
Mechanisms of Myofibre Death in Muscular Dystrophies: The Emergence of the Regulated Forms of Necrosis in Myology


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Myofibre Death in Muscular Dystrophies
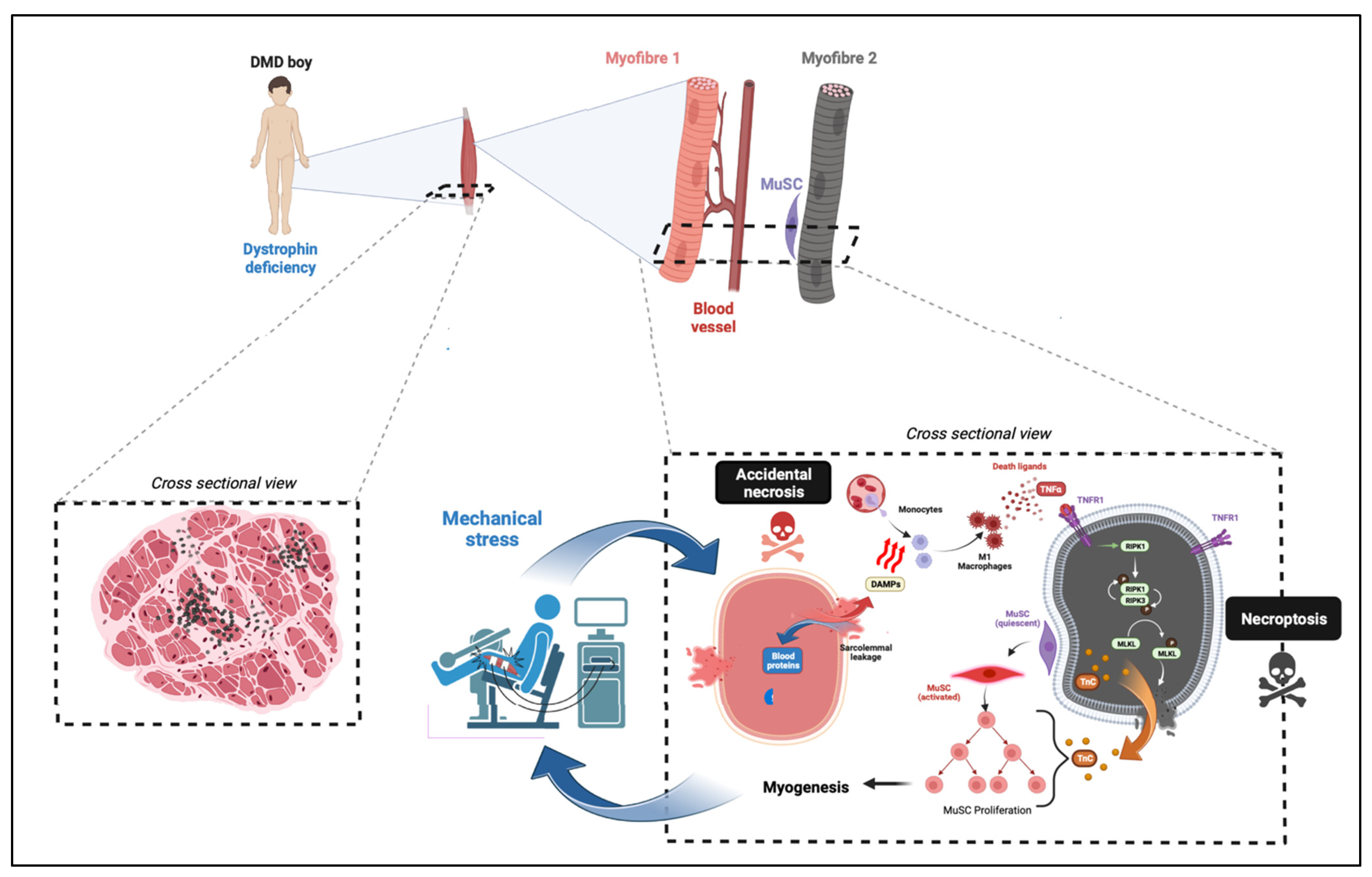
2.1. Myofibre Death in Dystrophinopathies
2.2. Sarcoglycanopathies
2.3. Dysferlinopathies
2.4. Merosinopathy
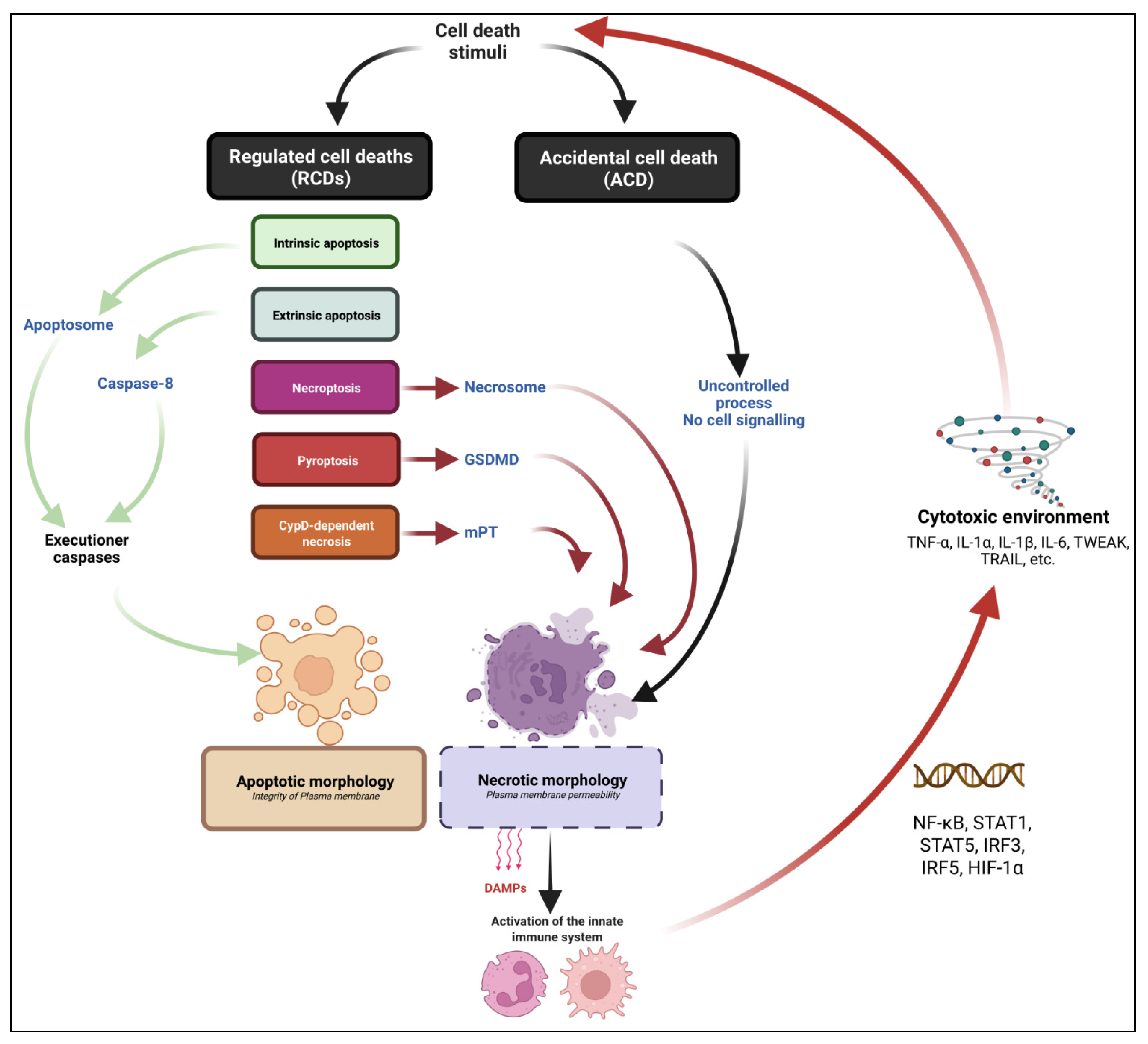
3. The Emergence of Non-Apoptotic Regulated Cell Death Pathways: A Conceptual Revolution
3.1. Defining Cell Death and the Different Cell Death Routes
3.2. Intrinsic and Extrinsic Apoptotic Pathways
3.3. Necrosis: One Word That Can Mean Many Death Pathways… or No Pathway at All
3.4. Necroptosis: A Backup Plan to Die When Apoptosis Fails
3.5. Pyroptosis
3.6. CypD-Dependent Necrosis
4. Regulated Cell Death in Muscular Dystrophies
4.1. Apoptosis in MDs
4.2. Necroptosis in MDs
4.3. CypD-Dependent Necrosis in MDs
4.4. Pyroptosis
5. Conclusions
5.1. Has Apoptosis Any Relevance in Myofibre Degeneration?
5.2. RCDs, New Therapeutic Targets for MDs?
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giudice, J.; Taylor, J.M. Muscle as a Paracrine and Endocrine Organ. Curr. Opin. Pharm. 2017, 34, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin Protects the Sarcolemma from Stresses Developed during Muscle Contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A. Satellite Cell of Skeletal Muscle Fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Relaix, F.; Bencze, M.; Borok, M.J.; Der Vartanian, A.; Gattazzo, F.; Mademtzoglou, D.; Perez-Diaz, S.; Prola, A.; Reyes-Fernandez, P.C.; Rotini, A.; et al. Perspectives on Skeletal Muscle Stem Cells. Nat. Commun. 2021, 12, 692. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G.; Villalta, S.A. Regulatory Interactions between Muscle and the Immune System during Muscle Regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1173–R1187. [Google Scholar] [CrossRef]
- Gordon, S.; Plüddemann, A. Tissue Macrophages: Heterogeneity and Functions. BMC Biol. 2017, 15, 53. [Google Scholar] [CrossRef]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory Monocytes Recruited after Skeletal Muscle Injury Switch into Antiinflammatory Macrophages to Support Myogenesis. J. Exp. Med. 2007, 204, 1057–1069. [Google Scholar] [CrossRef]
- Bencze, M.; Negroni, E.; Vallese, D.; Yacoub-Youssef, H.; Chaouch, S.; Wolff, A.; Aamiri, A.; Santo, J.P.D.; Chazaud, B.; Butler-Browne, G.; et al. Proinflammatory Macrophages Enhance the Regenerative Capacity of Human Myoblasts by Modifying Their Kinetics of Proliferation and Differentiation. Mol. Ther. 2012, 20, 2168–2179. [Google Scholar] [CrossRef]
- Cornelison, D.D.W.; Wilcox-Adelman, S.A.; Goetinck, P.F.; Rauvala, H.; Rapraeger, A.C.; Olwin, B.B. Essential and Separable Roles for Syndecan-3 and Syndecan-4 in Skeletal Muscle Development and Regeneration. Genes Dev. 2004, 18, 2231–2236. [Google Scholar] [CrossRef]
- Plant, D.R.; Colarossi, F.E.; Lynch, G.S. Notexin Causes Greater Myotoxic Damage and Slower Functional Repair in Mouse Skeletal Muscles than Bupivacaine. Muscle Nerve 2006, 34, 577–585. [Google Scholar] [CrossRef]
- Ownby, C.L.; Fletcher, J.E.; Colberg, T.R. Cardiotoxin 1 from Cobra (Naja Naja Atra) Venom Causes Necrosis of Skeletal Muscle in Vivo. Toxicon 1993, 31, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Hardy, D.; Besnard, A.; Latil, M.; Jouvion, G.; Briand, D.; Thépenier, C.; Pascal, Q.; Guguin, A.; Gayraud-Morel, B.; Cavaillon, J.-M.; et al. Comparative Study of Injury Models for Studying Muscle Regeneration in Mice. PLoS ONE 2016, 11, e0147198. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, N.P.; Yeung, E.W.; Allen, D.G. Muscle Damage in Mdx (Dystrophic) Mice: Role of Calcium and Reactive Oxygen Species. Clin. Exp. Pharm. Physiol. 2006, 33, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated Necrosis: The Expanding Network of Non-Apoptotic Cell Death Pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Borok, M.; Didier, N.; Gattazzo, F.; Ozturk, T.; Corneau, A.; Rouard, H.; Relaix, F. Progressive and Coordinated Mobilization of the Skeletal Muscle Niche throughout Tissue Repair Revealed by Single-Cell Proteomic Analysis. Cells 2021, 10, 744. [Google Scholar] [CrossRef]
- Conrad, M.; Angeli, J.P.F.; Vandenabeele, P.; Stockwell, B.R. Regulated Necrosis: Disease Relevance and Therapeutic Opportunities. Nat. Rev. Drug Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef]
- Mariot, V.; Joubert, R.; Le Gall, L.; Sidlauskaite, E.; Hourde, C.; Duddy, W.; Voit, T.; Bencze, M.; Dumonceaux, J. RIPK3-Mediated Cell Death Is Involved in DUX4-Mediated Toxicity in Facioscapulohumeral Dystrophy. J. Cachexia Sarcopenia Muscle 2021, 12, 2079–2090. [Google Scholar] [CrossRef]
- Morgan, J.E.; Prola, A.; Mariot, V.; Pini, V.; Meng, J.; Hourde, C.; Dumonceaux, J.; Conti, F.; Relaix, F.; Authier, F.-J.; et al. Necroptosis Mediates Myofibre Death in Dystrophin-Deficient Mice. Nat. Commun. 2018, 9, 3655. [Google Scholar] [CrossRef]
- Zhou, S.; Zhang, W.; Cai, G.; Ding, Y.; Wei, C.; Li, S.; Yang, Y.; Qin, J.; Liu, D.; Zhang, H.; et al. Myofiber Necroptosis Promotes Muscle Stem Cell Proliferation via Releasing Tenascin-C during Regeneration. Cell Res. 2020, 30, 1063–1077. [Google Scholar] [CrossRef]
- Kamiya, M.; Mizoguchi, F.; Kawahata, K.; Wang, D.; Nishibori, M.; Day, J.; Louis, C.; Wicks, I.P.; Kohsaka, H.; Yasuda, S. Targeting Necroptosis in Muscle Fibers Ameliorates Inflammatory Myopathies. Nat. Commun. 2022, 13, 166. [Google Scholar] [CrossRef]
- Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular Dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Cullen, M.J.; Mastaglia, F.L. Morphological Changes in Dystrophic Muscle. Br. Med. Bull. 1980, 36, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Straub, V.; Rafael, J.A.; Chamberlain, J.S.; Campbell, K.P. Animal Models for Muscular Dystrophy Show Different Patterns of Sarcolemmal Disruption. J. Cell Biol. 1997, 139, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Bencze, M.; Periou, B.; Baba-Amer, Y.; Authier, F.J. Immunolabelling Myofiber Degeneration in Muscle Biopsies. J. Vis. Exp. 2019, 154, e59754. [Google Scholar] [CrossRef]
- Lian, D.; Chen, M.-M.; Wu, H.; Deng, S.; Hu, X. The Role of Oxidative Stress in Skeletal Muscle Myogenesis and Muscle Disease. Antioxidants 2022, 11, 755. [Google Scholar] [CrossRef] [PubMed]
- Renjini, R.; Gayathri, N.; Nalini, A.; Srinivas Bharath, M.M. Oxidative Damage in Muscular Dystrophy Correlates with the Severity of the Pathology: Role of Glutathione Metabolism. Neurochem. Res. 2012, 37, 885–898. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Kanneganti, T.-D. Stressed-out ROS Take a Silent Death Route. Nat. Immunol. 2018, 19, 103–105. [Google Scholar] [CrossRef]
- Serrano, A.L.; Muñoz-Cánoves, P. Fibrosis Development in Early-Onset Muscular Dystrophies: Mechanisms and Translational Implications. Semin Cell Dev. Biol. 2017, 64, 181–190. [Google Scholar] [CrossRef]
- Bougé, A.-L.; Murauer, E.; Beyne, E.; Miro, J.; Varilh, J.; Taulan, M.; Koenig, M.; Claustres, M.; Tuffery-Giraud, S. Targeted RNA-Seq Profiling of Splicing Pattern in the DMD Gene: Exons Are Mostly Constitutively Spliced in Human Skeletal Muscle. Sci. Rep. 2017, 7, 39094. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Van Deutekom, J.C.T.; Fokkema, I.F.; Van Ommen, G.-J.B.; Den Dunnen, J.T. Entries in the Leiden Duchenne Muscular Dystrophy Mutation Database: An Overview of Mutation Types and Paradoxical Cases That Confirm the Reading-Frame Rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef]
- Wein, N.; Alfano, L.; Flanigan, K.M. Genetics and Emerging Treatments for Duchenne and Becker Muscular Dystrophy. Pediatr. Clin. N. Am. 2015, 62, 723–742. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Beggs, A.H.; Moyer, M.; Scherpf, S.; Heindrich, K.; Bettecken, T.; Meng, G.; Müller, C.R.; Lindlöf, M.; Kaariainen, H.; et al. The Molecular Basis for Duchenne versus Becker Muscular Dystrophy: Correlation of Severity with Type of Deletion. Am. J. Hum. Genet. 1989, 45, 498–506. [Google Scholar] [PubMed]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and Management of Duchenne Muscular Dystrophy, Part 1: Diagnosis, and Pharmacological and Psychosocial Management. Lancet Neurol 2010, 9, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and Management of Duchenne Muscular Dystrophy, Part 2: Respiratory, Cardiac, Bone Health, and Orthopaedic Management. Lancet Neurol 2018, 17, 347–361. [Google Scholar] [CrossRef]
- Gilbert, R.K.; Hawk, W.A. The Incidence of Necrosis of Muscle Fibers in Duchenne Type Muscular Dystrophy. Am. J. Pathol. 1963, 43, 107–122. [Google Scholar]
- Coulton, G.R.; Morgan, J.E.; Partridge, T.A.; Sloper, J.C. The Mdx Mouse Skeletal Muscle Myopathy: I. A Histological, Morphometric and Biochemical Investigation. Neuropathol. Appl. Neurobiol. 1988, 14, 53–70. [Google Scholar] [CrossRef]
- Fukada, S.; Morikawa, D.; Yamamoto, Y.; Yoshida, T.; Sumie, N.; Yamaguchi, M.; Ito, T.; Miyagoe-Suzuki, Y.; Takeda, S.; Tsujikawa, K.; et al. Genetic Background Affects Properties of Satellite Cells and Mdx Phenotypes. Am. J. Pathol. 2010, 176, 2414–2424. [Google Scholar] [CrossRef]
- Sharp, N.J.H.; Kornegay, J.N.; Van Camp, S.D.; Herbstreith, M.H.; Secore, S.L.; Kettle, S.; Hung, W.-Y.; Constantinou, C.D.; Dykstra, M.J.; Roses, A.D.; et al. An Error in Dystrophin MRNA Processing in Golden Retriever Muscular Dystrophy, an Animal Homologue of Duchenne Muscular Dystrophy. Genomics 1992, 13, 115–121. [Google Scholar] [CrossRef]
- Klymiuk, N.; Blutke, A.; Graf, A.; Krause, S.; Burkhardt, K.; Wuensch, A.; Krebs, S.; Kessler, B.; Zakhartchenko, V.; Kurome, M.; et al. Dystrophin-Deficient Pigs Provide New Insights into the Hierarchy of Physiological Derangements of Dystrophic Muscle. Hum. Mol. Genet. 2013, 22, 4368–4382. [Google Scholar] [CrossRef]
- Sui, T.; Lau, Y.S.; Liu, D.; Liu, T.; Xu, L.; Gao, Y.; Lai, L.; Li, Z.; Han, R. A Novel Rabbit Model of Duchenne Muscular Dystrophy Generated by CRISPR/Cas9. Dis Model. Mech. 2018, 11, dmm032201. [Google Scholar] [CrossRef]
- Nakamura, K.; Fujii, W.; Tsuboi, M.; Tanihata, J.; Teramoto, N.; Takeuchi, S.; Naito, K.; Yamanouchi, K.; Nishihara, M. Generation of Muscular Dystrophy Model Rats with a CRISPR/Cas System. Sci. Rep. 2014, 4, 5635. [Google Scholar] [CrossRef] [PubMed]
- Larcher, T.; Lafoux, A.; Tesson, L.; Remy, S.; Thepenier, V.; François, V.; Le Guiner, C.; Goubin, H.; Dutilleul, M.; Guigand, L.; et al. Characterization of Dystrophin Deficient Rats: A New Model for Duchenne Muscular Dystrophy. PLoS ONE 2014, 9, e110371. [Google Scholar] [CrossRef] [PubMed]
- Taglietti, V.; Kefi, K.; Bronisz-Budzyńska, I.; Mirciloglu, B.; Rodrigues, M.; Cardone, N.; Coulpier, F.; Periou, B.; Gentil, C.; Goddard, M.; et al. Duchenne Muscular Dystrophy Trajectory in R-DMDdel52 Preclinical Rat Model Identifies COMP as Biomarker of Fibrosis. Acta Neuropathol. Commun. 2022, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Hamer, P.W.; McGeachie, J.M.; Davies, M.J.; Grounds, M.D. Evans Blue Dye as an in Vivo Marker of Myofibre Damage: Optimising Parameters for Detecting Initial Myofibre Membrane Permeability. J. Anat. 2002, 200, 69. [Google Scholar] [CrossRef]
- Lovering, R.M.; De Deyne, P.G. Contractile Function, Sarcolemma Integrity, and the Loss of Dystrophin after Skeletal Muscle Eccentric Contraction-Induced Injury. Am. J. Physiol. Cell Physiol. 2004, 286, C230–C238. [Google Scholar] [CrossRef]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric Oxide Synthase Complexed with Dystrophin and Absent from Skeletal Muscle Sarcolemma in Duchenne Muscular Dystrophy. Cell 1995, 82, 743–752. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef]
- Mareedu, S.; Million, E.D.; Duan, D.; Babu, G.J. Abnormal Calcium Handling in Duchenne Muscular Dystrophy: Mechanisms and Potential Therapies. Front. Physiol. 2021, 12, 647010. [Google Scholar] [CrossRef]
- Rando, T.A.; Disatnik, M.H.; Yu, Y.; Franco, A. Muscle Cells from Mdx Mice Have an Increased Susceptibility to Oxidative Stress. Neuromuscul. Disord. 1998, 8, 14–21. [Google Scholar] [CrossRef]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne Muscular Dystrophy: Myonecrosis, Inflammation and Oxidative Stress. Dis. Model. Mech. 2020, 13, dmm043638. [Google Scholar] [CrossRef]
- Haycock, J.W.; MacNeil, S.; Jones, P.; Harris, J.B.; Mantle, D. Oxidative Damage to Muscle Protein in Duchenne Muscular Dystrophy. Neuroreport 1996, 8, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.; Morgan, J.; Bozyk, M.E. Effect of Pretreatment with Prednisolone on Enzyme Efflux from Isolated Skeletal and Heart Muscle. Res. Commun. Chem. Pathol. Pharm. 1975, 12, 363–377. [Google Scholar]
- Ricotti, V.; Ridout, D.A.; Scott, E.; Quinlivan, R.; Robb, S.A.; Manzur, A.Y.; Muntoni, F. NorthStar Clinical Network Long-Term Benefits and Adverse Effects of Intermittent versus Daily Glucocorticoids in Boys with Duchenne Muscular Dystrophy. J. Neurol. Neurosurg. Psychiatry 2013, 84, 698–705. [Google Scholar] [CrossRef]
- Mendell, J.R.; Moxley, R.T.; Griggs, R.C.; Brooke, M.H.; Fenichel, G.M.; Miller, J.P.; King, W.; Signore, L.; Pandya, S.; Florence, J. Randomized, Double-Blind Six-Month Trial of Prednisone in Duchenne’s Muscular Dystrophy. N. Engl. J. Med. 1989, 320, 1592–1597. [Google Scholar] [CrossRef]
- Fenichel, G.M.; Florence, J.M.; Pestronk, A.; Mendell, J.R.; Moxley, R.T.; Griggs, R.C.; Brooke, M.H.; Miller, J.P.; Robison, J.; King, W. Long-Term Benefit from Prednisone Therapy in Duchenne Muscular Dystrophy. Neurology 1991, 41, 1874–1877. [Google Scholar] [CrossRef] [PubMed]
- Conklin, L.S.; Damsker, J.M.; Hoffman, E.P.; Jusko, W.J.; Mavroudis, P.D.; Schwartz, B.D.; Mengle-Gaw, L.J.; Smith, E.C.; Mah, J.K.; Guglieri, M.; et al. Phase IIa Trial in Duchenne Muscular Dystrophy Shows Vamorolone Is a First-in-Class Dissociative Steroidal Anti-Inflammatory Drug. Pharmacol. Res. 2018, 136, 140–150. [Google Scholar] [CrossRef]
- Jacobs, S.C.; Bootsma, A.L.; Willems, P.W.; Bär, P.R.; Wokke, J.H. Prednisone Can Protect against Exercise-Induced Muscle Damage. J. Neurol. 1996, 243, 410–416. [Google Scholar] [CrossRef]
- Cohen, L.; Morgan, J.; Bozyk, M.E. Variable Effects of Corticosteroid Treatment of Serum Enzyme Activities in Duchenne’s Muscular Dystrophy. Res. Commun. Chem. Pathol. Pharm. 1977, 17, 529–538. [Google Scholar]
- Guerriero, V.; Florini, J.R. Dexamethasone Effects on Myoblast Proliferation and Differentiation. Endocrinology 1980, 106, 1198–1202. [Google Scholar] [CrossRef]
- Passaquin, A.C.; Metzinger, L.; Léger, J.J.; Warter, J.M.; Poindron, P. Prednisolone Enhances Myogenesis and Dystrophin-Related Protein in Skeletal Muscle Cell Cultures from Mdx Mouse. J. Neurosci. Res. 1993, 35, 363–372. [Google Scholar] [CrossRef]
- Quattrocelli, M.; Barefield, D.Y.; Warner, J.L.; Vo, A.H.; Hadhazy, M.; Earley, J.U.; Demonbreun, A.R.; McNally, E.M. Intermittent Glucocorticoid Steroid Dosing Enhances Muscle Repair without Eliciting Muscle Atrophy. J. Clin. Investig. 2017, 127, 2418–2432. [Google Scholar] [CrossRef] [PubMed]
- Londhe, P.; Guttridge, D.C. Inflammation Induced Loss of Skeletal Muscle. Bone 2015, 80, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Kepp, O.; Kroemer, G. Regulated Cell Death and Adaptive Stress Responses. Cell Mol. Life Sci. 2016, 73, 2405–2410. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-E.; Jin, B.; Li, Y.-P. TNF-Alpha Regulates Myogenesis and Muscle Regeneration by Activating P38 MAPK. Am. J. Physiol. Cell Physiol. 2007, 292, C1660–C1671. [Google Scholar] [CrossRef]
- Guttridge, D.C.; Mayo, M.W.; Madrid, L.V.; Wang, C.Y.; Baldwin, A.S. NF-KappaB-Induced Loss of MyoD Messenger RNA: Possible Role in Muscle Decay and Cachexia. Science 2000, 289, 2363–2366. [Google Scholar] [CrossRef]
- Palacios, D.; Mozzetta, C.; Consalvi, S.; Caretti, G.; Saccone, V.; Proserpio, V.; Marquez, V.E.; Valente, S.; Mai, A.; Forcales, S.V.; et al. TNF/P38α/Polycomb Signaling to Pax7 Locus in Satellite Cells Links Inflammation to the Epigenetic Control of Muscle Regeneration. Cell Stem Cell 2010, 7, 455–469. [Google Scholar] [CrossRef]
- Li, Y.-P. TNF-Alpha Is a Mitogen in Skeletal Muscle. Am. J. Physiol. Cell Physiol. 2003, 285, C370–C376. [Google Scholar] [CrossRef]
- Hodgetts, S.; Radley, H.; Davies, M.; Grounds, M.D. Reduced Necrosis of Dystrophic Muscle by Depletion of Host Neutrophils, or Blocking TNFalpha Function with Etanercept in Mdx Mice. Neuromuscul. Disord. 2006, 16, 591–602. [Google Scholar] [CrossRef]
- Grounds, M.D.; Torrisi, J. Anti-TNFα (Remicade®) Therapy Protects Dystrophic Skeletal Muscle from Necrosis. FASEB J. 2004, 18, 676–682. [Google Scholar] [CrossRef]
- Enwere, E.K.; Boudreault, L.; Holbrook, J.; Timusk, K.; Earl, N.; LaCasse, E.; Renaud, J.-M.; Korneluk, R.G. Loss of CIAP1 Attenuates Soleus Muscle Pathology and Improves Diaphragm Function in Mdx Mice. Hum. Mol. Genet. 2013, 22, 867–878. [Google Scholar] [CrossRef]
- Enwere, E.K.; LaCasse, E.C.; Adam, N.J.; Korneluk, R.G. Role of the TWEAK-Fn14-CIAP1-NF-ΚB Signaling Axis in the Regulation of Myogenesis and Muscle Homeostasis. Front. Immunol. 2014, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Enwere, E.K.; Holbrook, J.; Lejmi-Mrad, R.; Vineham, J.; Timusk, K.; Sivaraj, B.; Isaac, M.; Uehling, D.; Al-awar, R.; LaCasse, E.; et al. TWEAK and CIAP1 Regulate Myoblast Fusion through the Noncanonical NF-ΚB Signaling Pathway. Sci. Signal. 2012, 5, ra75. [Google Scholar] [CrossRef] [PubMed]
- Villalta, S.A.; Nguyen, H.X.; Deng, B.; Gotoh, T.; Tidball, J.G. Shifts in Macrophage Phenotypes and Macrophage Competition for Arginine Metabolism Affect the Severity of Muscle Pathology in Muscular Dystrophy. Hum. Mol. Genet. 2009, 18, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Lagrota-Candido, J.; Vasconcellos, R.; Cavalcanti, M.; Bozza, M.; Savino, W.; Quirico-Santos, T. Resolution of Skeletal Muscle Inflammation in Mdx Dystrophic Mouse Is Accompanied by Increased Immunoglobulin and Interferon-Gamma Production. Int. J. Exp. Pathol. 2002, 83, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Villalta, S.A.; Deng, B.; Rinaldi, C.; Wehling-Henricks, M.; Tidball, J.G. IFN-γ Promotes Muscle Damage in the Mdx Mouse Model of Duchenne Muscular Dystrophy by Suppressing M2 Macrophage Activation and Inhibiting Muscle Cell Proliferation. J. Immunol. 2011, 187, 5419–5428. [Google Scholar] [CrossRef] [PubMed]
- Londhe, P.; Davie, J.K. Gamma Interferon Modulates Myogenesis through the Major Histocompatibility Complex Class II Transactivator, CIITA. Mol. Cell Biol. 2011, 31, 2854–2866. [Google Scholar] [CrossRef]
- Hou, C.; Periou, B.; Gervais, M.; Berthier, J.; Baba-Amer, Y.; Souvannanorath, S.; Malfatti, E.; Relaix, F.; Bencze, M.; Authier, F.J. Interferon-Gamma Mediates Skeletal Muscle Lesions through JAK/STAT Pathway Activation in Inclusion Body Myositis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Giordano, C.; Mojumdar, K.; Liang, F.; Lemaire, C.; Li, T.; Richardson, J.; Divangahi, M.; Qureshi, S.; Petrof, B.J. Toll-like Receptor 4 Ablation in Mdx Mice Reveals Innate Immunity as a Therapeutic Target in Duchenne Muscular Dystrophy. Hum. Mol. Genet. 2015, 24, 2147–2162. [Google Scholar] [CrossRef]
- Zabłocka, B.; Górecki, D.C.; Zabłocki, K. Disrupted Calcium Homeostasis in Duchenne Muscular Dystrophy: A Common Mechanism behind Diverse Consequences. Int. J. Mol. Sci. 2021, 22, 11040. [Google Scholar] [CrossRef]
- Bartoli, M.; Richard, I. Calpains in Muscle Wasting. Int. J. Biochem. Cell. Biol. 2005, 37, 2115–2133. [Google Scholar] [CrossRef]
- Scholtes, C.; Bellemin, S.; Martin, E.; Carre-Pierrat, M.; Mollereau, B.; Gieseler, K.; Walter, L. DRP-1-Mediated Apoptosis Induces Muscle Degeneration in Dystrophin Mutants. Sci. Rep. 2018, 8, 7354. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G.; Albrecht, D.E.; Lokensgard, B.E.; Spencer, M.J. Apoptosis Precedes Necrosis of Dystrophin-Deficient Muscle. J. Cell Sci. 1995, 108 Pt 6, 2197–2204. [Google Scholar] [CrossRef]
- Spencer, M.J.; Walsh, C.M.; Dorshkind, K.A.; Rodriguez, E.M.; Tidball, J.G. Myonuclear Apoptosis in Dystrophic Mdx Muscle Occurs by Perforin-Mediated Cytotoxicity. J. Clin. Investig. 1997, 99, 2745–2751. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Salam, E.; Abdel-Meguid, I.; Korraa, S.S. Markers of Degeneration and Regeneration in Duchenne Muscular Dystrophy. Acta Myol. 2009, 28, 94–100. [Google Scholar] [PubMed]
- Adams, V.; Gielen, S.; Hambrecht, R.; Schuler, G. Apoptosis in Skeletal Muscle. Front. Biosci. 2001, 6, D1–D11. [Google Scholar] [CrossRef] [PubMed]
- Israeli, D.; Cosette, J.; Corre, G.; Amor, F.; Poupiot, J.; Stockholm, D.; Montus, M.; Gjata, B.; Richard, I. An AAV-SGCG Dose-Response Study in a γ-Sarcoglycanopathy Mouse Model in the Context of Mechanical Stress. Mol. Ther.-Methods Clin. Dev. 2019, 13, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C. LGMD. Identification, Description and Classification. Acta Myol. 2020, 39, 207–217. [Google Scholar] [CrossRef]
- Sánchez Riera, C.; Lozanoska-Ochser, B.; Testa, S.; Fornetti, E.; Bouché, M.; Madaro, L. Muscle Diversity, Heterogeneity, and Gradients: Learning from Sarcoglycanopathies. Int. J. Mol. Sci. 2021, 22, 2502. [Google Scholar] [CrossRef]
- Duclos, F.; Straub, V.; Moore, S.A.; Venzke, D.P.; Hrstka, R.F.; Crosbie, R.H.; Durbeej, M.; Lebakken, C.S.; Ettinger, A.J.; van der Meulen, J.; et al. Progressive Muscular Dystrophy in Alpha-Sarcoglycan-Deficient Mice. J. Cell Biol. 1998, 142, 1461–1471. [Google Scholar] [CrossRef]
- Araishi, K.; Sasaoka, T.; Imamura, M.; Noguchi, S.; Hama, H.; Wakabayashi, E.; Yoshida, M.; Hori, T.; Ozawa, E. Loss of the Sarcoglycan Complex and Sarcospan Leads to Muscular Dystrophy in Beta-Sarcoglycan-Deficient Mice. Hum. Mol. Genet. 1999, 8, 1589–1598. [Google Scholar] [CrossRef]
- Straub, V.; Duclos, F.; Venzke, D.P.; Lee, J.C.; Cutshall, S.; Leveille, C.J.; Campbell, K.P. Molecular Pathogenesis of Muscle Degeneration in the Delta-Sarcoglycan-Deficient Hamster. Am. J. Pathol. 1998, 153, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Hack, A.A.; Cordier, L.; Shoturma, D.I.; Lam, M.Y.; Sweeney, H.L.; McNally, E.M. Muscle Degeneration without Mechanical Injury in Sarcoglycan Deficiency. Proc. Natl. Acad. Sci. USA 1999, 96, 10723–10728. [Google Scholar] [CrossRef] [PubMed]
- Durbeej, M.; Cohn, R.D.; Hrstka, R.F.; Moore, S.A.; Allamand, V.; Davidson, B.L.; Williamson, R.A.; Campbell, K.P. Disruption of the β-Sarcoglycan Gene Reveals Pathogenetic Complexity of Limb-Girdle Muscular Dystrophy Type 2E. Mol. Cell 2000, 5, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.A.; Feng, H.; Tewari, M.; Acosta, P.; Kawana, M.; Sweeney, H.L.; Discher, D.E. γ-Sarcoglycan Deficiency Increases Cell Contractility, Apoptosis and MAPK Pathway Activation but Does Not Affect Adhesion. J. Cell Sci. 2005, 118, 1405–1416. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.Y.; Iwata, Y.; Sampaolesi, M.; Hanada, H.; Saito, N.; Artman, M.; Coetzee, W.A.; Shigekawa, M. Stretch-Activated Cation Channels in Skeletal Muscle Myotubes from Sarcoglycan-Deficient Hamsters. Am. J. Physiol. Cell Physiol. 2001, 281, C690–C699. [Google Scholar] [CrossRef]
- Iwata, Y.; Katanosaka, Y.; Arai, Y.; Komamura, K.; Miyatake, K.; Shigekawa, M. A Novel Mechanism of Myocyte Degeneration Involving the Ca2+-Permeable Growth Factor-Regulated Channel. J. Cell Biol. 2003, 161, 957–967. [Google Scholar] [CrossRef]
- Raffaghello, L.; Principi, E.; Baratto, S.; Panicucci, C.; Pintus, S.; Antonini, F.; Del Zotto, G.; Benzi, A.; Bruzzone, S.; Scudieri, P.; et al. P2X7 Receptor Antagonist Reduces Fibrosis and Inflammation in a Mouse Model of Alpha-Sarcoglycan Muscular Dystrophy. Pharmaceuticals 2022, 15, 89. [Google Scholar] [CrossRef]
- Sandonà, D.; Gastaldello, S.; Martinello, T.; Betto, R. Characterization of the ATP-Hydrolysing Activity of α-Sarcoglycan. Biochem. J. 2004, 381, 105–112. [Google Scholar] [CrossRef]
- Liu, J.; Aoki, M.; Illa, I.; Wu, C.; Fardeau, M.; Angelini, C.; Serrano, C.; Urtizberea, J.A.; Hentati, F.; Hamida, M.B.; et al. Dysferlin, a Novel Skeletal Muscle Gene, Is Mutated in Miyoshi Myopathy and Limb Girdle Muscular Dystrophy. Nat. Genet. 1998, 20, 31–36. [Google Scholar] [CrossRef]
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.-C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective Membrane Repair in Dysferlin-Deficient Muscular Dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef]
- Fanin, M.; Angelini, C. Muscle Pathology in Dysferlin Deficiency. Neuropathol. Appl. Neurobiol. 2002, 28, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Kesari, A.; Fukuda, M.; Knoblach, S.; Bashir, R.; Nader, G.A.; Rao, D.; Nagaraju, K.; Hoffman, E.P. Dysferlin Deficiency Shows Compensatory Induction of Rab27A/Slp2a That May Contribute to Inflammatory Onset. Am. J. Pathol. 2008, 173, 1476–1487. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, K.; Rawat, R.; Veszelovszky, E.; Thapliyal, R.; Kesari, A.; Sparks, S.; Raben, N.; Plotz, P.; Hoffman, E.P. Dysferlin Deficiency Enhances Monocyte Phagocytosis: A Model for the Inflammatory Onset of Limb-Girdle Muscular Dystrophy 2B. Am. J. Pathol. 2008, 172, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Rawat, R.; Cohen, T.V.; Ampong, B.; Francia, D.; Henriques-Pons, A.; Hoffman, E.P.; Nagaraju, K. Inflammasome Up-Regulation and Activation in Dysferlin-Deficient Skeletal Muscle. Am. J. Pathol. 2010, 176, 2891–2900. [Google Scholar] [CrossRef]
- Tzeng, H.-P.; Evans, S.; Gao, F.; Chambers, K.; Topkara, V.K.; Sivasubramanian, N.; Barger, P.M.; Mann, D.L. Dysferlin Mediates the Cytoprotective Effects of TRAF2 Following Myocardial Ischemia Reperfusion Injury. J. Am. Heart Assoc. 2014, 3, e000662. [Google Scholar] [CrossRef]
- Yao, W.; Li, H.; Han, X.; Chen, C.; Zhang, Y.; Tai, W.L.; Xia, Z.; Hei, Z. MG53 Anchored by Dysferlin to Cell Membrane Reduces Hepatocyte Apoptosis Which Induced by Ischaemia/Reperfusion Injury in Vivo and in Vitro. J. Cell Mol. Med. 2017, 21, 2503–2513. [Google Scholar] [CrossRef]
- Prelle, A.; Sciacco, M.; Tancredi, L.; Fagiolari, G.; Comi, G.P.; Ciscato, P.; Serafini, M.; Fortunato, F.; Zecca, C.; Gallanti, A.; et al. Clinical, Morphological and Immunological Evaluation of Six Patients with Dysferlin Deficiency. Acta Neuropathol. 2003, 105, 537–542. [Google Scholar] [CrossRef]
- Oliveira, J.; Gruber, A.; Cardoso, M.; Taipa, R.; Fineza, I.; Gonçalves, A.; Laner, A.; Winder, T.L.; Schroeder, J.; Rath, J.; et al. LAMA2 Gene Mutation Update: Toward a More Comprehensive Picture of the Laminin-A2 Variome and Its Related Phenotypes. Hum. Mutat. 2018, 39, 1314–1337. [Google Scholar] [CrossRef]
- Bönnemann, C.G.; Wang, C.H.; Quijano-Roy, S.; Deconinck, N.; Bertini, E.; Ferreiro, A.; Muntoni, F.; Sewry, C.; Béroud, C.; Mathews, K.D.; et al. Diagnostic Approach to the Congenital Muscular Dystrophies. Neuromuscul. Disord. 2014, 24, 289–311. [Google Scholar] [CrossRef]
- Pegoraro, E.; Mancias, P.; Swerdlow, S.H.; Raikow, R.B.; Garcia, C.; Marks, H.; Crawford, T.; Carver, V.; Di Cianno, B.; Hoffman, E.P. Congenital Muscular Dystrophy with Primary Laminin Alpha2 (Merosin) Deficiency Presenting as Inflammatory Myopathy. Ann. Neurol. 1996, 40, 782–791. [Google Scholar] [CrossRef]
- Gawlik, K.I.; Durbeej, M. A Family of Laminin A2 Chain-Deficient Mouse Mutants: Advancing the Research on LAMA2-CMD. Front. Mol. Neurosci. 2020, 13, 59. [Google Scholar] [CrossRef] [PubMed]
- Harandi, V.M.; Oliveira, B.M.S.; Allamand, V.; Friberg, A.; Fontes-Oliveira, C.C.; Durbeej, M. Antioxidants Reduce Muscular Dystrophy in the Dy2J/Dy2J Mouse Model of Laminin A2 Chain-Deficient Muscular Dystrophy. Antioxidants 2020, 9, 244. [Google Scholar] [CrossRef] [PubMed]
- Girgenrath, M.; Dominov, J.A.; Kostek, C.A.; Miller, J.B. Inhibition of Apoptosis Improves Outcome in a Model of Congenital Muscular Dystrophy. J. Clin. Investig. 2004, 114, 1635–1639. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef]
- Galluzzi, L.; Maiuri, M.C.; Vitale, I.; Zischka, H.; Castedo, M.; Zitvogel, L.; Kroemer, G. Cell Death Modalities: Classification and Pathophysiological Implications. Cell Death Differ. 2007, 14, 1237–1243. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular Definitions of Cell Death Subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus Accessory Aspects of Cell Death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Fernando, P.; Kelly, J.F.; Balazsi, K.; Slack, R.S.; Megeney, L.A. Caspase 3 Activity Is Required for Skeletal Muscle Differentiation. Proc. Natl. Acad. Sci. USA 2002, 99, 11025–11030. [Google Scholar] [CrossRef]
- Dehkordi, M.H.; Tashakor, A.; O’Connell, E.; Fearnhead, H.O. Apoptosome-Dependent Myotube Formation Involves Activation of Caspase-3 in Differentiating Myoblasts. Cell Death Dis. 2020, 11, 308. [Google Scholar] [CrossRef]
- Burgon, P.G.; Megeney, L.A. Caspase Signaling, a Conserved Inductive Cue for Metazoan Cell Differentiation. Semin Cell Dev. Biol. 2018, 82, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Shlomovitz, I.; Speir, M.; Gerlic, M. Flipping the Dogma – Phosphatidylserine in Non-Apoptotic Cell Death. Cell Commun. Signal. 2019, 17, 139. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.-N.; Crawford, J.C.; Heckmann, B.L.; Green, D.R. To the Edge of Cell Death and Back. FEBS J. 2019, 286, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.M.; Horvitz, H.R. Genetic Control of Programmed Cell Death in the Nematode C. Elegans. Cell 1986, 44, 817–829. [Google Scholar] [CrossRef]
- Lockshin, R.A.; Williams, C.M. Programmed Cell Death—II. Endocrine Potentiation of the Breakdown of the Intersegmental Muscles of Silkmoths. J. Insect Physiol. 1964, 10, 643–649. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wide-Ranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Kono, H.; Rock, K.L. How Dying Cells Alert the Immune System to Danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological Aspects of Cancer Chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef]
- Fridman, J.S.; Lowe, S.W. Control of Apoptosis by P53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef]
- Speidel, D. Transcription-Independent P53 Apoptosis: An Alternative Route to Death. Trends Cell Biol. 2010, 20, 14–24. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Berghe, T.V.; Vanlangenakker, N.; Parthoens, E.; Deckers, W.; Devos, M.; Festjens, N.; Guerin, C.J.; Brunk, U.T.; Declercq, W.; Vandenabeele, P. Necroptosis, Necrosis and Secondary Necrosis Converge on Similar Cellular Disintegration Features. Cell Death Differ. 2010, 17, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical Inhibitor of Nonapoptotic Cell Death with Therapeutic Potential for Ischemic Brain Injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 Kinase as a Specific Cellular Target of Necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Festjens, N.; Vanden Berghe, T.; Cornelis, S.; Vandenabeele, P. RIP1, a Kinase on the Crossroads of a Cell’s Decision to Live or Die. Cell Death Differ. 2007, 14, 400–410. [Google Scholar] [CrossRef]
- Vanlangenakker, N.; Vanden Berghe, T.; Bogaert, P.; Laukens, B.; Zobel, K.; Deshayes, K.; Vucic, D.; Fulda, S.; Vandenabeele, P.; Bertrand, M.J.M. CIAP1 and TAK1 Protect Cells from TNF-Induced Necrosis by Preventing RIP1/RIP3-Dependent Reactive Oxygen Species Production. Cell Death Differ. 2011, 18, 656–665. [Google Scholar] [CrossRef]
- Upton, J.W.; Chan, F.K.-M. Staying Alive: Cell Death in Antiviral Immunity. Mol. Cell 2014, 54, 273–280. [Google Scholar] [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K.-M. Phosphorylation-Driven Assembly of the RIP1-RIP3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef]
- Zhang, D.-W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.-J.; Lin, S.-C.; Dong, M.-Q.; Han, J. RIP3, an Energy Metabolism Regulator That Switches TNF-Induced Cell Death from Apoptosis to Necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef]
- Dovey, C.M.; Diep, J.; Clarke, B.P.; Hale, A.T.; McNamara, D.E.; Guo, H.; Brown, N.W.; Cao, J.Y.; Grace, C.R.; Gough, P.J.; et al. MLKL Requires the Inositol Phosphate Code to Execute Necroptosis. Mol. Cell 2018, 70, 936–948.e7. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.-F.; Wang, F.-S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.L.; Zhang, Y.; Geoghegan, N.D.; Gavin, X.J.; Davies, K.A.; Mlodzianoski, M.J.; Whitehead, L.W.; Frank, D.; Garnish, S.E.; Fitzgibbon, C.; et al. MLKL Trafficking and Accumulation at the Plasma Membrane Control the Kinetics and Threshold for Necroptosis. Nat. Commun. 2020, 11, 3151. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Hassannia, B.; Vandenabeele, P. An Outline of Necrosome Triggers. Cell. Mol. Life Sci. 2016, 73, 2137–2152. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.J.; Basagoudanavar, S.H.; Nogusa, S.; Irrinki, K.; Mallilankaraman, K.; Slifker, M.J.; Beg, A.A.; Madesh, M.; Balachandran, S. NF-ΚB Protects Cells from Gamma Interferon-Induced RIP1-Dependent Necroptosis. Mol. Cell. Biol. 2011, 31, 2934–2946. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like Receptor 3-Mediated Necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef]
- He, S.; Liang, Y.; Shao, F.; Wang, X. Toll-like Receptors Activate Programmed Necrosis in Macrophages through a Receptor-Interacting Kinase-3-Mediated Pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 20054–20059. [Google Scholar] [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 Complexes with RIP3 to Mediate Virus-Induced Programmed Necrosis That Is Targeted by Murine Cytomegalovirus VIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. The Inflammasome Turns 15. Nature 2017, 548, 534–535. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and Diseases. Signal. Transduct. Target. 2021, 6, 128. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of ProIL-Beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.K.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Liston, A.; Masters, S.L. Homeostasis-Altering Molecular Processes as Mechanisms of Inflammasome Activation. Nat. Rev. Immunol. 2017, 17, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial Membrane Permeabilization in Cell Death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondrial Regulation of Cell Death: A Phylogenetically Conserved Control. Microb. Cell 2016, 3, 101–108. [Google Scholar] [CrossRef]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D Is a Component of Mitochondrial Permeability Transition and Mediates Neuronal Cell Death after Focal Cerebral Ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar] [CrossRef]
- Wang, J.; Walsh, K. Resistance to Apoptosis Conferred by Cdk Inhibitors during Myocyte Differentiation. Science 1996, 273, 359–361. [Google Scholar] [CrossRef]
- Xiao, R.; Ferry, A.L.; Dupont-Versteegden, E.E. Cell Death-Resistance of Differentiated Myotubes Is Associated with Enhanced Anti-Apoptotic Mechanisms Compared to Myoblasts. Apoptosis 2011, 16, 221–234. [Google Scholar] [CrossRef]
- Salucci, S.; Burattini, S.; Baldassarri, V.; Battistelli, M.; Canonico, B.; Valmori, A.; Papa, S.; Falcieri, E. The Peculiar Apoptotic Behavior of Skeletal Muscle Cells. Histol. Histopathol. 2013, 28, 1073–1087. [Google Scholar] [CrossRef]
- Tews, D.S. Muscle-Fiber Apoptosis in Neuromuscular Diseases. Muscle Nerve 2005, 32, 443–458. [Google Scholar] [CrossRef]
- Battistelli, M.; Salucci, S.; Burattini, S.; Falcieri, E. Further Considerations on in Vitro Skeletal Muscle Cell Death. Muscles Ligaments Tendons J. 2013, 3, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Oláh, G.; Szczesny, B.; Brunyánszki, A.; López-García, I.A.; Gerö, D.; Radák, Z.; Szabo, C. Differentiation-Associated Downregulation of Poly(ADP-Ribose) Polymerase-1 Expression in Myoblasts Serves to Increase Their Resistance to Oxidative Stress. PLoS ONE 2015, 10, e0134227. [Google Scholar] [CrossRef] [PubMed]
- Borisov, A.B.; Carlson, B.M. Cell Death in Denervated Skeletal Muscle Is Distinct from Classical Apoptosis. Anat. Rec. 2000, 258, 305–318. [Google Scholar] [CrossRef]
- Sandri, M.; El Meslemani, A.H.; Sandri, C.; Schjerling, P.; Vissing, K.; Andersen, J.L.; Rossini, K.; Carraro, U.; Angelini, C. Caspase 3 Expression Correlates with Skeletal Muscle Apoptosis in Duchenne and Facioscapulo Human Muscular Dystrophy. A Potential Target for Pharmacological Treatment? J. Neuropathol. Exp. Neurol. 2001, 60, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Minetti, C.; Pedemonte, M.; Carraro, U. Apoptotic Myonuclei in Human Duchenne Muscular Dystrophy. Lab. Investig. 1998, 78, 1005–1016. [Google Scholar]
- Ortiz-Cordero, C.; Bincoletto, C.; Dhoke, N.R.; Selvaraj, S.; Magli, A.; Zhou, H.; Kim, D.-H.; Bang, A.G.; Perlingeiro, R.C.R. Defective Autophagy and Increased Apoptosis Contribute toward the Pathogenesis of FKRP-Associated Muscular Dystrophies. Stem. Cell Rep. 2021, 16, 2752–2767. [Google Scholar] [CrossRef]
- Langenbach, K.J.; Rando, T.A. Inhibition of Dystroglycan Binding to Laminin Disrupts the PI3K/AKT Pathway and Survival Signaling in Muscle Cells. Muscle Nerve 2002, 26, 644–653. [Google Scholar] [CrossRef]
- Baghdiguian, S.; Martin, M.; Richard, I.; Pons, F.; Astier, C.; Bourg, N.; Hay, R.T.; Chemaly, R.; Halaby, G.; Loiselet, J.; et al. Calpain 3 Deficiency Is Associated with Myonuclear Apoptosis and Profound Perturbation of the IkappaB Alpha/NF-KappaB Pathway in Limb-Girdle Muscular Dystrophy Type 2A. Nat. Med. 1999, 5, 503–511. [Google Scholar] [CrossRef]
- Irwin, W.A.; Bergamin, N.; Sabatelli, P.; Reggiani, C.; Megighian, A.; Merlini, L.; Braghetta, P.; Columbaro, M.; Volpin, D.; Bressan, G.M.; et al. Mitochondrial Dysfunction and Apoptosis in Myopathic Mice with Collagen VI Deficiency. Nat. Genet. 2003, 35, 367–371. [Google Scholar] [CrossRef]
- Dominov, J.A.; Kravetz, A.J.; Ardelt, M.; Kostek, C.A.; Beermann, M.L.; Miller, J.B. Muscle-Specific BCL2 Expression Ameliorates Muscle Disease in Laminin {alpha}2-Deficient, but Not in Dystrophin-Deficient, Mice. Hum. Mol. Genet. 2005, 14, 1029–1040. [Google Scholar] [CrossRef][Green Version]
- Davies, J.E.; Wang, L.; Garcia-Oroz, L.; Cook, L.J.; Vacher, C.; O’Donovan, D.G.; Rubinsztein, D.C. Doxycycline Attenuates and Delays Toxicity of the Oculopharyngeal Muscular Dystrophy Mutation in Transgenic Mice. Nat. Med. 2005, 11, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Wallace, L.M.; Garwick, S.E.; Mei, W.; Belayew, A.; Coppee, F.; Ladner, K.J.; Guttridge, D.; Yang, J.; Harper, S.Q. DUX4, a Candidate Gene for Facioscapulohumeral Muscular Dystrophy, Causes P53-Dependent Myopathy in Vivo. Ann. Neurol. 2011, 69, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Kowaljow, V.; Marcowycz, A.; Ansseau, E.; Conde, C.B.; Sauvage, S.; Mattéotti, C.; Arias, C.; Corona, E.D.; Nuñez, N.G.; Leo, O.; et al. The DUX4 Gene at the FSHD1A Locus Encodes a Pro-Apoptotic Protein. Neuromuscul. Disord. 2007, 17, 611–623. [Google Scholar] [CrossRef]
- Jones, T.I.; Chew, G.-L.; Barraza-Flores, P.; Schreier, S.; Ramirez, M.; Wuebbles, R.D.; Burkin, D.J.; Bradley, R.K.; Jones, P.L. Transgenic Mice Expressing Tunable Levels of DUX4 Develop Characteristic Facioscapulohumeral Muscular Dystrophy-like Pathophysiology Ranging in Severity. Skelet Muscle 2020, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.; Leeuwenburgh, C. Muscle Fiber Specific Apoptosis and TNF-Alpha Signaling in Sarcopenia Are Attenuated by Life-Long Calorie Restriction. FASEB J. 2005, 19, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Siu, P.M.; Pistilli, E.E.; Butler, D.C.; Alway, S.E. Aging Influences Cellular and Molecular Responses of Apoptosis to Skeletal Muscle Unloading. Am. J. Physiol.-Cell Physiol. 2005, 288, C338–C349. [Google Scholar] [CrossRef]
- Dupont-Versteegden, E.E. Apoptosis in Muscle Atrophy: Relevance to Sarcopenia. Exp. Gerontol. 2005, 40, 473–481. [Google Scholar] [CrossRef]
- Shi, J.; Tang, M.; Zhou, S.; Xu, D.; Zhao, J.; Wu, C.; Wang, Q.; Tian, X.; Li, M.; Zeng, X. Programmed Cell Death Pathways in the Pathogenesis of Idiopathic Inflammatory Myopathies. Front. Immunol. 2021, 12, 783616. [Google Scholar] [CrossRef]
- Danielsson, O.; Häggqvist, B.; Gröntoft, L.; Öllinger, K.; Ernerudh, J. Apoptosis in Idiopathic Inflammatory Myopathies with Partial Invasion; a Role for CD8+ Cytotoxic T Cells? PLoS ONE 2020, 15, e0239176. [Google Scholar] [CrossRef]
- Turner, P.R.; Westwood, T.; Regen, C.M.; Steinhardt, R.A. Increased Protein Degradation Results from Elevated Free Calcium Levels Found in Muscle from Mdx Mice. Nature 1988, 335, 735–738. [Google Scholar] [CrossRef]
- Spencer, M.J.; Croall, D.E.; Tidball, J.G. Calpains Are Activated in Necrotic Fibers from Mdx Dystrophic Mice. J. Biol. Chem. 1995, 270, 10909–10914. [Google Scholar] [CrossRef]
- Alderton, J.M.; Steinhardt, R.A. How Calcium Influx through Calcium Leak Channels Is Responsible for the Elevated Levels of Calcium-Dependent Proteolysis in Dystrophic Myotubes. Trends Cardiovasc. Med. 2000, 10, 268–272. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of Cell Death: The Calcium–Apoptosis Link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Fernando, P.; Megeney, L.A. Is Caspase-Dependent Apoptosis Only Cell Differentiation Taken to the Extreme? FASEB J. 2007, 21, 8–17. [Google Scholar] [CrossRef]
- Luedde, M.; Lutz, M.; Carter, N.; Sosna, J.; Jacoby, C.; Vucur, M.; Gautheron, J.; Roderburg, C.; Borg, N.; Reisinger, F.; et al. RIP3, a Kinase Promoting Necroptotic Cell Death, Mediates Adverse Remodelling after Myocardial Infarction. Cardiovasc. Res. 2014, 103, 206–216. [Google Scholar] [CrossRef]
- Bencze, M.; Hou, C.; Periou, B.; Agbulut, O.; Gervais, M.; Ternacle, J.; Derumeaux, G.; Tiret, L.; Relaix, F.; Authier, F.-J. Receptor Interacting Protein Kinase-3 Promotes Both Myopathy and Cardiomyopathy in Dystrophin-Deficient Mice. bioRxiv 2022. [Google Scholar] [CrossRef]
- Ramani Sattiraju, S.; Jama, A.; Alshudukhi, A.A.; Edward Townsend, N.; Reynold Miranda, D.; Reese, R.R.; Voss, A.A.; Ren, H. Loss of Membrane Integrity Drives Myofiber Death in Lipin1-Deficient Skeletal Muscle. Physiol. Rep. 2020, 8, e14620. [Google Scholar] [CrossRef]
- Picon, C.; Jayaraman, A.; James, R.; Beck, C.; Gallego, P.; Witte, M.E.; van Horssen, J.; Mazarakis, N.D.; Reynolds, R. Neuron-Specific Activation of Necroptosis Signaling in Multiple Sclerosis Cortical Grey Matter. Acta Neuropathol. 2021, 141, 585–604. [Google Scholar] [CrossRef]
- Chehade, L.; Deguise, M.-O.; De Repentigny, Y.; Yaworski, R.; Beauvais, A.; Gagnon, S.; Hensel, N.; Kothary, R. Suppression of the Necroptotic Cell Death Pathways Improves Survival in Smn 2B/- Mice. Front. Cell Neurosci. 2022, 16, 972029. [Google Scholar] [CrossRef]
- Re, D.B.; Le Verche, V.; Yu, C.; Amoroso, M.W.; Politi, K.A.; Phani, S.; Ikiz, B.; Hoffmann, L.; Koolen, M.; Nagata, T.; et al. Necroptosis Drives Motor Neuron Death in Models of Both Sporadic and Familial ALS. Neuron 2014, 81, 1001–1008. [Google Scholar] [CrossRef]
- Ofengeim, D.; Ito, Y.; Najafov, A.; Zhang, Y.; Shan, B.; DeWitt, J.P.; Ye, J.; Zhang, X.; Chang, A.; Vakifahmetoglu-Norberg, H.; et al. Activation of Necroptosis in Multiple Sclerosis. Cell Rep. 2015, 10, 1836–1849. [Google Scholar] [CrossRef]
- Yuan, J.; Amin, P.; Ofengeim, D. Necroptosis and RIPK1-Mediated Neuroinflammation in CNS Diseases. Nat. Rev. Neurosci. 2019, 20, 19–33. [Google Scholar] [CrossRef]
- Dermentzaki, G.; Politi, K.A.; Lu, L.; Mishra, V.; Pérez-Torres, E.J.; Sosunov, A.A.; McKhann, G.M.; Lotti, F.; Shneider, N.A.; Przedborski, S. Deletion of Ripk3 Prevents Motor Neuron Death In Vitro but Not In Vivo. eNeuro 2019, 6, ENEURO.0308-18.2018. [Google Scholar] [CrossRef]
- Wang, T.; Perera, N.D.; Chiam, M.D.F.; Cuic, B.; Wanniarachchillage, N.; Tomas, D.; Samson, A.L.; Cawthorne, W.; Valor, E.N.; Murphy, J.M.; et al. Necroptosis Is Dispensable for Motor Neuron Degeneration in a Mouse Model of ALS. Cell Death Differ. 2020, 27, 1728–1739. [Google Scholar] [CrossRef]
- Canafax, D.M.; Ascher, N.L. Cyclosporine Immunosuppression. Clin. Pharm. 1983, 2, 515–524. [Google Scholar] [CrossRef]
- Schiavone, M.; Zulian, A.; Menazza, S.; Petronilli, V.; Argenton, F.; Merlini, L.; Sabatelli, P.; Bernardi, P. Alisporivir Rescues Defective Mitochondrial Respiration in Duchenne Muscular Dystrophy. Pharmacol. Res. 2017, 125, 122–131. [Google Scholar] [CrossRef]
- Reutenauer, J.; Dorchies, O.M.; Patthey-Vuadens, O.; Vuagniaux, G.; Ruegg, U.T. Investigation of Debio 025, a Cyclophilin Inhibitor, in the Dystrophic Mdx Mouse, a Model for Duchenne Muscular Dystrophy. Br. J. Pharm. 2008, 155, 574–584. [Google Scholar] [CrossRef]
- Burr, A.R.; Molkentin, J.D. Genetic Evidence in the Mouse Solidifies the Calcium Hypothesis of Myofiber Death in Muscular Dystrophy. Cell Death Differ. 2015, 22, 1402–1412. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne Muscular Dystrophy Is Associated with the Inhibition of Calcium Uniport in Mitochondria and an Increased Sensitivity of the Organelles to the Calcium-Induced Permeability Transition. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and Pharmacologic Inhibition of Mitochondrial-Dependent Necrosis Attenuates Muscular Dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Starinets, V.S.; Talanov, E.Y.; Mikheeva, I.B.; Belosludtseva, N.V.; Belosludtsev, K.N. Alisporivir Improves Mitochondrial Function in Skeletal Muscle of Mdx Mice but Suppresses Mitochondrial Dynamics and Biogenesis. Int. J. Mol. Sci. 2021, 22, 9780. [Google Scholar] [CrossRef] [PubMed]
- Ryffel, B. The Carcinogenicity of Ciclosporin. Toxicology 1992, 73, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Rocher, L.L.; McQuillan, M.A.; Schmaltz, S.; Palella, T.D.; Fox, I.H. Cyclosporine-Induced Hyperuricemia and Gout. N. Engl. J. Med. 1989, 321. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Effect of Alisporivir on Calcium Ion Transport and Mitophagy in Skeletal Muscle and Heart Mitochondria in Dystrophin-Deficient Mice. Bull. Exp. Biol. Med. 2022, 172, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.S.; Zhao, L.; Zhang, C.; Li, H.; Han, R. Genetic Disruption of the Inflammasome Adaptor ASC Has Minimal Impact on the Pathogenesis of Duchenne Muscular Dystrophy in Mdx Mice. Life Sci. 2020, 257, 118069. [Google Scholar] [CrossRef] [PubMed]
- Boursereau, R.; Abou-Samra, M.; Lecompte, S.; Noel, L.; Brichard, S.M. Downregulation of the NLRP3 Inflammasome by Adiponectin Rescues Duchenne Muscular Dystrophy. BMC Biol. 2018, 16, 33. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical Role for Calcium Mobilization in Activation of the NLRP3 Inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- Nalbandian, A.; Khan, A.A.; Srivastava, R.; Llewellyn, K.J.; Tan, B.; Shukr, N.; Fazli, Y.; Kimonis, V.E.; BenMohamed, L. Activation of the NLRP3 Inflammasome Is Associated with Valosin-Containing Protein Myopathy. Inflammation 2017, 40, 21–41. [Google Scholar] [CrossRef]
- Grasl-Kraupp, B.; Ruttkay-Nedecky, B.; Koudelka, H.; Bukowska, K.; Bursch, W.; Schulte-Hermann, R. In Situ Detection of Fragmented DNA (TUNEL Assay) Fails to Discriminate among Apoptosis, Necrosis, and Autolytic Cell Death: A Cautionary Note. Hepatology 1995, 21, 1465–1468. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The Molecular Machinery of Regulated Cell Death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Brouckaert, G.; Kalai, M.; Krysko, D.V.; Saelens, X.; Vercammen, D.; Ndlovu, M.; Haegeman, G.; D’Herde, K.; Vandenabeele, P. Phagocytosis of Necrotic Cells by Macrophages Is Phosphatidylserine Dependent and Does Not Induce Inflammatory Cytokine Production. Mol. Biol. Cell 2004, 15, 1089–1100. [Google Scholar] [CrossRef]
- Bell, R.A.V.; Megeney, L.A. Evolution of Caspase-Mediated Cell Death and Differentiation: Twins Separated at Birth. Cell Death Differ. 2017, 24, 1359–1368. [Google Scholar] [CrossRef]
- Bell, R.A.V.; Al-Khalaf, M.H.; Brunette, S.; Alsowaida, D.; Chu, A.; Bandukwala, H.; Dechant, G.; Apostolova, G.; Dilworth, F.J.; Megeney, L.A. Chromatin Reorganization during Myoblast Differentiation Involves the Caspase-Dependent Removal of SATB2. Cells 2022, 11, 966. [Google Scholar] [CrossRef]
- Dick, S.A.; Chang, N.C.; Dumont, N.A.; Bell, R.A.V.; Putinski, C.; Kawabe, Y.; Litchfield, D.W.; Rudnicki, M.A.; Megeney, L.A. Caspase 3 Cleavage of Pax7 Inhibits Self-Renewal of Satellite Cells. Proc. Natl. Acad. Sci. USA 2015, 112, E5246–E5252. [Google Scholar] [CrossRef]
- Yamazaki, T.; Galluzzi, L. BAX and BAK Dynamics Control Mitochondrial DNA Release during Apoptosis. Cell Death Differ. 2022, 29, 1296–1298. [Google Scholar] [CrossRef]
- Ge, Y.; Huang, M.; Yao, Y. Efferocytosis and Its Role in Inflammatory Disorders. Front. Cell Dev. Biol. 2022, 10, 839248. [Google Scholar] [CrossRef]
- Thorp, E.; Subramanian, M.; Tabas, I. The Role of Macrophages and Dendritic Cells in the Clearance of Apoptotic Cells in Advanced Atherosclerosis. Eur. J. Immunol. 2011, 41, 2515–2518. [Google Scholar] [CrossRef]
- Hao, Q.; Idell, S.; Tang, H. M1 Macrophages Are More Susceptible to Necroptosis. J. Cell Immunol. 2021, 3, 97–102. [Google Scholar] [CrossRef]
- Bruusgaard, J.C.; Johansen, I.B.; Egner, I.M.; Rana, Z.A.; Gundersen, K. Myonuclei Acquired by Overload Exercise Precede Hypertrophy and Are Not Lost on Detraining. Proc. Natl. Acad. Sci. USA 2010, 107, 15111–15116. [Google Scholar] [CrossRef]
- Brack, A.S.; Bildsoe, H.; Hughes, S.M. Evidence That Satellite Cell Decrement Contributes to Preferential Decline in Nuclear Number from Large Fibres during Murine Age-Related Muscle Atrophy. J. Cell Sci. 2005, 118, 4813–4821. [Google Scholar] [CrossRef]
- Kirby, T.J.; Dupont-Versteegden, E.E. Cross Talk Proposal: Myonuclei Are Lost with Ageing and Atrophy. J. Physiol. 2022, 600, 2077–2080. [Google Scholar] [CrossRef]
- McLoon, L.K.; Rowe, J.; Wirtschafter, J.; McCormick, K.M. Continuous Myofiber Remodeling in Uninjured Extraocular Myofibers: Myonuclear Turnover and Evidence for Apoptosis. Muscle Nerve 2004, 29, 707–715. [Google Scholar] [CrossRef]
- McLoon, L.K.; Wirtschafter, J. Activated Satellite Cells in Extraocular Muscles of Normal Adult Monkeys and Humans. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1927–1932. [Google Scholar] [CrossRef]
- Newton, K.; Wickliffe, K.E.; Dugger, D.L.; Maltzman, A.; Roose-Girma, M.; Dohse, M.; Kőműves, L.; Webster, J.D.; Dixit, V.M. Cleavage of RIPK1 by Caspase-8 Is Crucial for Limiting Apoptosis and Necroptosis. Nature 2019, 574, 428–431. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Vanden Berghe, T.; Festjens, N. Caspase Inhibitors Promote Alternative Cell Death Pathways. Sci. STKE 2006, 2006, pe44. [Google Scholar] [CrossRef]
- Fischer, U.; Schulze-Osthoff, K. Apoptosis-Based Therapies and Drug Targets. Cell Death Differ. 2005, 12, 942–961. [Google Scholar] [CrossRef]
- Eskandari, E.; Eaves, C.J. Paradoxical Roles of Caspase-3 in Regulating Cell Survival, Proliferation, and Tumorigenesis. J. Cell Biol. 2022, 221, e202201159. [Google Scholar] [CrossRef]
- Julien, O.; Wells, J.A. Caspases and Their Substrates. Cell Death Differ. 2017, 24, 1380–1389. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bencze, M. Mechanisms of Myofibre Death in Muscular Dystrophies: The Emergence of the Regulated Forms of Necrosis in Myology. Int. J. Mol. Sci. 2023, 24, 362. https://doi.org/10.3390/ijms24010362
Bencze M. Mechanisms of Myofibre Death in Muscular Dystrophies: The Emergence of the Regulated Forms of Necrosis in Myology. International Journal of Molecular Sciences. 2023; 24(1):362. https://doi.org/10.3390/ijms24010362
Chicago/Turabian StyleBencze, Maximilien. 2023. "Mechanisms of Myofibre Death in Muscular Dystrophies: The Emergence of the Regulated Forms of Necrosis in Myology" International Journal of Molecular Sciences 24, no. 1: 362. https://doi.org/10.3390/ijms24010362
APA StyleBencze, M. (2023). Mechanisms of Myofibre Death in Muscular Dystrophies: The Emergence of the Regulated Forms of Necrosis in Myology. International Journal of Molecular Sciences, 24(1), 362. https://doi.org/10.3390/ijms24010362