
Targeting Energy Protection as a Novel Strategy to Disclose Di’ao Xinxuekang against the Cardiotoxicity Caused by Doxorubicin
 and
and
Abstract
1. Introduction
2. Results
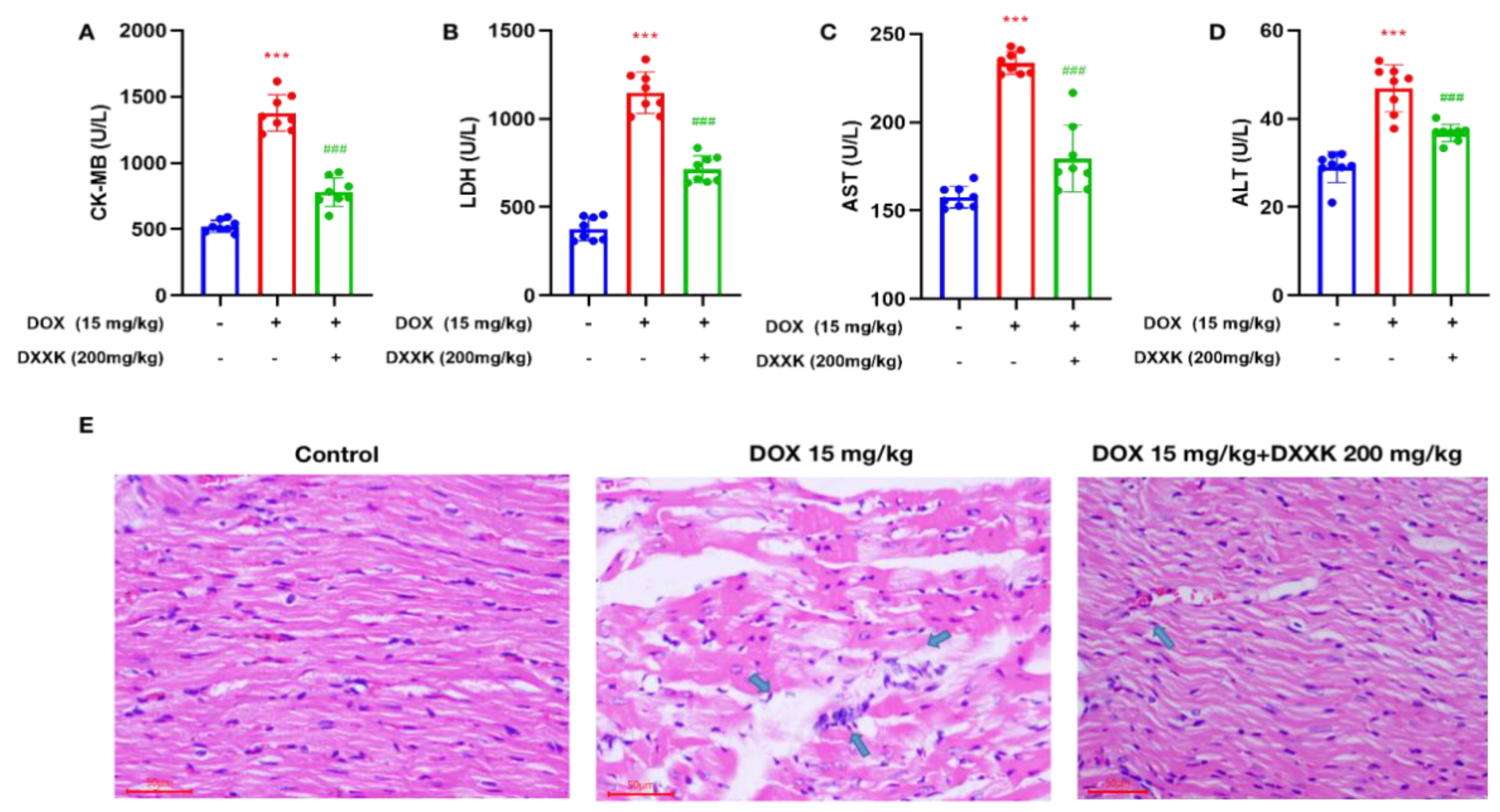
2.1. DXXK Protects against Heart Injury Caused by DOX
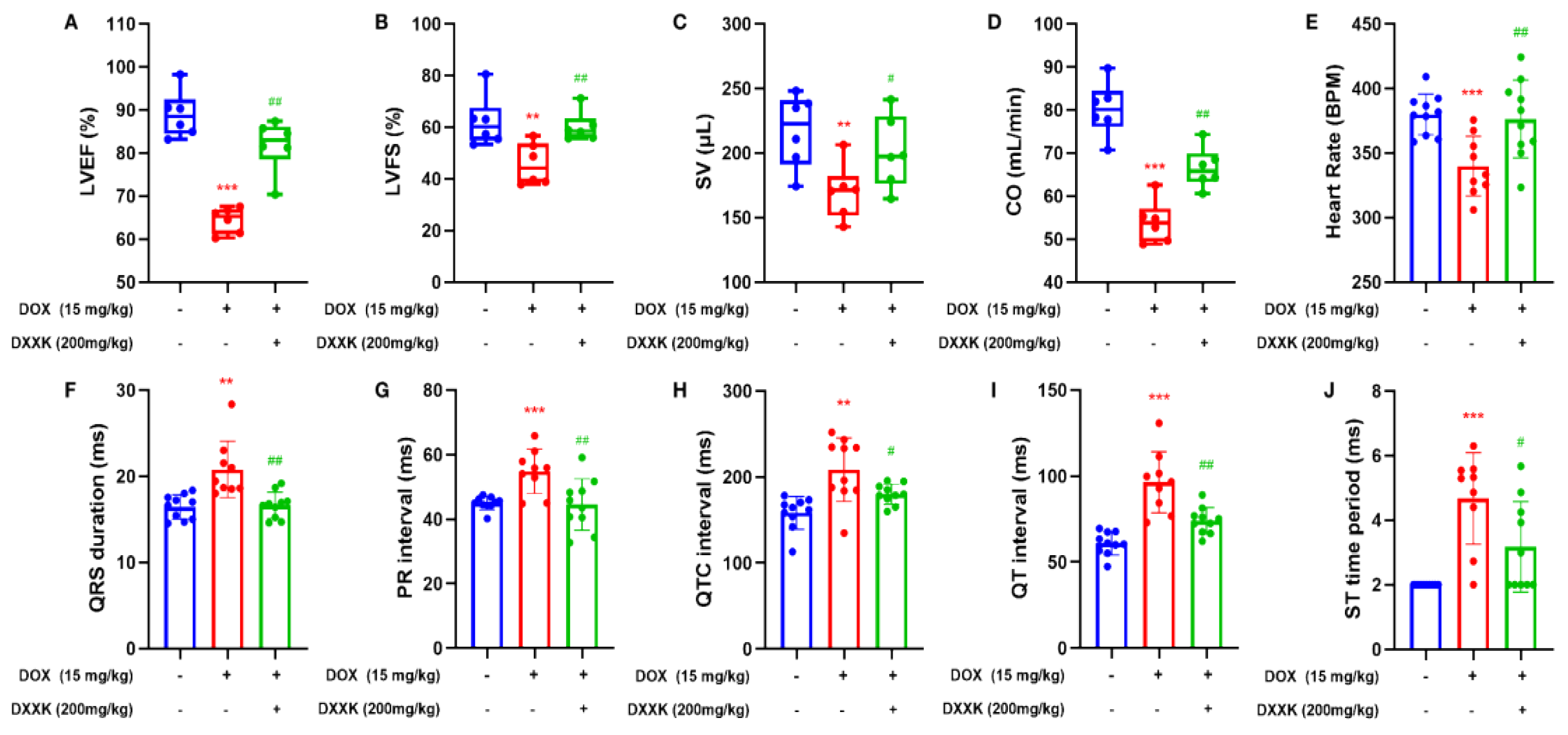
2.2. DXXK Improves DOX-Induced Cardiac Function Decline and Electrical Signal Conduction Block in Rats
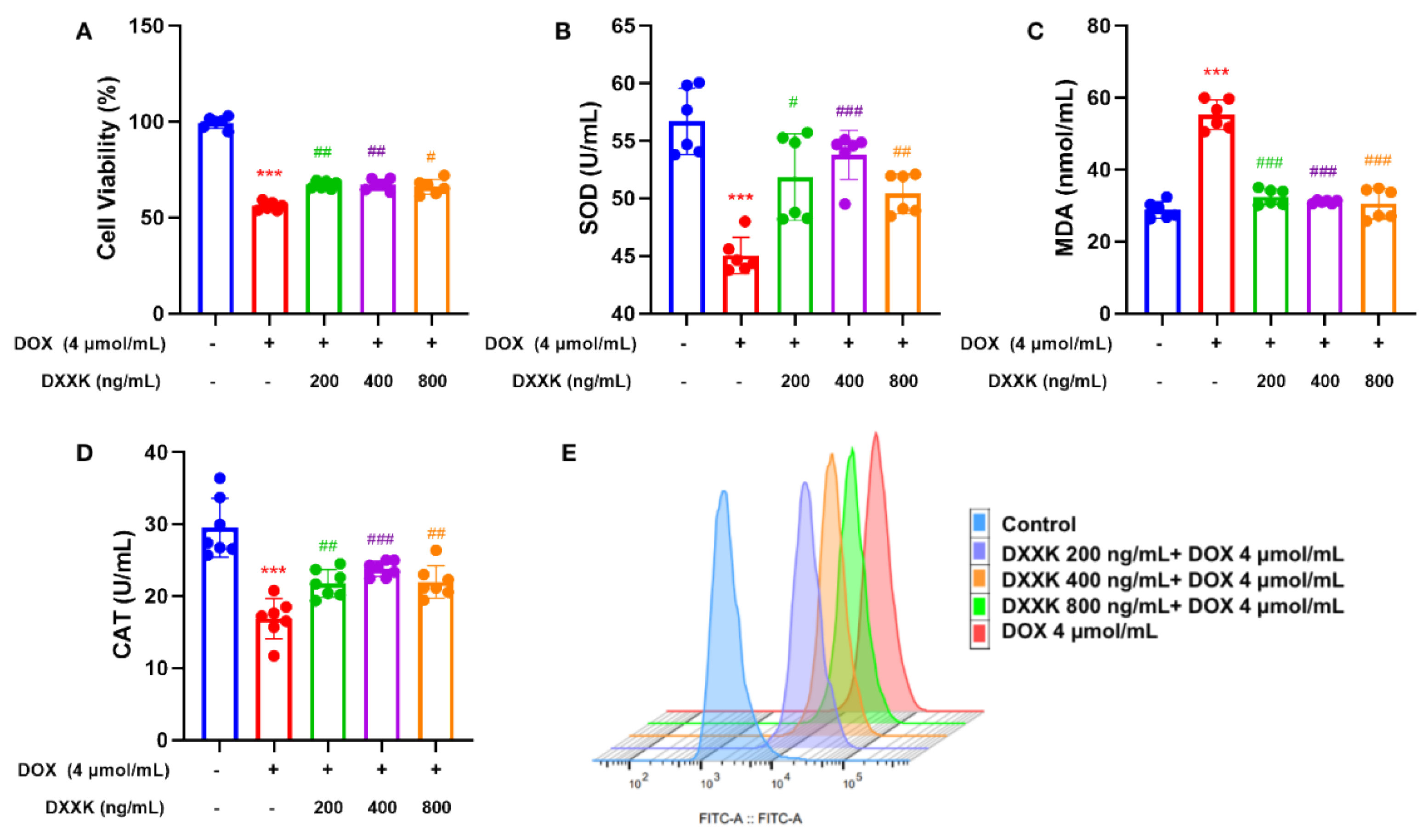
2.3. DXXK Protects H9c2 Cells from DOX-Induced Injury
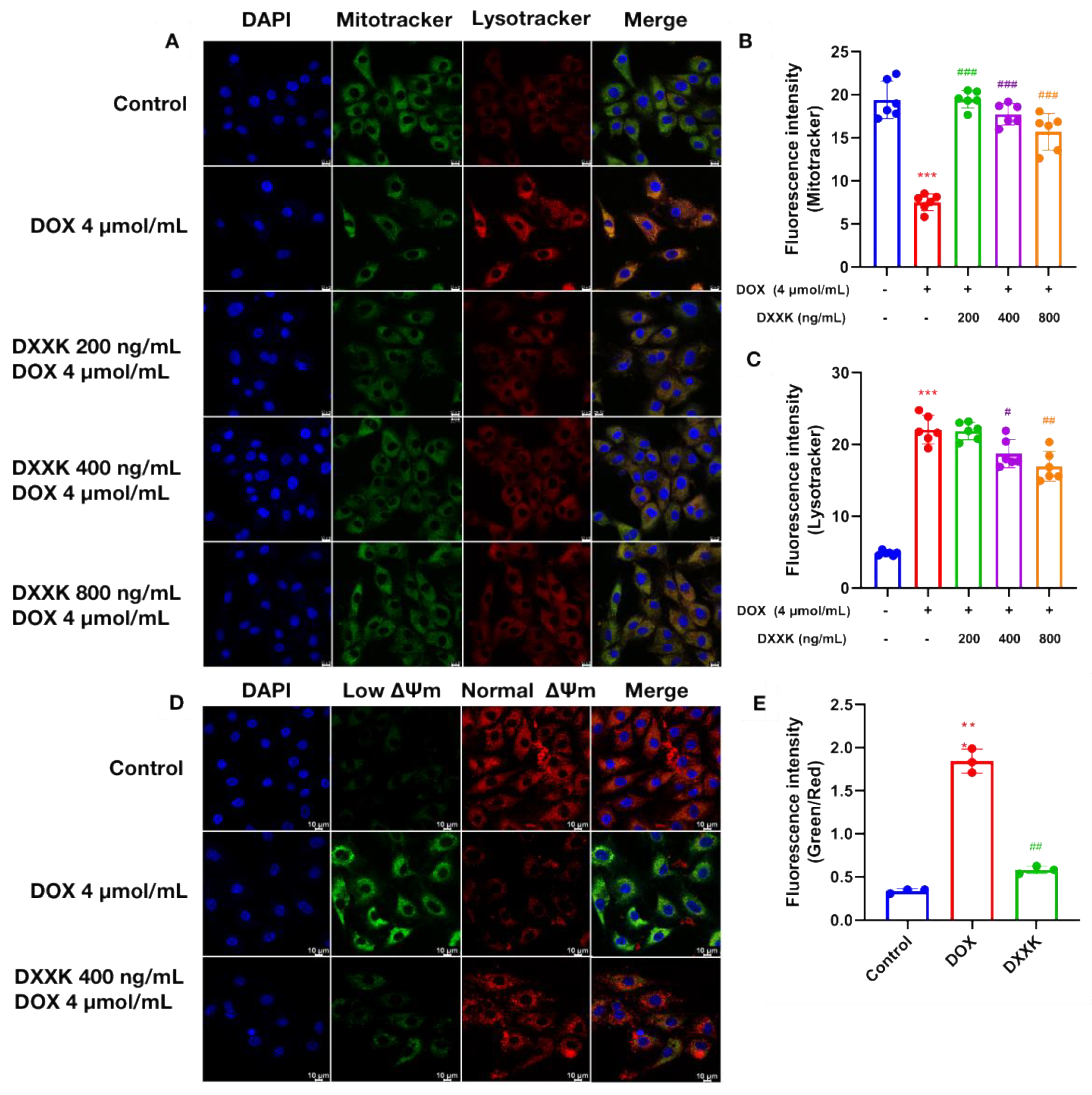
2.4. DXXK Protects Myocardial Mitochondria from DOX-Induced Damage
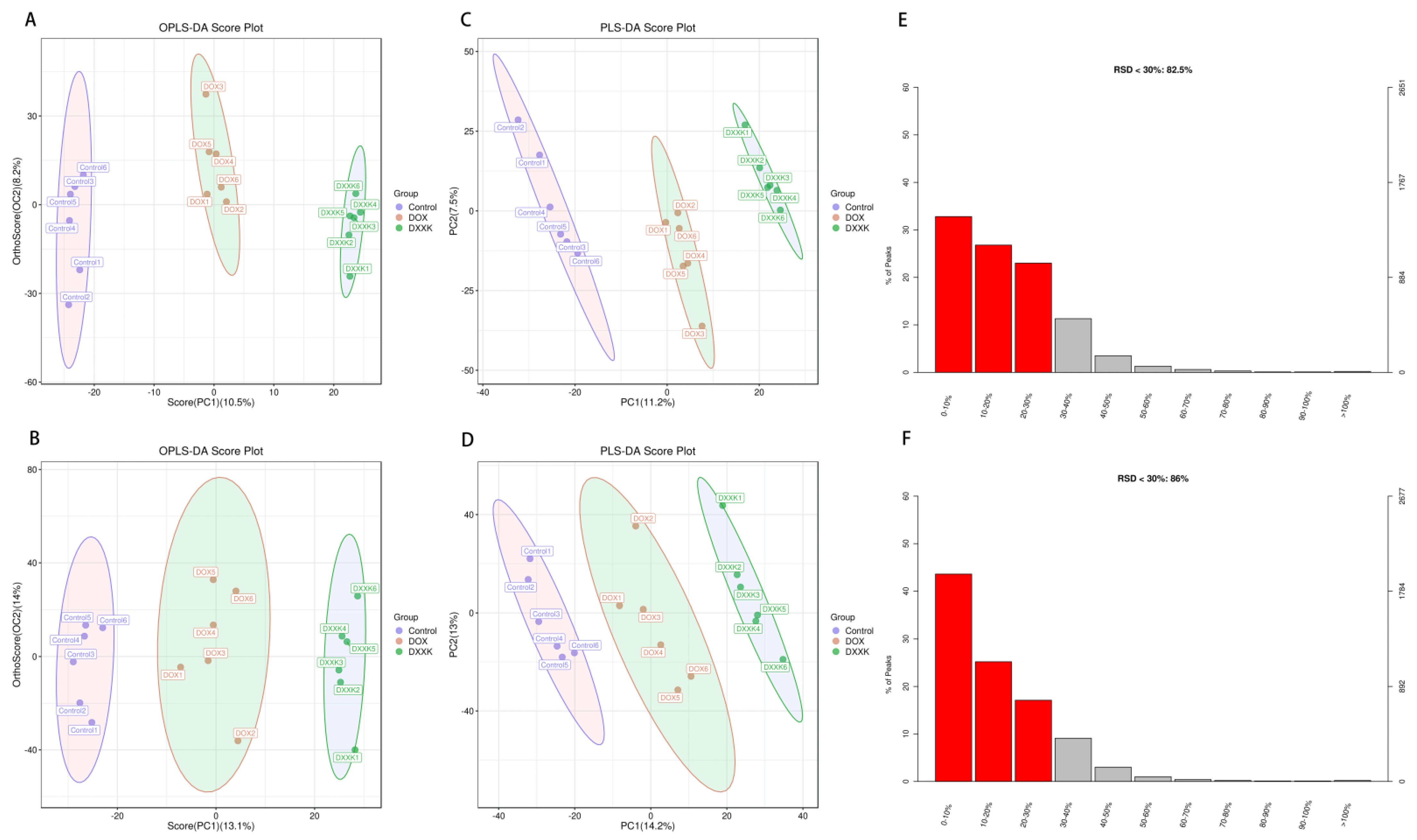
2.5. DOX Induces Metabolic Disorder in H9c2 Cells
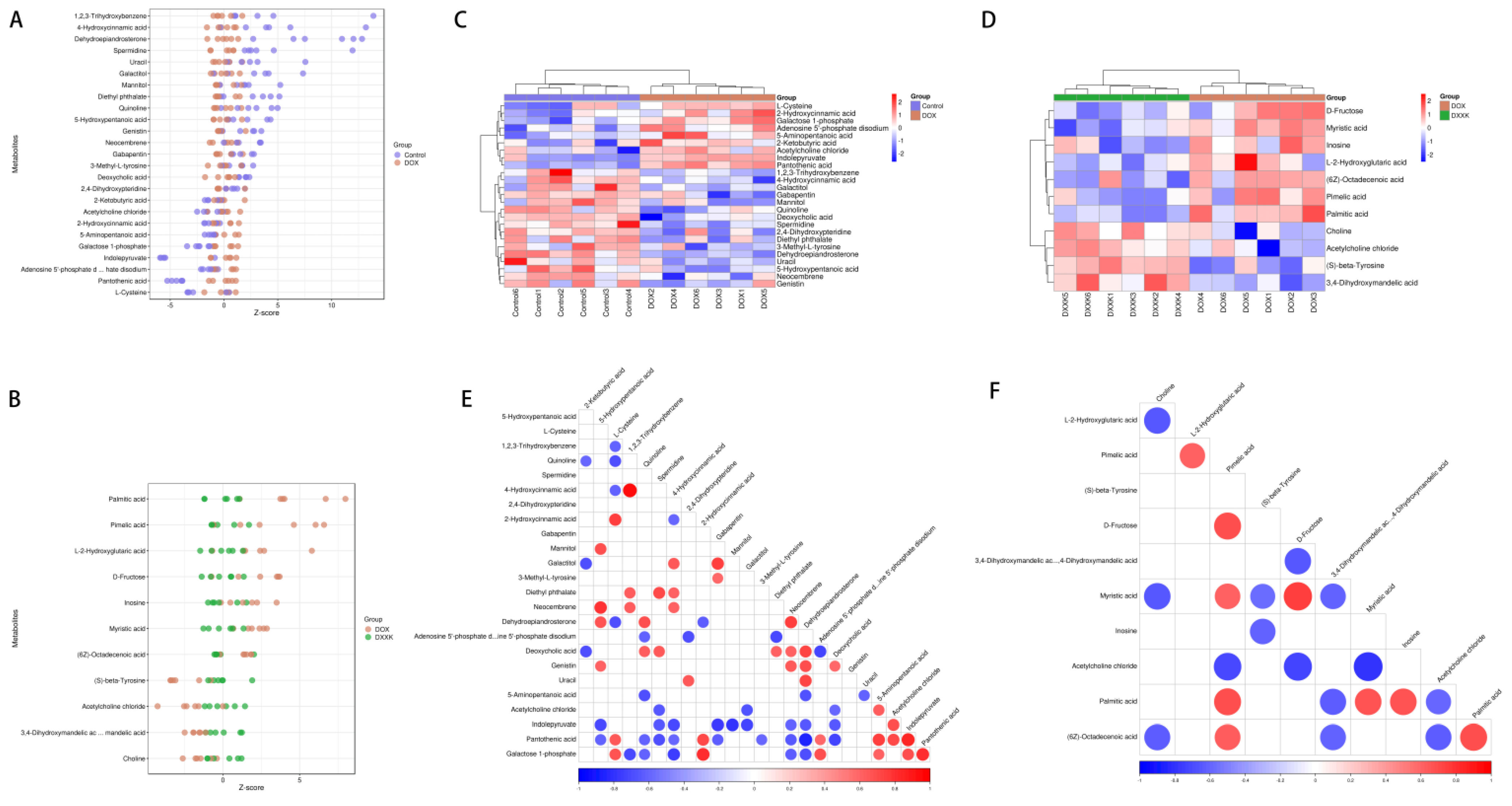
2.6. Identification of Potential Biomarkers of DOX-Induced Cardiotoxicity and DXXK Cardioprotection
2.7. Pathway Analysis of DXXK Protecting Myocardial Cells from DOX-Induced Injury
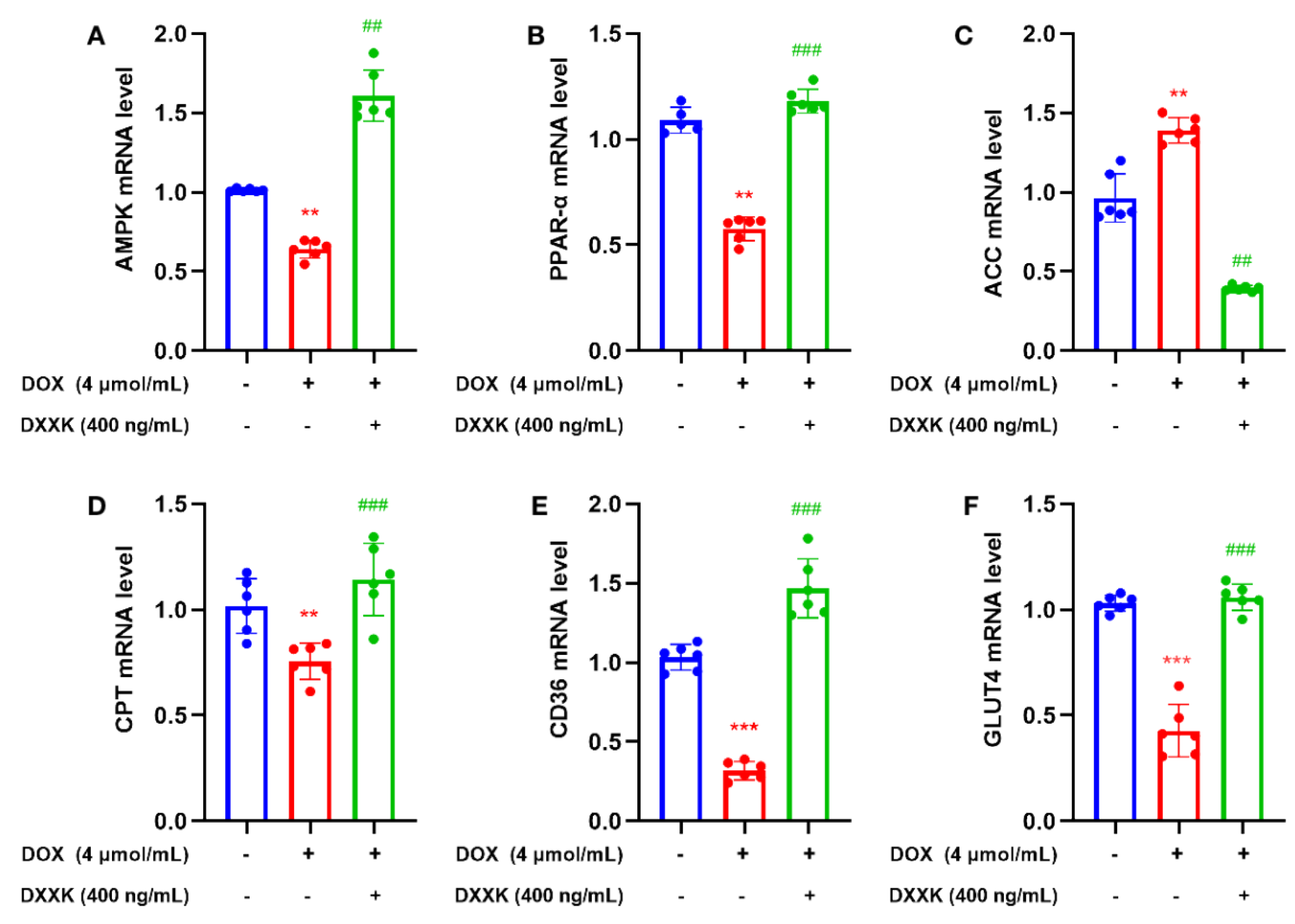
2.8. DXXK Promotes the Expression of Key Target mRNAs in the Myocardial Energy Metabolism Pathway
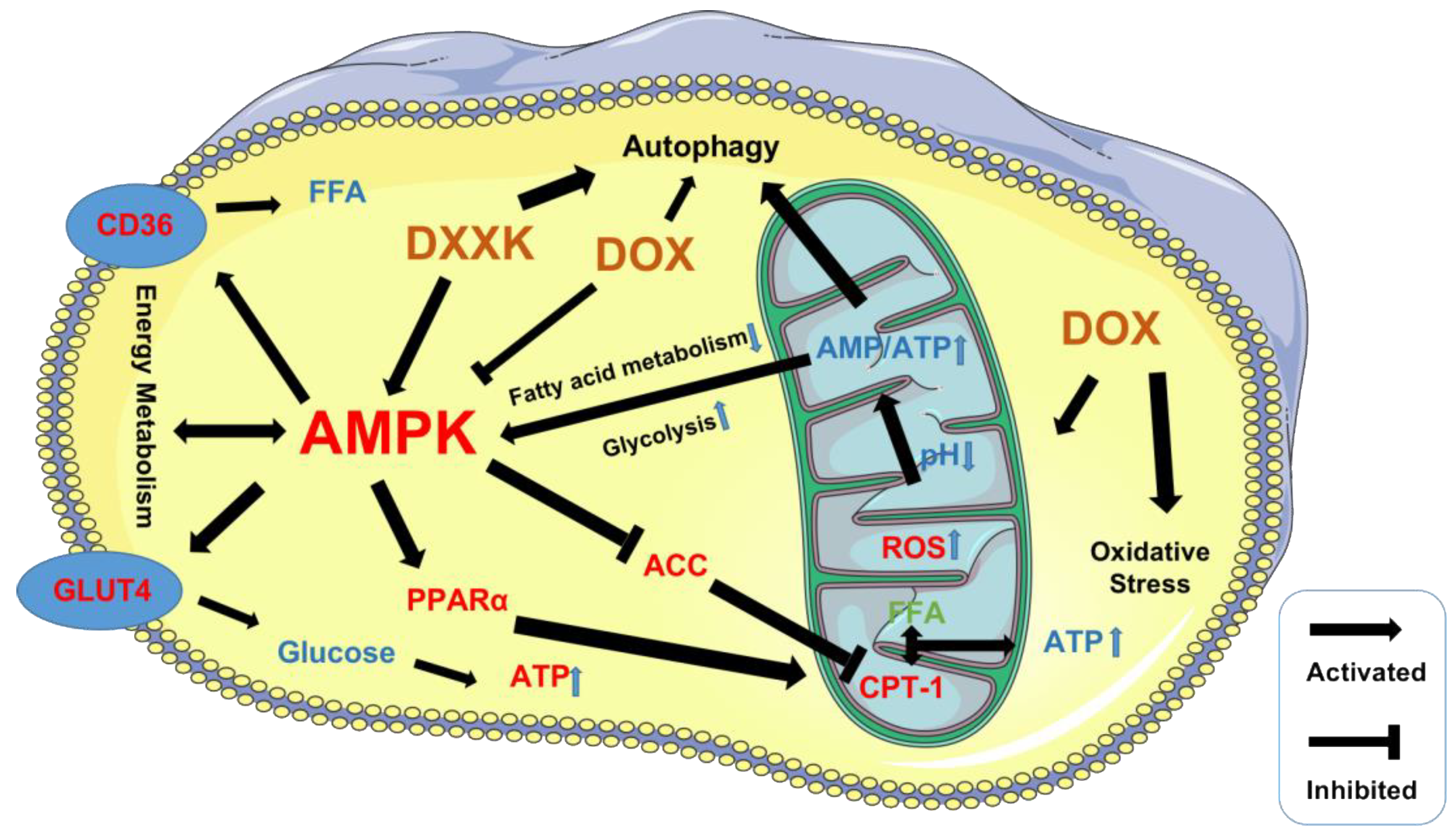
3. Discussion
4. Materials and Methods
4.1. Animal Handling
4.2. Detection of Pharmacodynamic Indexes
4.3. Reagents and Antibodies
4.4. Cell Survival Rate
4.5. Oxidative Stress Detection
4.6. Detection of Mitochondria and Lysosomes by Laser Confocal Method
4.7. Extraction of Intracellular Metabolites
4.8. RUPLC-Q-TOF-MS Conditions
4.9. Data Statistics and Analysis
4.10. PCR Detection
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin—A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef] [PubMed]
- Joachim, B.; Steen, M.; Peter, E.L. Cumulative anthracycline exposure and risk of cardiotoxicity; a danish nationwide cohort study of 2440 lymphoma patients treated with or without anthracyclines. Br. J. Haematol. 2018, 183, 717–726. [Google Scholar] [CrossRef]
- Skalicka, H.; Pudil, R.; Gregor, P. Summary of the 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC committee for practice guidelines: Prepared by the czech society of cardiology. Cor Et Vasa 2017, 59, e181–e195. [Google Scholar] [CrossRef]
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.R.; Ait-Oudhia, S. Insights into doxorubicin-induced cardiotoxicity: Molecular mechanisms, preventive strategies, and early monitoring. Mol. Pharmacol. 2019, 96, 219–232. [Google Scholar] [CrossRef]
- He, L.; Liu, F.; Li, J. Mitochondrial sirtuins and doxorubicin-induced cardiotoxicity. Cardiovasc. Toxicol. 2021, 21, 179–191. [Google Scholar] [CrossRef]
- Li, J.; Chang, H.; Banchs, J. Detection of subclinical cardiotoxicity in sarcoma patients receiving continuous doxorubicin infusion or pre-treatment with dexrazoxane before bolus doxorubicin. Cardio-Oncology 2020, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Koleini, N.; Nickel, B.E.; Edel, A.L. Oxidized phospholipids in doxorubicin-induced cardiotoxicity. Chem. Biol. Interact. 2019, 303, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Clayton, Z.S.; Brunt, V.E.; Hutton, D.A. Doxorubicin-Induced oxidative stress and endothelial dysfunction in conduit arteries is prevented by mitochondrial-specific antioxidant treatment. JACC CardioOncol. 2020, 2, 475–488. [Google Scholar] [CrossRef]
- Yu, J.; Wang, C.; Kong, Q. Recent progress in doxorubicin-induced cardiotoxicity and protective potential of natural products. Phytomedicine 2018, 40, 125–139. [Google Scholar] [CrossRef]
- Furqan, S.; Lee, D.L.; Sarah, A.l. Cardioprotection and second malignant neoplasms associated with dexrazoxane in children receiving anthracycline chemotherapy: A systematic review and meta-analysis. J. Natl. Cancer. Inst. 2016, 4, 46–69. [Google Scholar] [CrossRef]
- Chi, K.L.; Wu, J.C. Clinical trial in a dish: Using patient-derived induced pluripotent stem cells to identify risks of drug-Induced cardiotoxicity. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1019–1031. [Google Scholar] [CrossRef]
- Wu, B.B.; Kam, T.L.; Ellen, N. Mitochondrial-targeted therapy for doxorubicin-induced cardiotoxicity. Int. J. Mol. Sci. 2022, 23, 1912. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R. Cardiac energy metabolism in heart failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef] [PubMed]
- Greenwell, A.A.; Gopal, K.; Ussher, J.R. Myocardial energy metabolism in non-ischemic cardiomyopathy. Front. Physiol. 2020, 11, 570421. [Google Scholar] [CrossRef] [PubMed]
- Daniels, L.J.; Varma, U.; Annandale, M. Myocardial energy stress, autophagy induction, and cardiomyocyte functional responses. Antioxid. Redox. Signal. 2018, 31, 472–486. [Google Scholar] [CrossRef]
- Rawat, P.S.; Jaiswal, A.; Khurana, A.; Bhatti, J.S.; Navik, U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021, 139, 111708. [Google Scholar] [CrossRef]
- Wu, Y.P.; Zhang, S.; Xin, Y.F. Evidences for the mechanism of shenmai injection antagonizing doxorubicin-induced cardiotoxicity. Phytomedicine 2021, 88, 153597. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, V.W.; Cole, L.K.; Sparagna, G.C.; Hatch, G.M. Cardiac mitochondrial energy metabolism in heart failure: Role of cardiolipin and sirtuins. Biochim. Biophys. Acta 2016, 1861, 1544–1554. [Google Scholar] [CrossRef]
- He, Y.; Huang, W.; Zhang, C. Energy metabolism disorders and potential therapeutic drugs in heart failure. Acta Pharm. Sin. B. 2021, 11, 1098–1116. [Google Scholar] [CrossRef]
- Piquereau, J.; Boitard, S.E.; Ventura-Clapier, R.; Mericskay, M. Metabolic Therapy of heart failure: Is there a future for B vitamins? Int. J. Mol. Sci. 2021, 23, 30. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, T.; Ikeda, M.; Ide, T. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI. Insight. 2020, 5, e132747. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, S.; Autschbach, R. Doxorubicin in experimental and clinical heart failure. Eur. J. Cardiothorac. Surg. 2006, 30, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, H.; Han, D. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.Y.; Kang, Y.; Zhang, Z.; Yu, L.; Liu, X. Protective mechanism of DXXK on hypoxia/reoxygenation injury of cultured H9c2 cardiomyocytes. Pharmacol. Clin. Chin. Mater. Med. 2011, 27, 37–39. [Google Scholar] [CrossRef]
- Fang, X.J.; Jiao, J.J.; Zhao, Y.Q. Effect of dioscin on myocardial energy metabolism after ischemia reperfusion in rats. J. Tianjin Medical Univ. 2008, 4, 409–411+429. [Google Scholar] [CrossRef]
- Kang, Y.; Liu, X. Protective effect of dioscin on myocardial ischemia reperfusion injury and its mechanism. Chin. J. Pathophysiol. 2010, 10, 1990. [Google Scholar]
- Guo, S.; Zhang, Y.Y.; Peng, J.J.; Li, Y.Q.; Liu, W.N.; Tang, M.X.; Zhang, X.J.; Yang, J.; Peng, J.; Luo, X.J. Natural compound methyl protodioscin protects rat brain from ischemia/reperfusion injury through regulation of Mul1/SOD2 pathway. Eur. J. Pharmacol. 2019, 849, 50–58. [Google Scholar] [CrossRef]
- Zhao, L.S.; Tao, X.F.; Qi, Y.; Xu, L.N.; Yin, L.H.; Peng, J.Y. Protective effect of dioscin against doxorubicin-induced cardiotoxicity via adjusting microRNA-140-5p-mediated myocardial oxidative stress. Redox. Biol. 2018, 16, 189–198. [Google Scholar] [CrossRef]
- Song, Y.X.; Ou, Y.M.; Zhou, J.Y. Gracillin inhibits apoptosis and inflammation induced by lipopolysaccharide (LPS) to alleviate cardiac injury in mice via improving miR-29a. Biochem. Biophys. Res. Commun. 2020, 523, 580–587. [Google Scholar] [CrossRef]
- Neubauer, S. The failing heart an engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Al-Malky, H.S.; Al Harthi, S.E.; Osman, A.M. Major obstacles to doxorubicin therapy: Cardiotoxicity and drug resistance. J. Oncol. Pharm. Pract. 2020, 26, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.H.; Chen, C.; Wang, D.W. AMPK-α2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef]
- Guo, C.A.; Guo, S. Insulin receptor substrate signaling controls cardiac energy metabolism and heart failure. J. Endocrinol. 2017, 233, R131–R143. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.C.; Nabben, M.; Young, M.E.; Schulze, P.C.; Taegtmeyer, H.; Luiken, J.J.F.P. Re-balancing cellular energy substrate metabolism to mend the failing heart. Biochim. Biophys. Acta 2019, 1866, 165579. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, J.; Lu, Q.; Ren, D.; Sun, X.; Rousselle, T.; Tan, Y.; Li, J. AMPK: A therapeutic target of heart failure—Not only metabolism regulation. Biosci. Rep. 2018, 39, BSR20181767. [Google Scholar] [CrossRef] [PubMed]
- Qi, D.; Young, L. AMPK: Energy sensor and survival mechanism in the ischemic heart. Trends Endocrinol. Metab. 2015, 26, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Dyck, J. Is AMPK the savior of the failing heart? Trends Endocrinol. Metab. 2015, 26, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, S.; Pan, G.; Lin, L.; Liu, D.; Liu, Z.; Mei, S.; Zhang, L.; Hu, Z.; Chen, J.; et al. Modulatory effect of metformin on cardiotoxicity induced by doxorubicin via the MAPK and AMPK pathways. Life Sci. 2020, 249, 117498. [Google Scholar] [CrossRef]
- Liu, D.; Ma, Z.; Xu, L.; Zhang, X.; Qiao, S.; Yuan, J. PGC1-α activation by pterostilbene ameliorates acute doxorubicin cardiotoxicity by reducing oxidative stress via enhancing AMPK and SIRT1 cascades. Aging 2019, 11, 10061–10073. [Google Scholar] [CrossRef]
- Liu, D.; Ma, Z.; Di, S.; Yang, Y.; Yang, J.; Xu, L.; Reiter, R.J.; Qiao, S.; Yuan, J. AMPK/PGC1-α activation by melatonin attenuates acute doxorubicin cardiotoxicity via alleviating mitochondrial oxidative damage and apoptosis. Free Radic. Biol. Med. 2018, 129, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, L.; Xie, Y.; Wang, J.; Chen, X.; Zheng, S.; Li, Y.; Dang, Y. Cardiac contractility modulation ameliorates myocardial metabolic remodeling in a rabbit model of chronic heart failure through activation of AMPK and PPAR-α pathway. Open Med. (Wars) 2022, 17, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, S.; Liu, Q.; Shan, T.; Wang, X.; Feng, J.; Wang, Y. AMPK facilitates intestinal long-chain fatty acid uptake by manipulating CD36 expression and translocation. FASEB J. 2020, 34, 4852–4869. [Google Scholar] [CrossRef] [PubMed]
- Lepropre, S.; Kautbally, S.; Octave, M.; Ginion, A.; Onselaer, M.B.; Steinberg, G.R.; Kemp, B.E.; Hego, A.; Wéra, O.; Brouns, S.; et al. AMPK-ACC signaling modulates platelet phospholipids and potentiates thrombus formation. Blood 2018, 32, 1180–1192. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Mattos, A.; Shao, D.; Li, T.; Nabben, M.; Kim, M.; Wang, W. Preservation of myocardial fatty acid oxidation prevents diastolic dysfunction in mice subjected to angiotensin II infusion. J. Mol. Cell Cardiol. 2016, 100, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Riehle, C.; Abel, E. PGC-1 proteins and heart failure. Trends Cardiovasc. Med. 2012, 22, 98–105. [Google Scholar] [CrossRef]
- Qi, H.; Ren, J.; Ba, L. MSTN attenuates cardiac hypertrophy through inhibition of excessive cardiac autophagy by blocking AMPK /mTOR and miR-128/PPARγ/NF-κB. Mol. Ther. Nucleic Acids 2019, 19, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Y. AMPK and autophagy. Adv. Exp. Med. Biol. 2019, 1206, 85–108. [Google Scholar] [CrossRef]
- Madhavi, Y.V.; Gaikwad, N.; Yerra, V.G.; Kalvala, A.K.; Nanduri, S.; Kumar, A. Targeting AMPK in diabetes and diabetic complications: Energy homeostasis, autophagy and mitochondrial health. Curr. Med. Chem. 2018, 26, 5207–5229. [Google Scholar] [CrossRef]
- Tong, X.; Roman, R.; Liu, Z. AMP-activated protein kinase (AMPK) regulates autophagy, inflammation and immunity and contributes to osteoclast differentiation and functionabs. Biol. Cell 2020, 12, 251–264. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Name | m/z | rt | ppm | Formula | KEGG |
|---|---|---|---|---|---|---|
| M121T90 | L-Cysteine | 120.9811 | 89.7 | 0.740305618 | C3H7NO2S | C00097 |
| M218T245 | Pantothenic acid | 218.1026 | 245.1 | 1.300497289 | C9H17NO5 | C00864 |
| M111T750 | Uracil | 111.0181 | 750.2 | 5.46386555 | C4H4N2O2 | C00106 |
| M146T958_2 | Spermidine | 145.9846 | 957.7 | 14.71119161 | C7H19N3 | C00315 |
| M183T469 | Galactitol | 183.0864 | 469 | 0.485376675 | C6H14O6 | C01697 |
| M102T90_2 | 2-Ketobutyric acid | 102.0342 | 89.7 | 0.014065686 | C4H6O3 | C00109 |
| M117T877 | 5-Aminopentanoic acid | 116.9259 | 876.8 | 10.96036025 | C5H11NO2 | C00431 |
| M164T198 | 2,4-Dihydroxypteridine | 163.9405 | 197.8 | 2.624555689 | C6H4N4O2 | C03212 |
| M182T979 | Mannitol | 181.978 | 978.7 | 4.796869979 | C6H14O6 | C00392 |
| M288T647 | Dehydroepiandrosterone | 288.2897 | 647.2 | 0.540973302 | C19H28O2 | C01227 |
| M432T719 | Genistin | 432.238 | 718.6 | 0.472864253 | C21H20O10 | C09126 |
| M119T339 | 5-Hydroxypentanoic acid | 119.0689 | 339 | 11.55633419 | C5H10O3 | C02804 |
| M202T784 | Indolepyruvate | 201.9045 | 784 | 4.770939835 | C11H9NO3 | C00331 |
| M127T114 | 1,2,3-Trihydroxybenzene | 127.0389 | 113.6 | 0.598241956 | C6H6O3 | C01108 |
| M259T91 | Galactose 1-phosphate | 259.0217 | 91.3 | 2.702476279 | C6H13O9P | C00103 |
| ID | Name | m/z | rt | ppm | Formula | KEGG |
|---|---|---|---|---|---|---|
| M181T782 | D-Fructose | 181.0146 | 782.3 | 3.073909779 | C6H12O6 | C00095 |
| M104T817 | Choline | 104.1079 | 817.4 | 3.842167597 | C5H14NO | C00114 |
| M269T264 | Inosine | 269.0873 | 263.6 | 2.601386242 | C10H12N4O5 | C00294 |
| M160T81 | Pimelic acid | 159.9675 | 80.9 | 10.19875742 | C7H12O4 | C02656 |
| M229T648_1 | Myristic acid | 229.1802 | 647.7 | 3.967308328 | C14H28O2 | C06424 |
| M149T215 | L-2-Hydroxyglutaric acid | 149.0249 | 214.5 | 9.097060136 | C5H8O5 | C03196 |
| M185T82 | 3,4-Dihydroxymandelic acid | 184.9844 | 82.3 | 6.298831399 | C8H8O5 | C05580 |
| Gene | Sequence (5′-3′) | Product Size | ID |
|---|---|---|---|
| AMPK | TACCTCGCCTCCAGTCC GTGCTTTGGGGCTGTCT | 121 | 78,975 |
| PPAR-α | TCATCACCCGAGAGTTCC TCCAGTTCGAGGGCATT | 100 | 25,747 |
| CD36 | GAACCAGGCCACATAGAAAG ACCAATAACGGCTCCAGTAA | 138 | 29,184 |
| ACC | TTGGACAACGCCTTCAC GCAGCCCATTACTTCATCA | 102 | 60,581 |
| CPT-1 | TGTGGCTTGCTGTATTTGA TGACTGGGTGGGATTAGAA | 172 | 25,757 |
| GLUT4 | CAGGATGAAGGAAACAGCA GCAGGACGGGAGAAAAG | 150 | 25,139 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Yuan, C.; Liu, J.; Deng, L.; Li, W.; He, J.; Liu, H.; Qu, L.; Wu, J.; Zou, W. Targeting Energy Protection as a Novel Strategy to Disclose Di’ao Xinxuekang against the Cardiotoxicity Caused by Doxorubicin. Int. J. Mol. Sci. 2023, 24, 897. https://doi.org/10.3390/ijms24020897
Wang T, Yuan C, Liu J, Deng L, Li W, He J, Liu H, Qu L, Wu J, Zou W. Targeting Energy Protection as a Novel Strategy to Disclose Di’ao Xinxuekang against the Cardiotoxicity Caused by Doxorubicin. International Journal of Molecular Sciences. 2023; 24(2):897. https://doi.org/10.3390/ijms24020897
Chicago/Turabian StyleWang, Tao, Chuqiao Yuan, Jia Liu, Liangyan Deng, Wei Li, Junling He, Honglin Liu, Liping Qu, Jianming Wu, and Wenjun Zou. 2023. "Targeting Energy Protection as a Novel Strategy to Disclose Di’ao Xinxuekang against the Cardiotoxicity Caused by Doxorubicin" International Journal of Molecular Sciences 24, no. 2: 897. https://doi.org/10.3390/ijms24020897
APA StyleWang, T., Yuan, C., Liu, J., Deng, L., Li, W., He, J., Liu, H., Qu, L., Wu, J., & Zou, W. (2023). Targeting Energy Protection as a Novel Strategy to Disclose Di’ao Xinxuekang against the Cardiotoxicity Caused by Doxorubicin. International Journal of Molecular Sciences, 24(2), 897. https://doi.org/10.3390/ijms24020897