
Crossing Bacterial Genomic Features and Methylation Patterns with MeStudio: An Epigenomic Analysis Tool

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Tool-Wide Comparison
2.2. The Sinorhizobium Case Study
3. Materials and Methods
3.1. Bacterial Strains and Culture Conditions
3.2. DNA Extraction and SMRT Sequencing
3.3. Sequence Analysis and Annotation
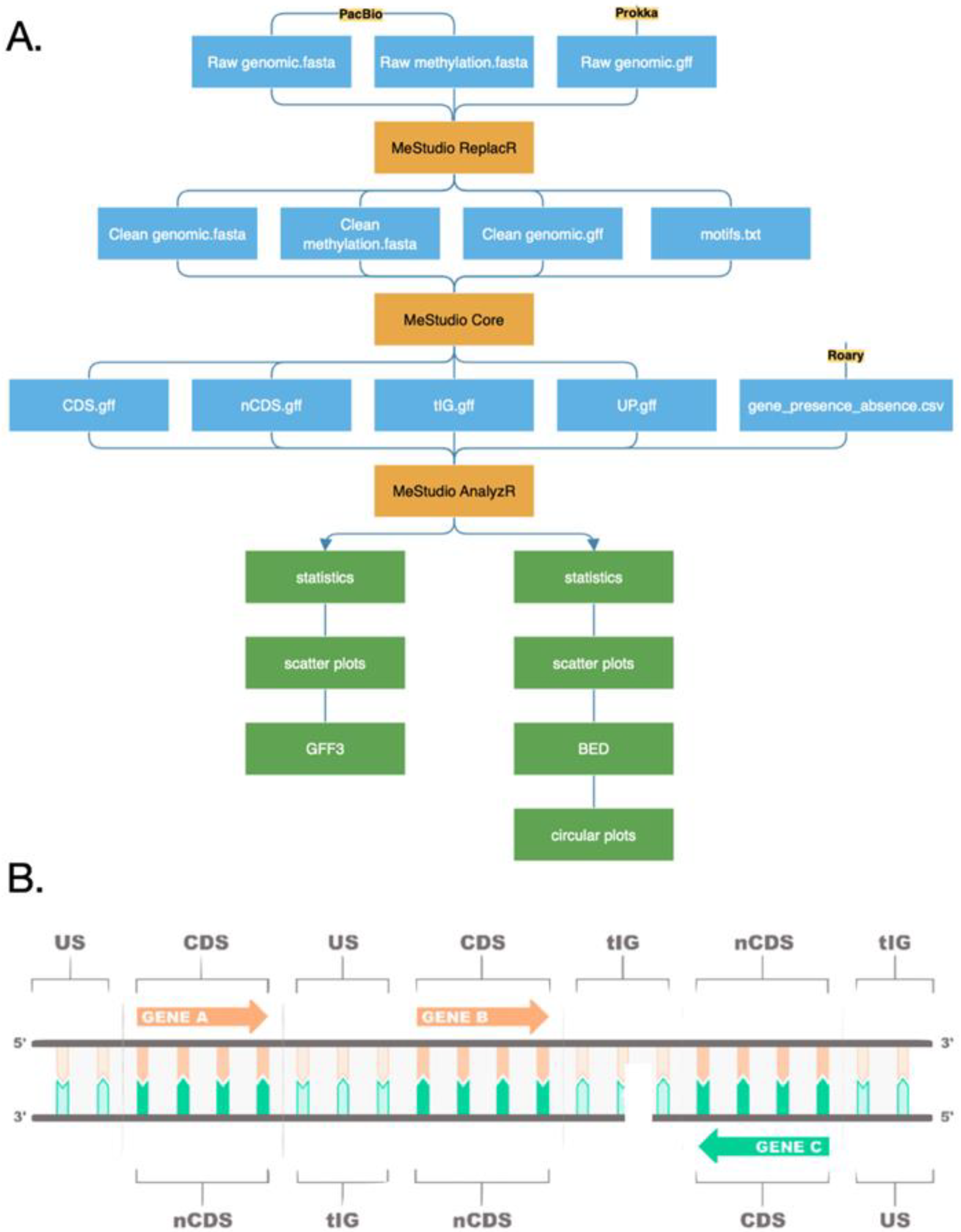
3.4. Software Design and Implementation
3.5. Preprocessing
3.6. Core Processing
3.7. Postprocessing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Moler, E.R.V.; Abakir, A.; Eleftheriou, M.; Johnson, J.S.; Krutovsky, K.V.; Lewis, L.C.; Ruzov, A.; Whipple, A.V.; Rajora, O.P. Population Epigenomics: Advancing Understanding of Phenotypic Plasticity, Acclimation, Adaptation and Diseases. In Population Genomics; Rajora, O.P., Ed.; Population Genomics; Springer International Publishing: Cham, Switzerland, 2018; pp. 179–260. ISBN 978-3-030-04587-6. [Google Scholar]
- Jones, P.A. Functions of DNA Methylation: Islands, Start Sites, Gene Bodies and Beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Bandoy, D.J.D.; Weimer, B.C. Bacterial Epigenomics: Epigenetics in the Age of Population Genomics. In The Pangenome: Diversity, Dynamics and Evolution of Genomes; Tettelin, H., Medini, D., Eds.; Springer: Cham, Switzerland, 2020; ISBN 978-3-030-38280-3. [Google Scholar]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct Detection of DNA Methylation during Single-Molecule, Real-Time Sequencing. Nat. Methods 2010, 7, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Fang, G.; Munera, D.; Friedman, D.I.; Mandlik, A.; Chao, M.C.; Banerjee, O.; Feng, Z.; Losic, B.; Mahajan, M.C.; Jabado, O.J.; et al. Genome-Wide Mapping of Methylated Adenine Residues in Pathogenic Escherichia Coli Using Single-Molecule Real-Time Sequencing. Nat. Biotechnol. 2012, 30, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.; Wu, H.-C.; Jayasinghe, L.; Patel, A.; Reid, S.; Bayley, H. Continuous Base Identification for Single-Molecule Nanopore DNA Sequencing. Nat. Nanotechnol. 2009, 4, 265–270. [Google Scholar] [CrossRef]
- Simpson, J.T.; Workman, R.E.; Zuzarte, P.C.; David, M.; Dursi, L.J.; Timp, W. Detecting DNA Cytosine Methylation Using Nanopore Sequencing. Nat. Methods 2017, 14, 407–410. [Google Scholar] [CrossRef]
- Gouil, Q.; Keniry, A. Latest Techniques to Study DNA Methylation. Essays Biochem. 2019, 63, 639–648. [Google Scholar] [CrossRef]
- Sánchez-Romero, M.A.; Casadesús, J. The Bacterial Epigenome. Nat. Rev. Microbiol. 2020, 18, 7–20. [Google Scholar] [CrossRef]
- Rand, A.C.; Jain, M.; Eizenga, J.M.; Musselman-Brown, A.; Olsen, H.E.; Akeson, M.; Paten, B. Mapping DNA Methylation with High-Throughput Nanopore Sequencing. Nat. Methods 2017, 14, 411–413. [Google Scholar] [CrossRef]
- Sánchez-Romero, M.A.; Casadesús, J. Waddington’s Landscapes in the Bacterial World. Front. Microbiol. 2021, 12, 685080. [Google Scholar] [CrossRef]
- diCenzo, G.C.; Cangioli, L.; Nicoud, Q.; Cheng, J.H.T.; Blow, M.J.; Shapiro, N.; Woyke, T.; Biondi, E.G.; Alunni, B.; Mengoni, A.; et al. DNA Methylation in Ensifer Species during Free-Living Growth and during Nitrogen-Fixing Symbiosis with Medicago Spp. mSystems 2022. [CrossRef]
- Müller, F.; Scherer, M.; Assenov, Y.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. RnBeads 2.0: Comprehensive Analysis of DNA Methylation Data. Genome Biol. 2019, 20, 55. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.-S.; Wu, B.-H.; Yen, M.-R.; Chen, P.-Y. MethGET: Web-Based Bioinformatics Software for Correlating Genome-Wide DNA Methylation and Gene Expression. BMC Genom. 2020, 21, 375. [Google Scholar] [CrossRef] [PubMed]
- Hillary, R.F.; Marioni, R.E. MethylDetectR: A Software for Methylation-Based Health Profiling 2021. Available online: https://wellcomeopenresearch.org/articles/5-283/v2 (accessed on 14 September 2022).
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A Flexible and Comprehensive Bioconductor Package for the Analysis of Infinium DNA Methylation Microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Reither, S.; Mikeska, T.; Paulsen, M.; Walter, J.; Lengauer, T. BiQ Analyzer: Visualization and Quality Control for DNA Methylation Data from Bisulfite Sequencing. Bioinformatics 2005, 21, 4067–4068. [Google Scholar] [CrossRef]
- Nunn, A.; Otto, C.; Stadler, P.F.; Langenberger, D. Comprehensive Benchmarking of Software for Mapping Whole Genome Bisulfite Data: From Read Alignment to DNA Methylation Analysis. Brief. Bioinform. 2021, 22, bbab021. [Google Scholar] [CrossRef]
- Su, S.; Gouil, Q.; Blewitt, M.E.; Cook, D.; Hickey, P.F.; Ritchie, M.E. NanoMethViz: An R/Bioconductor Package for Visualizing Long-Read Methylation Data. PLOS Comput. Biol. 2021, 17, e1009524. [Google Scholar] [CrossRef]
- Leger, A. A-Slide/PycoMeth. 2020. Available online: https://zenodo.org/record/4110144#.Y6LUYxVBxPY (accessed on 3 August 2022). [CrossRef]
- De Coster, W.; Stovner, E.B.; Strazisar, M. Methplotlib: Analysis of Modified Nucleotides from Nanopore Sequencing. Bioinformatics 2020, 36, 3236–3238. [Google Scholar] [CrossRef]
- Geddes, B.A.; Oresnik, I.J. Physiology, Genetics, and Biochemistry of Carbon Metabolism in the Alphaproteobacterium Sinorhizobium Meliloti. Can. J. Microbiol. 2014, 60, 491–507. [Google Scholar] [CrossRef]
- Fioravanti, A.; Fumeaux, C.; Mohapatra, S.S.; Bompard, C.; Brilli, M.; Frandi, A.; Castric, V.; Villeret, V.; Viollier, P.H.; Biondi, E.G. DNA Binding of the Cell Cycle Transcriptional Regulator GcrA Depends on N6-Adenosine Methylation in Caulobacter Crescentus and Other Alphaproteobacteria. PLoS Genet. 2013, 9, e1003541. [Google Scholar] [CrossRef]
- Beringer, J.E. R Factor Transfer in Rhizobium Leguminosarum. Microbiology 1974, 84, 188–198. [Google Scholar] [CrossRef]
- Bianco, C.; Andreozzi, A.; Romano, S.; Fagorzi, C.; Cangioli, L.; Prieto, P.; Cisse, F.; Niangado, O.; Sidibé, A.; Pianezze, S.; et al. Endophytes from African Rice (Oryza glaberrima L.) Efficiently Colonize Asian Rice (Oryza sativa L.) Stimulating the Activity of Its Antioxidant Enzymes and Increasing the Content of Nitrogen, Carbon, and Chlorophyll. Microorganisms 2021, 9, 1714. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Mouammine, A.; Collier, J. The Impact of DNA Methylation in Alphaproteobacteria. Mol. Microbiol. 2018, 110, 1–10. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tool | Programming Language | Motif Recognition | Motif Matching with Respect to Genomic Features | Graphical Outputs | Reference |
|---|---|---|---|---|---|
| MeStudio | Python, C | Yes | Yes | Yes | This study |
| GenomicRanges | R, C | No | No | Yes | Bioconductor package |
| motifmatchr | R, C++ | Yes | Yes (only providing genomic ranges) | Yes | Bioconductor package |
| Meta-epigenomics | Python | Yes | No | No | https://github.com/hoonjeseoho/Meta-epigenomics (accessed on 19 June 2022) |
| Methplotlib | Python, Bash | No | No | Yes | De Coster et al. (2020) [21] |
| a-slide/pycoMeth | Python, Bash | No | No | Yes | Leger (2020) [20] |
| NanoMethViz | Python, Bash | No | No | Yes | Su et al. (2021) [19] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riccardi, C.; Passeri, I.; Cangioli, L.; Fagorzi, C.; Fondi, M.; Mengoni, A. Crossing Bacterial Genomic Features and Methylation Patterns with MeStudio: An Epigenomic Analysis Tool. Int. J. Mol. Sci. 2023, 24, 159. https://doi.org/10.3390/ijms24010159
Riccardi C, Passeri I, Cangioli L, Fagorzi C, Fondi M, Mengoni A. Crossing Bacterial Genomic Features and Methylation Patterns with MeStudio: An Epigenomic Analysis Tool. International Journal of Molecular Sciences. 2023; 24(1):159. https://doi.org/10.3390/ijms24010159
Chicago/Turabian StyleRiccardi, Christopher, Iacopo Passeri, Lisa Cangioli, Camilla Fagorzi, Marco Fondi, and Alessio Mengoni. 2023. "Crossing Bacterial Genomic Features and Methylation Patterns with MeStudio: An Epigenomic Analysis Tool" International Journal of Molecular Sciences 24, no. 1: 159. https://doi.org/10.3390/ijms24010159
APA StyleRiccardi, C., Passeri, I., Cangioli, L., Fagorzi, C., Fondi, M., & Mengoni, A. (2023). Crossing Bacterial Genomic Features and Methylation Patterns with MeStudio: An Epigenomic Analysis Tool. International Journal of Molecular Sciences, 24(1), 159. https://doi.org/10.3390/ijms24010159