

G Protein-Coupled Receptor 15 Expression Is Associated with Myocardial Infarction
 , , ,
, , ,  ,
,  , , , ,
, , , ,  and
and 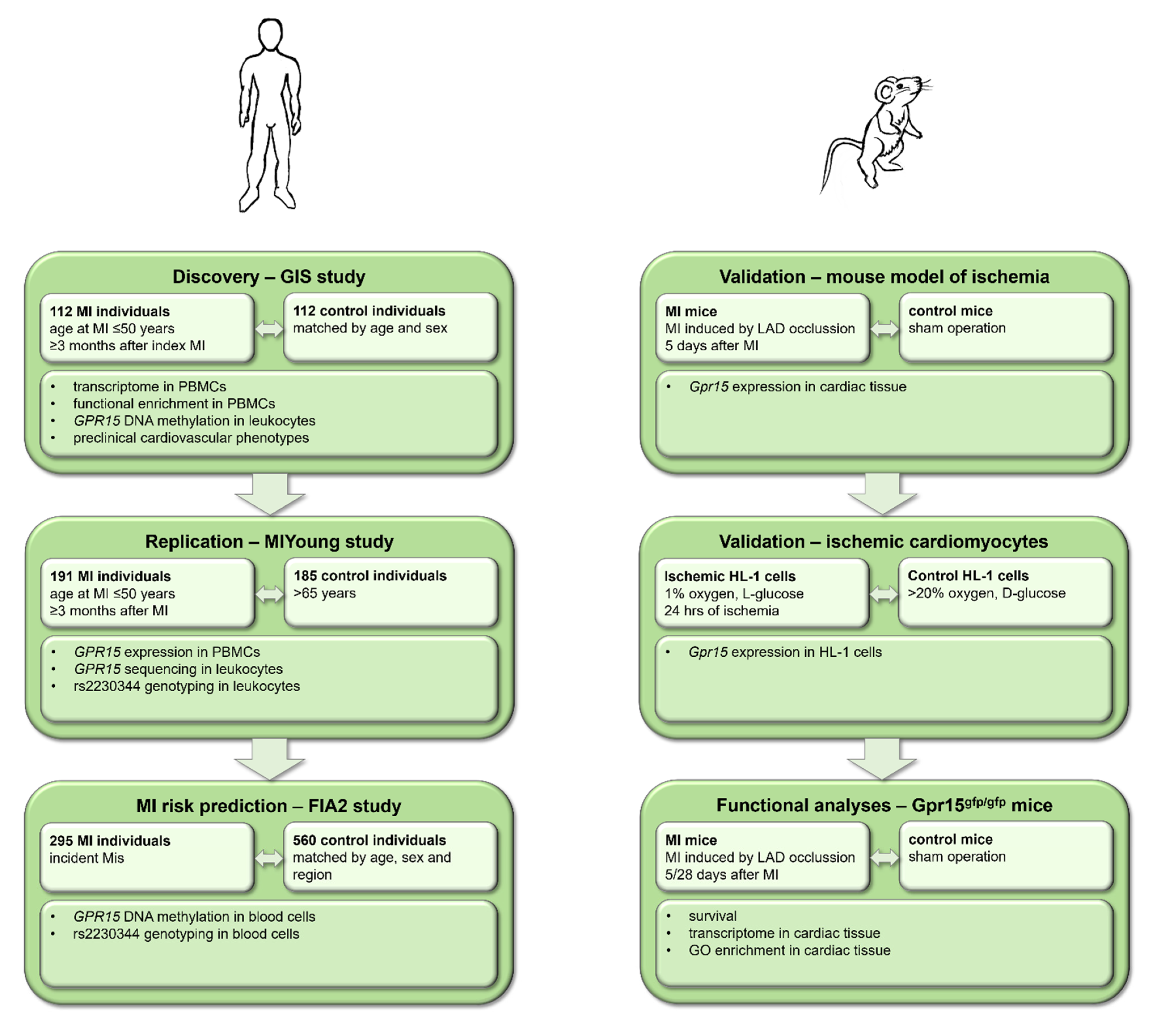{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
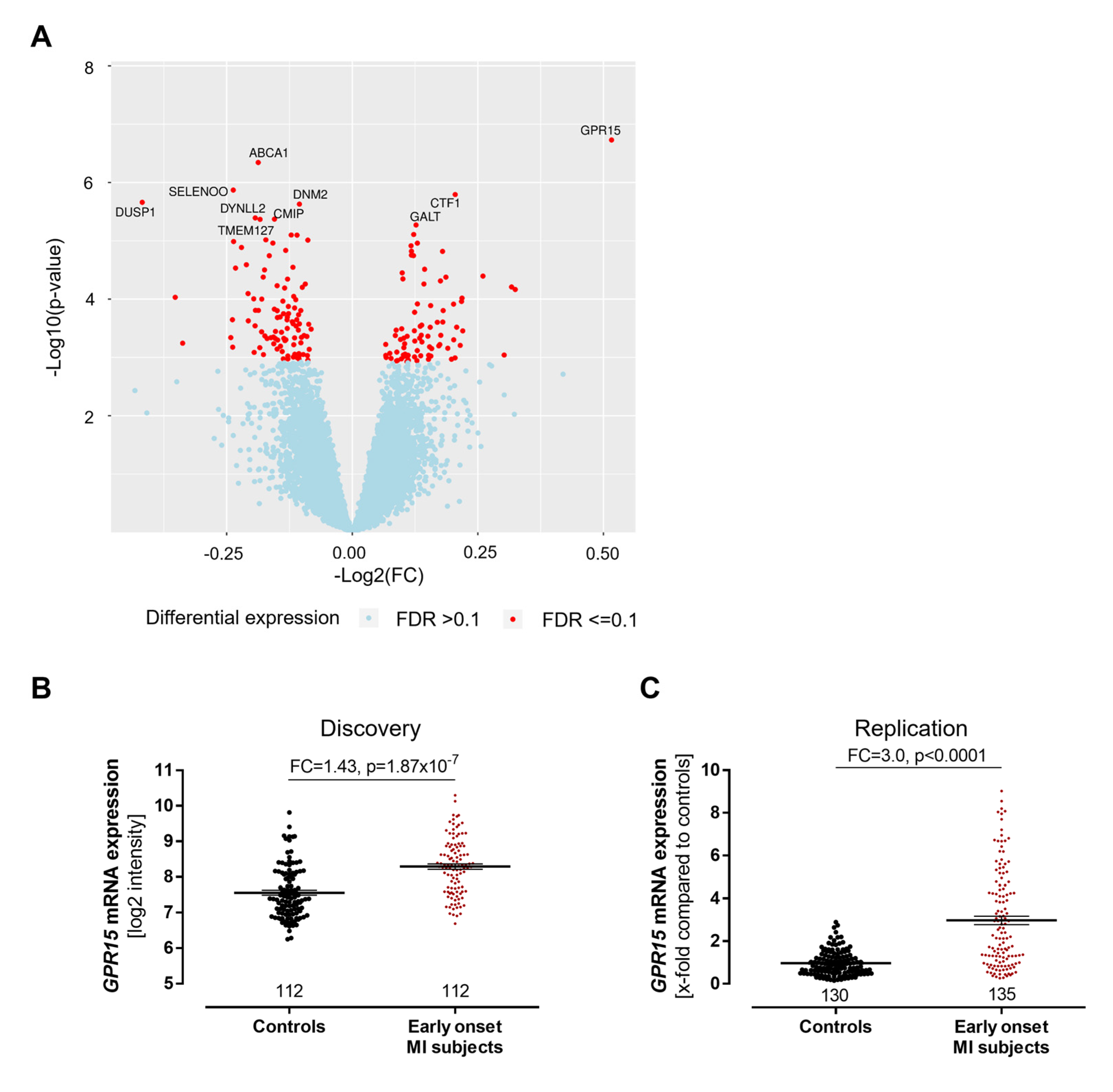
2.1. Expression of GRP15 Is Increased in Early-Onset Myocardial Infarction
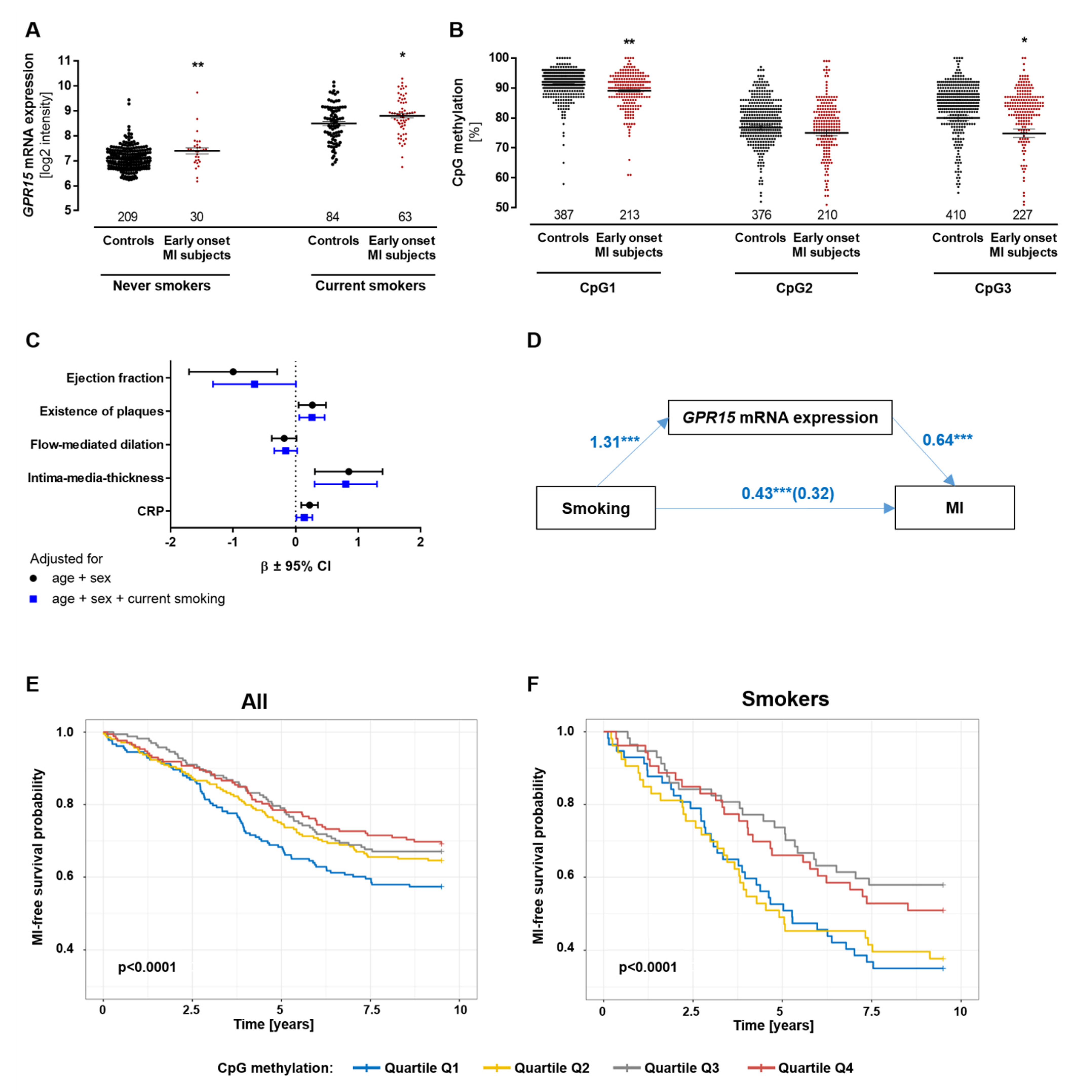
2.2. Relation of GPR15 Expression with DNA Methylation, Myocardial Infarction and Smoking
2.3. GPR15 DNA Methylation Can Predict Risk of Myocardial Infarction
2.4. GPR15 SNP rs2230344 Associates with Early-Onset Myocardial Infarction
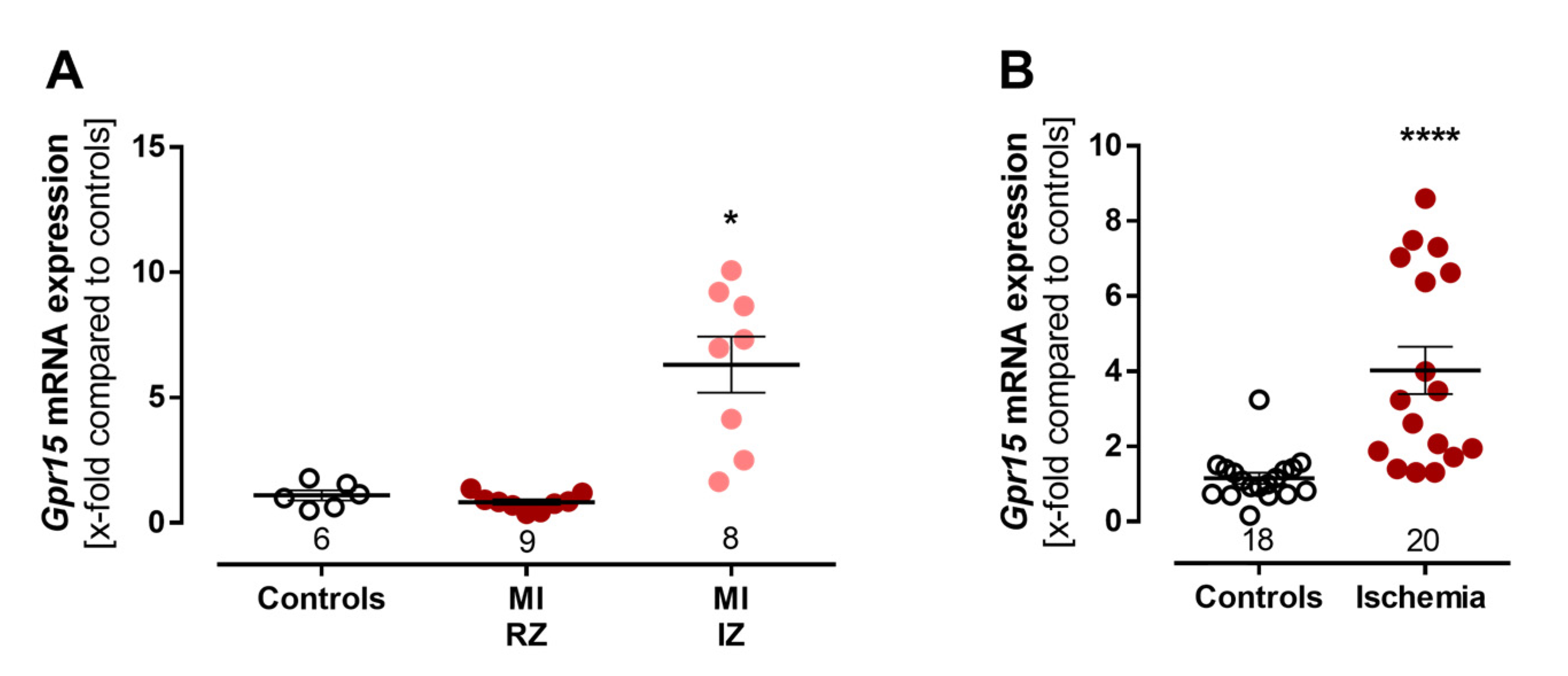
2.5. Experimental Validation of Increased Gpr15 Expression after Ischemia
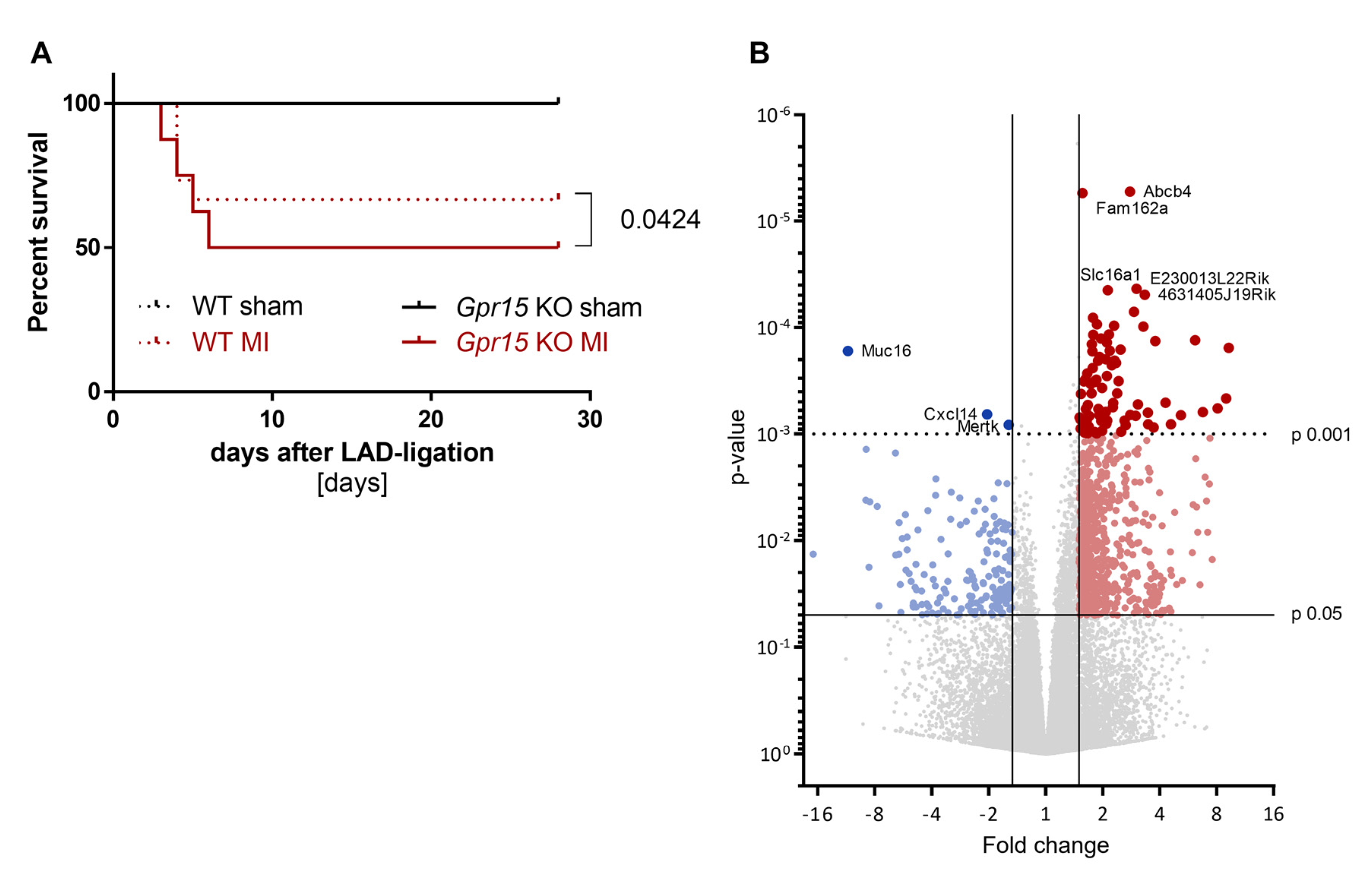
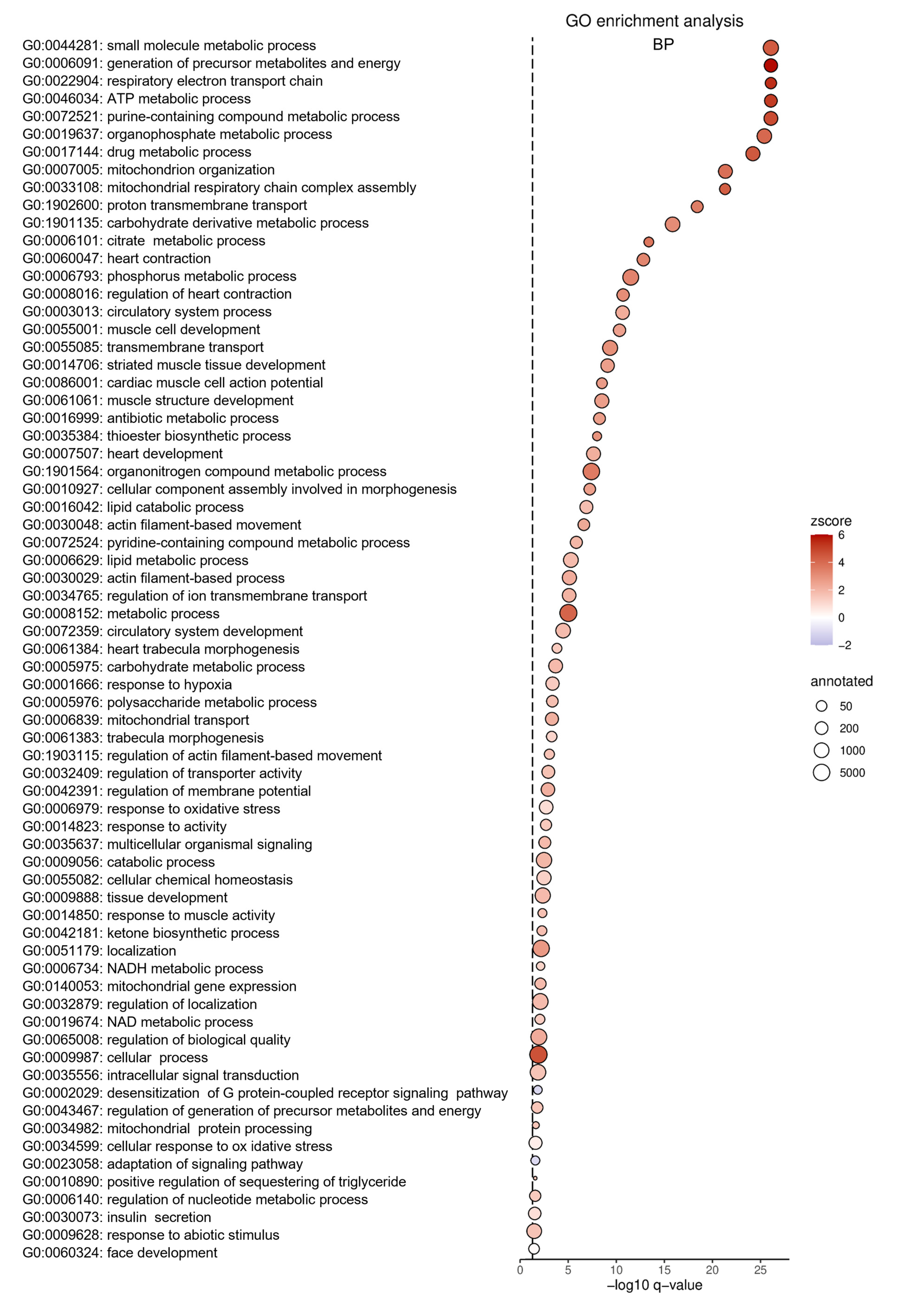
2.6. Gpr15 Knockout Affects Survival and Cardiac Remodeling in a Murine Model of Myocardial Infarction
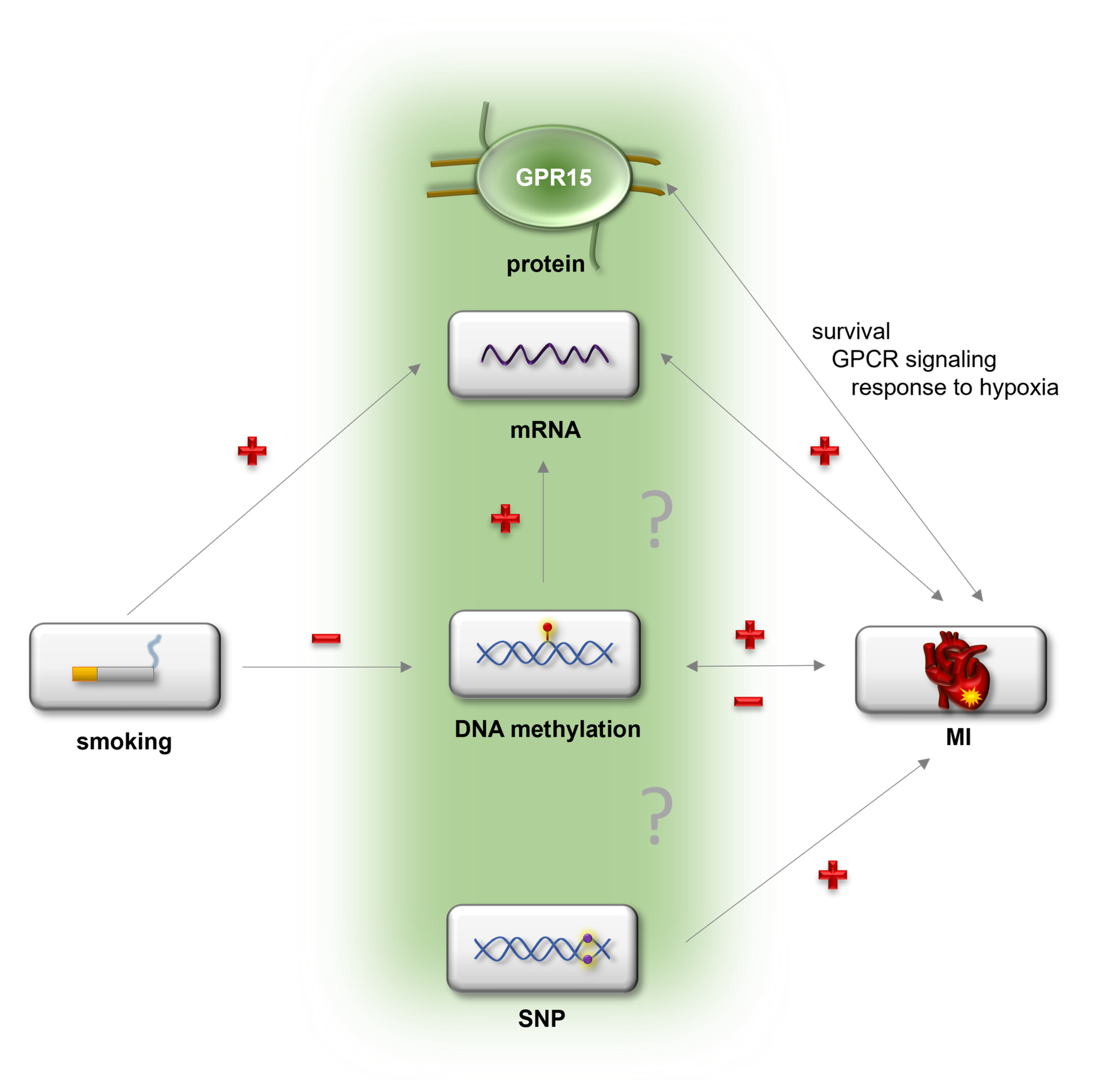
3. Discussion
4. Materials and Methods
4.1. Human Cohorts
4.1.1. Gutenberg Young Myocardial Infarction Study (GIS)
4.1.2. Young Myocardial Infarction Study (MIYoung)
4.1.3. First-Ever Myocardial Infarction Study 2 (FIA2)
4.2. Mouse Models
4.3. Cell Culture
4.4. Gene Expression Analysis
4.5. Analyses of DNA Methylation
4.6. Statistical and Bioinformatical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2015 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 315 diseases and injuries and healthy life expectancy (HALE), 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1603–1658. [Google Scholar] [CrossRef]
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Abdolmaleki, F.; Gheibi Hayat, S.M.; Bianconi, V.; Johnston, T.P.; Sahebkar, A. Atherosclerosis and immunity: A perspective. Trends Cardiovasc. Med. 2019, 29, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Yusuf, S.; Hawken, S.; Ounpuu, S.; Dans, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J.; et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): Case-control study. Lancet 2004, 364, 937–952. [Google Scholar] [CrossRef]
- Timmis, A.; Townsend, N.; Gale, C.; Grobbee, R.; Maniadakis, N.; Flather, M.; Wilkins, E.; Wright, L.; Vos, R.; Bax, J.; et al. European Society of Cardiology: Cardiovascular Disease Statistics 2017. Eur. Heart J. 2018, 39, 508–579. [Google Scholar] [CrossRef]
- Doughty, M.; Mehta, R.; Bruckman, D.; Das, S.; Karavite, D.; Tsai, T.; Eagle, K. Acute myocardial infarction in the young—The University of Michigan experience. Am. Heart J. 2002, 143, 56–62. [Google Scholar] [CrossRef]
- Brscic, E.; Bergerone, S.; Gagnor, A.; Colajanni, E.; Matullo, G.; Scaglione, L.; Cassader, M.; Gaschino, G.; Di Leo, M.; Brusca, A.; et al. Acute myocardial infarction in young adults: Prognostic role of angiotensin-converting enzyme, angiotensin II type I receptor, apolipoprotein E, endothelial constitutive nitric oxide synthase, and glycoprotein IIIa genetic polymorphisms at medium-term follow-up. Am. Heart J. 2000, 139, 979–984. [Google Scholar] [CrossRef]
- Marenberg, M.E.; Risch, N.; Berkman, L.F.; Floderus, B.; de Faire, U. Genetic susceptibility to death from coronary heart disease in a study of twins. N. Engl. J. Med. 1994, 330, 1041–1046. [Google Scholar] [CrossRef]
- Schunkert, H.; von Scheidt, M.; Kessler, T.; Stiller, B.; Zeng, L.; Vilne, B. Genetics of coronary artery disease in the light of genome-wide association studies. Clin. Res. Cardiol. 2018, 107, 2–9. [Google Scholar] [CrossRef]
- Myocardial Infarction Genetics Consortium; Kathiresan, S.; Voight, B.F.; Purcell, S.; Musunuru, K.; Ardissino, D.; Mannucci, P.M.; Anand, S.; Engert, J.C.; Samani, N.J.; et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat. Genet. 2009, 41, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, J.; Kessler, T.; Munoz Venegas, L.; Schunkert, H. A decade of genome-wide association studies for coronary artery disease: The challenges ahead. Cardiovasc. Res. 2018, 114, 1241–1257. [Google Scholar] [CrossRef] [PubMed]
- Tyndall, J.D.; Sandilya, R. GPCR agonists and antagonists in the clinic. Med. Chem. 2005, 1, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Hendriks-Balk, M.C.; Peters, S.L.; Michel, M.C.; Alewijnse, A.E. Regulation of G protein-coupled receptor signalling: Focus on the cardiovascular system and regulator of G protein signalling proteins. Eur. J. Pharmacol. 2008, 585, 278–291. [Google Scholar] [CrossRef]
- Wang, J.; Gareri, C.; Rockman, H.A. G-Protein-Coupled Receptors in Heart Disease. Circ. Res. 2018, 123, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Wan, E.S.; Qiu, W.; Baccarelli, A.; Carey, V.J.; Bacherman, H.; Rennard, S.I.; Agusti, A.; Anderson, W.; Lomas, D.A.; Demeo, D.L. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum. Mol. Genet. 2012, 21, 3073–3082. [Google Scholar] [CrossRef]
- Haase, T.; Muller, C.; Krause, J.; Rothemeier, C.; Stenzig, J.; Kunze, S.; Waldenberger, M.; Munzel, T.; Pfeiffer, N.; Wild, P.S.; et al. Novel DNA Methylation Sites Influence GPR15 Expression in Relation to Smoking. Biomolecules 2018, 8, 74. [Google Scholar] [CrossRef]
- Teo, K.K.; Ounpuu, S.; Hawken, S.; Pandey, M.R.; Valentin, V.; Hunt, D.; Diaz, R.; Rashed, W.; Freeman, R.; Jiang, L.; et al. Tobacco use and risk of myocardial infarction in 52 countries in the INTERHEART study: A case-control study. Lancet 2006, 368, 647–658. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Kim, S.V.; Xiang, W.V.; Kwak, C.; Yang, Y.; Lin, X.W.; Ota, M.; Sarpel, U.; Rifkin, D.B.; Xu, R.; Littman, D.R. GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science 2013, 340, 1456–1459. [Google Scholar] [CrossRef]
- Cartwright, A.; Schmutz, C.; Askari, A.; Kuiper, J.H.; Middleton, J. Orphan receptor GPR15/BOB is up-regulated in rheumatoid arthritis. Cytokine 2014, 67, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Zundler, S.; Atreya, R.; Rath, T.; Voskens, C.; Hirschmann, S.; Lopez-Posadas, R.; Watson, A.; Becker, C.; Schuler, G.; et al. Differential effects of alpha4beta7 and GPR15 on homing of effector and regulatory T cells from patients with UC to the inflamed gut in vivo. Gut 2016, 65, 1642–1664. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, A.; Gageik, D.; Frede, A.; Pastille, E.; Hansen, W.; Rueffer, A.; Buer, J.; Buning, J.; Langhorst, J.; Westendorf, A.M. Differential expression of GPR15 on T cells during ulcerative colitis. JCI Insight 2017, 2, e90585. [Google Scholar] [CrossRef] [PubMed]
- Ammitzboll, C.; von Essen, M.R.; Bornsen, L.; Petersen, E.R.; McWilliam, O.; Ratzer, R.; Jeppe, R.C.; Oturai, A.B.; Sondergaard, H.B.; Sellebjerg, F. GPR15(+) T cells are Th17 like, increased in smokers and associated with multiple sclerosis. J. Autoimmun. 2019, 97, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M. The Role of GPR15 Function in Blood and Vasculature. Int. J. Mol. Sci. 2021, 22, 824. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef]
- Jjingo, D.; Conley, A.B.; Yi, S.V.; Lunyak, V.V.; Jordan, I.K. On the presence and role of human gene-body DNA methylation. Oncotarget 2012, 3, 462–474. [Google Scholar] [CrossRef]
- Teissandier, A.; Bourc’his, D. Gene body DNA methylation conspires with H3K36me3 to preclude aberrant transcription. EMBO J. 2017, 36, 1471–1473. [Google Scholar] [CrossRef]
- Bauer, M.; Hackermuller, J.; Schor, J.; Schreiber, S.; Fink, B.; Pierzchalski, A.; Herberth, G. Specific induction of the unique GPR15 expression in heterogeneous blood lymphocytes by tobacco smoking. Biomarkers 2019, 24, 217–224. [Google Scholar] [CrossRef]
- Dogan, M.V.; Xiang, J.; Beach, S.R.; Cutrona, C.; Gibbons, F.X.; Simons, R.L.; Brody, G.H.; Stapleton, J.T.; Philibert, R.A. Ethnicity and Smoking-Associated DNA Methylation Changes at HIV Co-Receptor GPR15. Front. Psychiatry 2015, 6, 132. [Google Scholar] [CrossRef]
- Bilsborough, J.; Viney, J.L. GPR15: A tale of two species. Nat. Immunol. 2015, 16, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Ambroziak, M.; Niewczas-Wieprzowska, K.; Maicka, A.; Budaj, A. Younger age of patients with myocardial infarction is associated with a higher number of relatives with a history of premature atherosclerosis. BMC Cardiovasc. Disord. 2020, 20, 410. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.S.; Zeller, T.; Beutel, M.; Blettner, M.; Dugi, K.A.; Lackner, K.J.; Pfeiffer, N.; Munzel, T.; Blankenberg, S. The Gutenberg Health Study. Bundesgesundheitsblatt Gesundh. Gesundh. 2012, 55, 824–829. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.S.; Sinning, C.R.; Roth, A.; Wilde, S.; Schnabel, R.B.; Lubos, E.; Zeller, T.; Keller, T.; Lackner, K.J.; Blettner, M.; et al. Distribution and categorization of left ventricular measurements in the general population: Results from the population-based Gutenberg Heart Study. Circ. Cardiovasc. Imaging 2010, 3, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic. Acids. Res. 1988, 16, 1215. [Google Scholar] [CrossRef]
- Zeller, T.; Wild, P.; Szymczak, S.; Rotival, M.; Schillert, A.; Castagne, R.; Maouche, S.; Germain, M.; Lackner, K.; Rossmann, H.; et al. Genetics and beyond—The transcriptome of human monocytes and disease susceptibility. PLoS ONE 2010, 5, e10693. [Google Scholar] [CrossRef]
- Ekblom, K.; Marklund, S.L.; Jansson, J.H.; Hallmans, G.; Weinehall, L.; Hultdin, J. Iron stores and HFE genotypes are not related to increased risk of first-time myocardial infarction: A prospective nested case-referent study. Int. J. Cardiol. 2011, 150, 169–172. [Google Scholar] [CrossRef]
- Crawford, A.A.; Soderberg, S.; Kirschbaum, C.; Murphy, L.; Eliasson, M.; Ebrahim, S.; Davey Smith, G.; Olsson, T.; Sattar, N.; Lawlor, D.A.; et al. Morning plasma cortisol as a cardiovascular risk factor: Findings from prospective cohort and Mendelian randomization studies. Eur. J. Endocrinol. 2019, 181, 429–438. [Google Scholar] [CrossRef]
- Bayne, K. Revised Guide for the Care and Use of Laboratory Animals available. American Physiological Society. Physiologist 1996, 39, 208–211. [Google Scholar]
- Hinrichs, S.; Scherschel, K.; Kruger, S.; Neumann, J.T.; Schwarzl, M.; Yan, I.; Warnke, S.; Ojeda, F.M.; Zeller, T.; Karakas, M.; et al. Precursor proadrenomedullin influences cardiomyocyte survival and local inflammation related to myocardial infarction. Proc. Natl. Acad. Sci. USA 2018, 115, E8727–E8736. [Google Scholar] [CrossRef]
- Bacmeister, L.; Schwarzl, M.; Warnke, S.; Stoffers, B.; Blankenberg, S.; Westermann, D.; Lindner, D. Inflammation and fibrosis in murine models of heart failure. Basic Res. Cardiol. 2019, 114, 19. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kunze, S. Quantitative Region-Specific DNA Methylation Analysis by the EpiTYPER Technology. Methods Mol. Biol. 2018, 1708, 515–535. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- R Core Team. nlme: Linear and Nonlinear Mixed Effects Models; R Core Team: Vienna, Austria, 2013. [Google Scholar]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Bass, A.J. Qvalue: Q-Value Estimation for False Discovery Rate Control; Bioconductor: Boston, MA, USA, 2013. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Wickham, H. Elegant graphics for data analysis. Media 2009, 35, 10–1007. [Google Scholar]
- Tingley, D.; Yamamoto, T.; Hirose, K.; Keele, L.; Imai, K. Mediation: R package for causal mediation analysis. JSS J. Stat. Softw. 2014, 59. Available online: http://hdl.handle.net/1721.1/91154 (accessed on 4 November 2021).







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haase, T.; Müller, C.; Stoffers, B.; Kirn, P.; Waldenberger, M.; Kaiser, F.J.; Karakas, M.; Kim, S.V.; Voss, S.; Wild, P.S.; et al. G Protein-Coupled Receptor 15 Expression Is Associated with Myocardial Infarction. Int. J. Mol. Sci. 2023, 24, 180. https://doi.org/10.3390/ijms24010180
Haase T, Müller C, Stoffers B, Kirn P, Waldenberger M, Kaiser FJ, Karakas M, Kim SV, Voss S, Wild PS, et al. G Protein-Coupled Receptor 15 Expression Is Associated with Myocardial Infarction. International Journal of Molecular Sciences. 2023; 24(1):180. https://doi.org/10.3390/ijms24010180
Chicago/Turabian StyleHaase, Tina, Christian Müller, Bastian Stoffers, Philipp Kirn, Melanie Waldenberger, Frank J. Kaiser, Mahir Karakas, Sangwon V. Kim, Svenja Voss, Philipp S. Wild, and et al. 2023. "G Protein-Coupled Receptor 15 Expression Is Associated with Myocardial Infarction" International Journal of Molecular Sciences 24, no. 1: 180. https://doi.org/10.3390/ijms24010180
APA StyleHaase, T., Müller, C., Stoffers, B., Kirn, P., Waldenberger, M., Kaiser, F. J., Karakas, M., Kim, S. V., Voss, S., Wild, P. S., Lackner, K. J., Andersson, J., Söderberg, S., Lindner, D., & Zeller, T. (2023). G Protein-Coupled Receptor 15 Expression Is Associated with Myocardial Infarction. International Journal of Molecular Sciences, 24(1), 180. https://doi.org/10.3390/ijms24010180