human melanoma A375 and A2058 cells, and B16F10 cells in male C57BL model | ↓ Proliferation ↑ Apoptosis ↓ Migration and Invasive ↓ Metastasis | ↓ STAT3 signalling Interfered with STAT3 phosphorylation ↓ STAT3 nuclear localization ↓ A375 tumour growth ↓ STAT3 signalling | In vitro/In vivo | [161] | |
| A375SM and A375P human melanoma cells | ↓ Viability and proliferation of A375SM cells No effect on A375P cells ↓ A375SM tumour volume ↑ Apoptosis | ↑ Expression of Bax, phospho-JNK, phospho-p38 and phospho-ERK1/2 Cleaved poly-ADP ribose polymerases ↓ Bcl-2 | In vitro/In vivo | [162] | |
| B16 | ↓ Proliferation ↓ Proportion of cells in S and G2/M stages of the cell cycle ↑ SubG1 population of treated cells | Cell cycle disruption | In vitro | [163] | |
| ↓ HGF ↓ Cell migration ↓ Metastasis | ↓ HGF-induced melanoma cell migration ↓ c-Met homo-dimerization and phosphorylation ↓ c-Met protein expression ↓ Activation of c-Met and downstream Gabl, FAK and PAK Suppression of the HGF/c-Met signaling pathway | [164] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schanknecht, E.; Bachari, A.; Nassar, N.; Piva, T.; Mantri, N. Phytochemical Constituents and Derivatives of Cannabis sativa; Bridging the Gap in Melanoma Treatment. Int. J. Mol. Sci. 2023, 24, 859. https://doi.org/10.3390/ijms24010859
Schanknecht E, Bachari A, Nassar N, Piva T, Mantri N. Phytochemical Constituents and Derivatives of Cannabis sativa; Bridging the Gap in Melanoma Treatment. International Journal of Molecular Sciences. 2023; 24(1):859. https://doi.org/10.3390/ijms24010859
Chicago/Turabian StyleSchanknecht, Ellen, Ava Bachari, Nazim Nassar, Terrence Piva, and Nitin Mantri. 2023. "Phytochemical Constituents and Derivatives of Cannabis sativa; Bridging the Gap in Melanoma Treatment" International Journal of Molecular Sciences 24, no. 1: 859. https://doi.org/10.3390/ijms24010859
APA StyleSchanknecht, E., Bachari, A., Nassar, N., Piva, T., & Mantri, N. (2023). Phytochemical Constituents and Derivatives of Cannabis sativa; Bridging the Gap in Melanoma Treatment. International Journal of Molecular Sciences, 24(1), 859. https://doi.org/10.3390/ijms24010859