
A 3D, Compartmental Tumor-Stromal Microenvironment Model of Patient-Derived Bone Metastasis

 , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results and Discussion
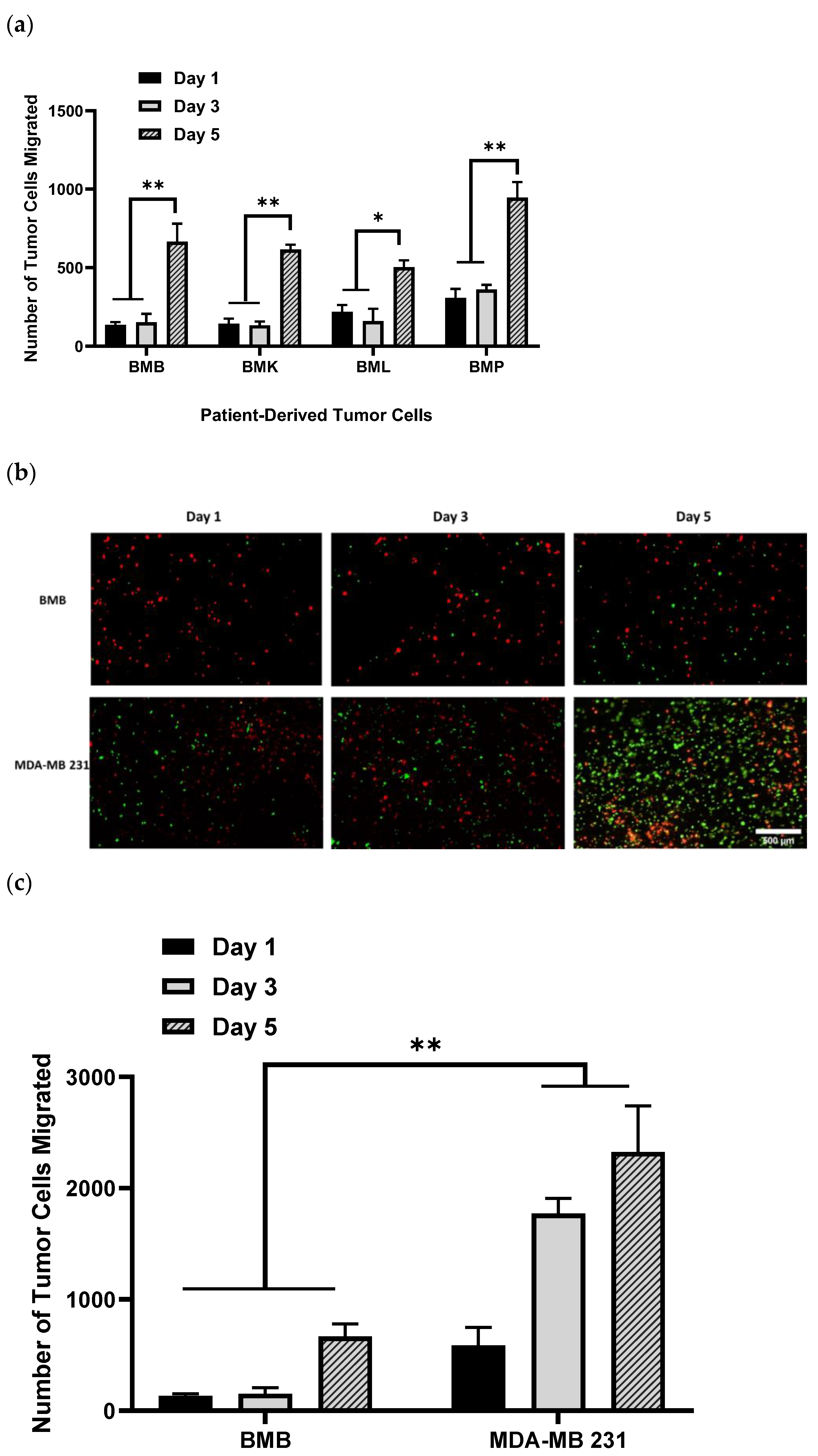
2.1. Invasion Behavior of Patient-Derived Spine-Metastasized Cells in the PP-3D-S Model
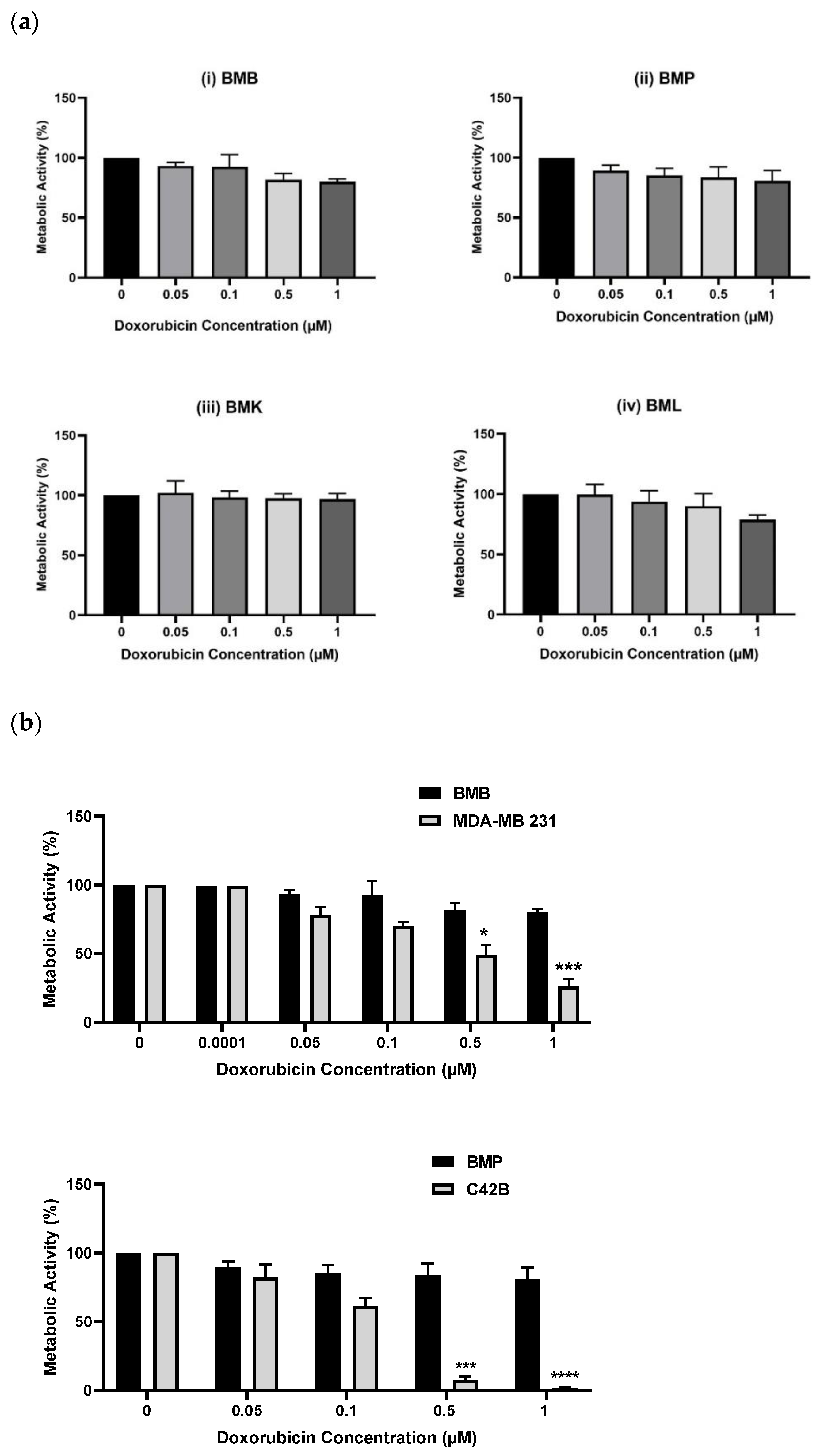
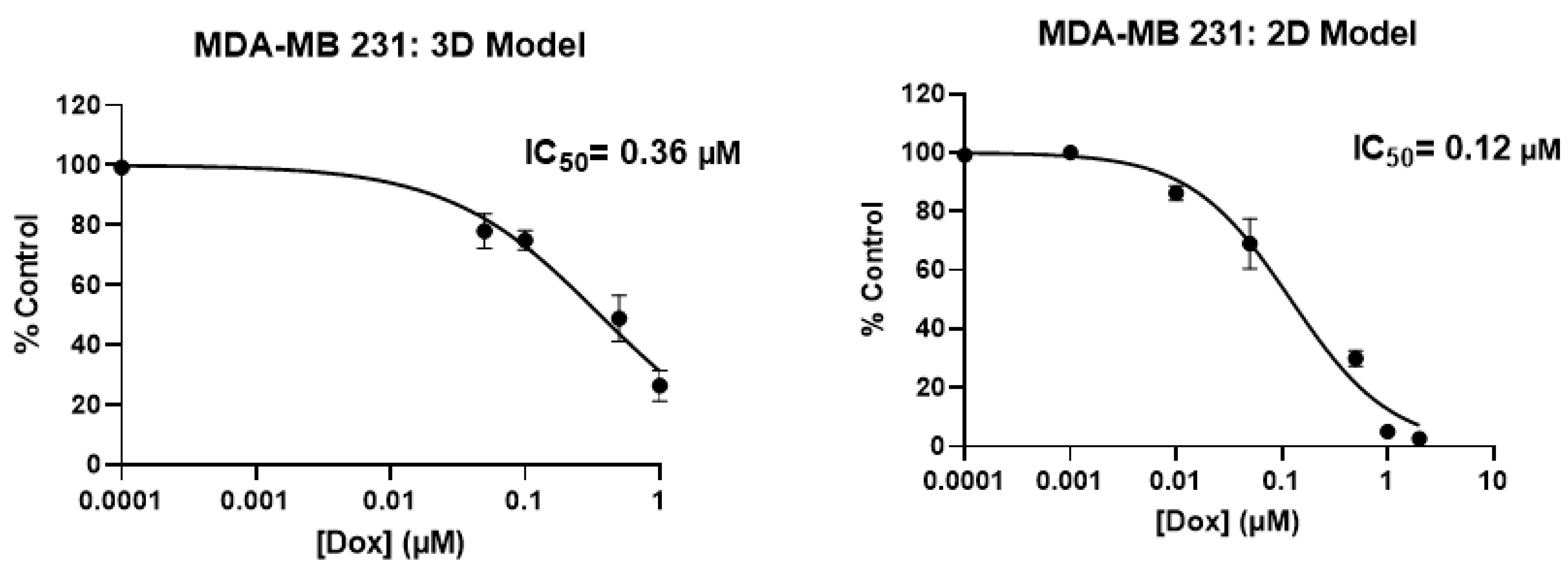
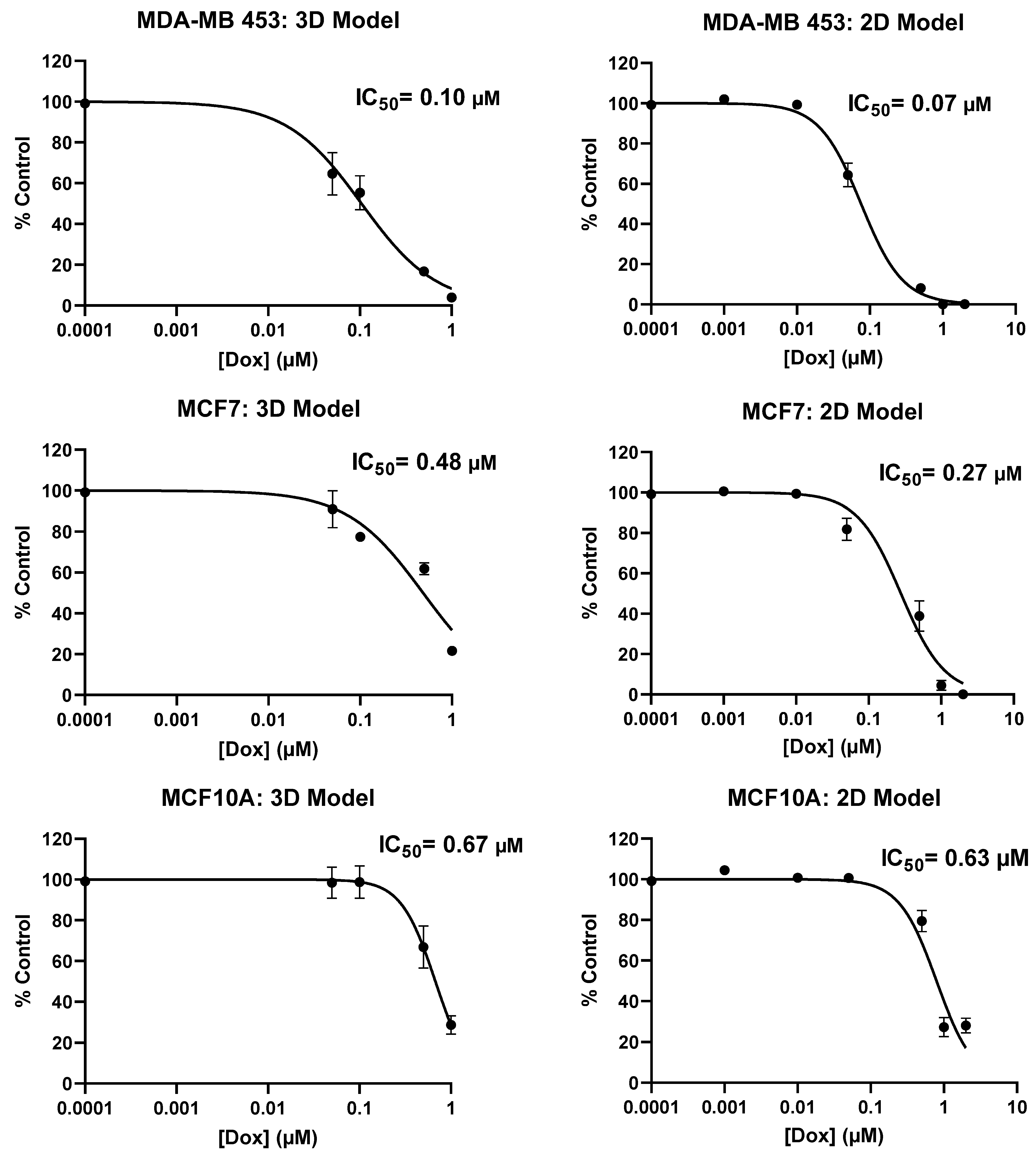
2.2. Screening of Patient-Derived Bone-Metastasized Cells with Doxorubicin
2.3. Treatment of Bone-Metastasized Secondary to Breast Tumor Cells with Different Chemotherapy Drugs
2.4. Gene Expression Profile of Breast Cancer Cell Lines in the PP-3D-S Model
3. Materials and Methods
3.1. Fabrication of Plasma-Treated Electrospun Nanofibrous Scaffold
3.1.1. Electrospinning of 3D Electrospun Mat
3.1.2. Surface Treatment of 3D Scaffolds by Plasma Functionalization
3.2. Biological Experiments
3.2.1. Cell Culture and Seeding in Hybrid 3D Scaffolds
3.2.2. Observation and Quantification of Tumor Cell Migration
3.2.3. Drug Screening Experiment
3.2.4. Metabolic Activity Measurement
3.2.5. Gene Expression Analysis: Real-Time qPCR
3.3. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
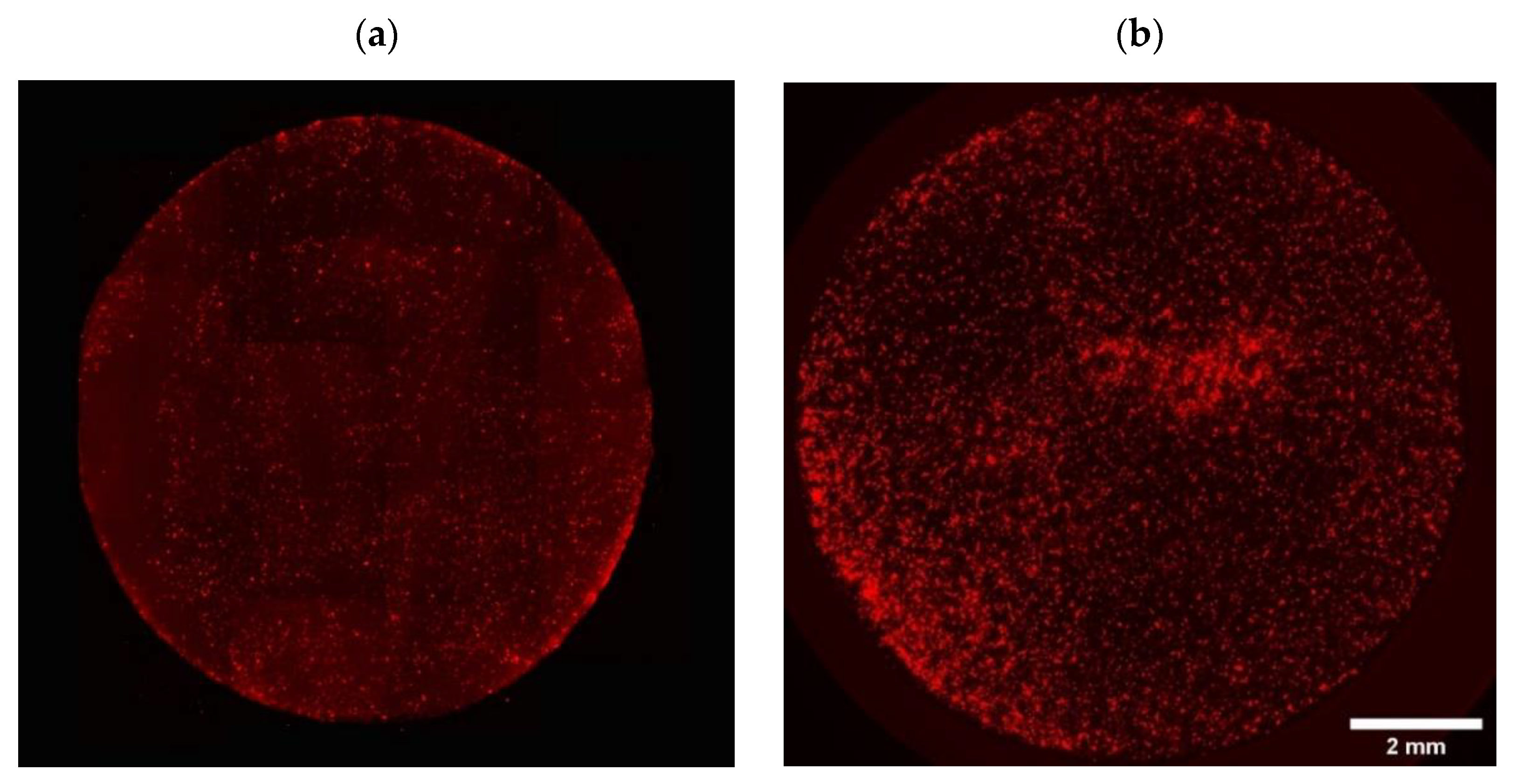
- Stromal cell distribution on and inside the mats:
- By florescent microscopy on the top surface of the mats:

- 2.
- By confocal imaging through the depth of mats at day 0 (penetration depth: max 60 μm after 30 min incubation with cells) and at day 7 (penetration depth: max 120 μm):


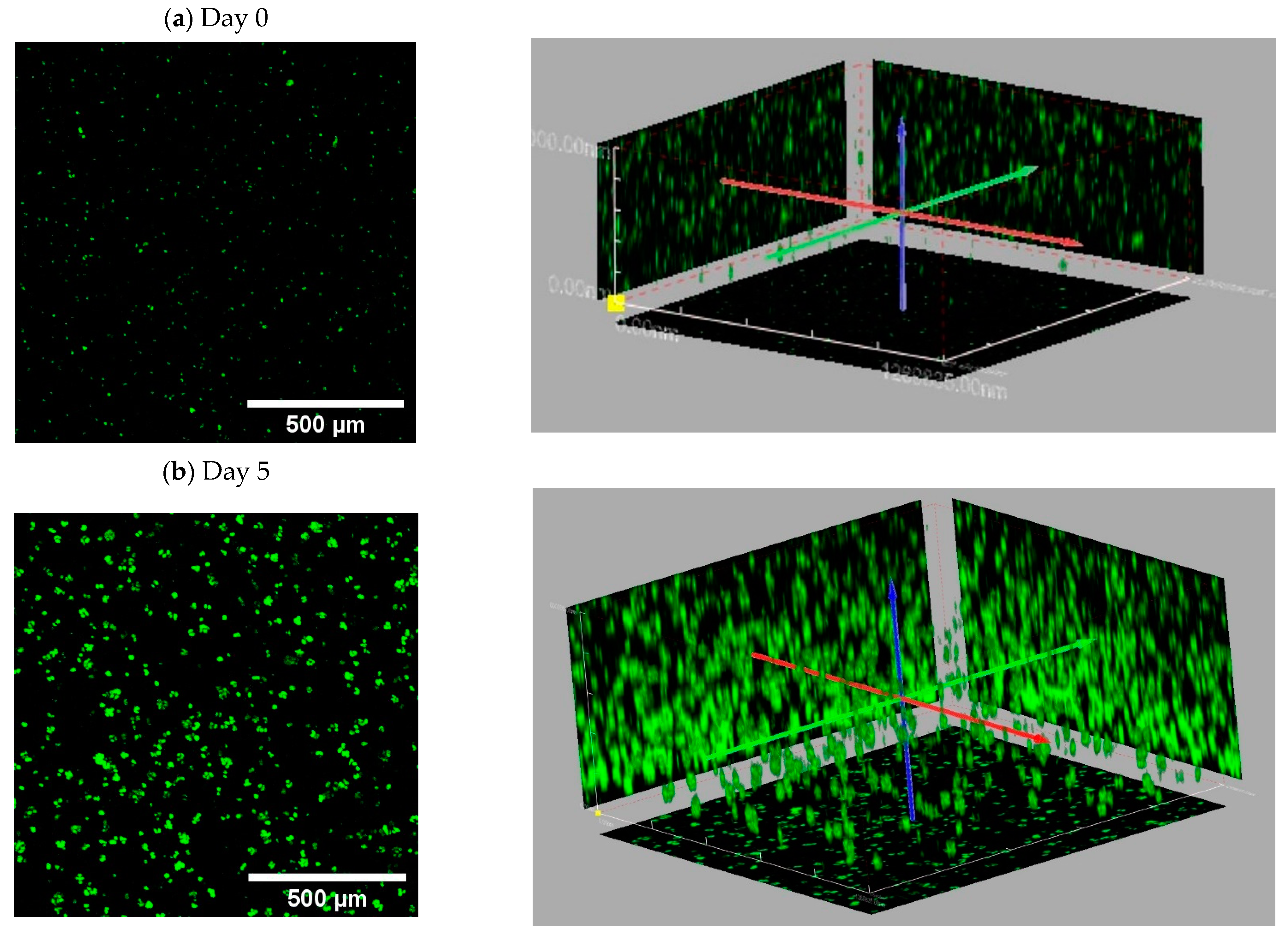
- Confocal images showing tumor cell distribution inside A1G7 hydrogel over 5 days:

- Evaluation of cell viability on plasma-treated mesh and inside hydrogel



- Doxorubicin screening tests in PP-3D-S versus 2D model:


- Imaging tumor cells migrated onto the mats with and without Doxorubicin treatment:

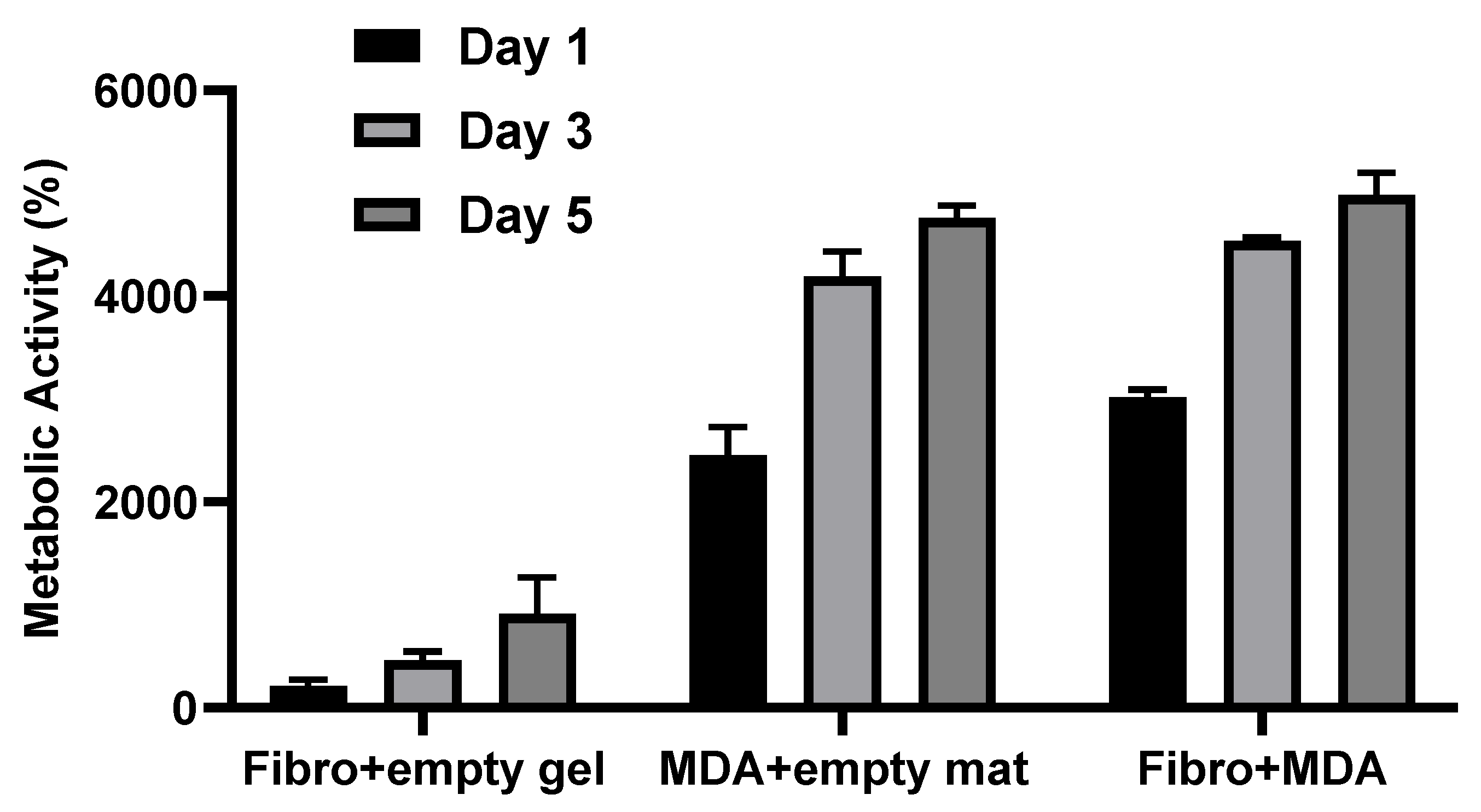
- Actual metabolic activity of tumor cells compared with stromal cells cultured in PP-3D-S model over the course of 5 days:

References
- Coleman, R. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar] [CrossRef]
- Coleman, R.E. Skeletal complications of malignancy. Cancer 1997, 80, 1588–1594. [Google Scholar] [CrossRef]
- Ahangar, P.; Aziz, M.; Rosenzweig, D.H.; Weber, M.H. Advances in personalized treatment of metastatic spine disease. Ann. Transl. Med. 2019, 7, 223. [Google Scholar] [CrossRef] [PubMed]
- Akoury, E.; Ahangar, P.; Nour, A.; Lapointe, J.; Guérard, K.-P.; Haglund, L.; Rosenzweig, D.H.; Weber, M.H. Low-dose zoledronate for the treatment of bone metastasis secondary to prostate cancer. Cancer Cell Int. 2019, 19, 28. [Google Scholar] [CrossRef]
- Akoury, E.; Ahangar, P.; Luna, A.S.R.; Nour, A.; Weber, M.; Rosenzweig, D. 3D-printed scaffolds loaded with therapeutics for the treatment of bone metastases. In Orthopaedic Proceedings; no. SUPP_7; The British Editorial Society of Bone & Joint Surgery: London, UK, 2020; Volume 102, p. 101. [Google Scholar]
- Akoury, E.; Luna, A.S.R.G.; Ahangar, P.; Gao, X.; Zolotarov, P.; Weber, M.H.; Rosenzweig, D.H. Anti-tumor effects of low dose zoledronate on lung cancer-induced spine metastasis. J. Clin. Med. 2019, 8, 1212. [Google Scholar] [CrossRef]
- Akoury, E.; Weber, M.H.; Rosenzweig, D.H. 3D-Printed nanoporous scaffolds impregnated with zoledronate for the treatment of spinal bone metastases. MRS Adv. 2019, 4, 1245–1251. [Google Scholar] [CrossRef]
- C. S. C. American Cancer Society. Available online: http://cancerstatisticscenter.cancer.org (accessed on 2 November 2022).
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Gonçalves, F. Bone metastases: An overview. Oncol. Rev. 2017, 11, 43. [Google Scholar]
- Liu, D.; Kuai, Y.; Zhu, R.; Zhou, C.; Tao, Y.; Han, W.; Chen, Q. Prognosis of prostate cancer and bone metastasis pattern of patients: A SEER-based study and a local hospital-based study from China. Sci. Rep. 2020, 10, 9104. [Google Scholar] [CrossRef]
- Krzyszczyk, P.; Acevedo, A.; Davidoff, E.J.; Timmins, L.M.; Marrero-Berrios, I.; Patel, M.; White, C.; Lowe, C.; Sherba, J.J.; Hartmanshenn, C.; et al. The growing role of precision and personalized medicine for cancer treatment. Technology 2018, 6, 79–100. [Google Scholar] [CrossRef]
- Vogenberg, F.R.; Barash, C.I.; Pursel, M. Personalized medicine: Part 1: Evolution and development into theranostics. Pharm. Ther. 2010, 35, 560. [Google Scholar]
- Jain, K.K. Personalized medicine. Curr. Opin. Mol. Ther. 2002, 4, 548–558. [Google Scholar] [PubMed]
- Gambardella, V.; Tarazona, N.; Cejalvo, J.M.; Lombardi, P.; Huerta, M.; Roselló, S.; Fleitas, T.; Roda, D.; Cervantes, A. Personalized medicine: Recent progress in cancer therapy. Cancers 2020, 12, 1009. [Google Scholar] [CrossRef]
- Langhans, S.A. Three-dimensional in vitro cell culture models in drug discovery and drug repositioning. Front. Pharmacol. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- van Norman, G.A. Limitations of animal studies for predicting toxicity in clinical trials: Is it time to rethink our current approach? JACC Basic Transl. Sci. 2019, 4, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, J.; Zhu, W.; Tang, M.; Lawrence, N.; Yu, C.; Gou, M.; Chen, S. 3D bioprinting of functional tissue models for personalized drug screening and in vitro disease modeling. Adv. Drug Deliv. Rev. 2018, 132, 235–251. [Google Scholar] [CrossRef]
- Maloney, E.; Clark, C.; Sivakumar, H.; Yoo, K.; Aleman, J.; Rajan, S.A.P.; Forsythe, S.; Mazzocchi, A.; Laxton, A.W.; Tatter, S.B.; et al. Immersion bioprinting of tumor organoids in multi-well plates for increasing chemotherapy screening throughput. Micromachines 2020, 11, 208. [Google Scholar] [CrossRef]
- Mao, S.; Pang, Y.; Liu, T.; Shao, Y.; He, J.; Yang, H.; Mao, Y.; Sun, W. Bioprinting of in vitro tumor models for personalized cancer treatment: A review. Biofabrication 2020, 12, 042001. [Google Scholar] [CrossRef]
- Nie, J.; Gao, Q.; Fu, J.; He, Y. Grafting of 3D bioprinting to in vitro drug screening: A review. Adv. Healthc. Mater. 2020, 9, 1901773. [Google Scholar] [CrossRef]
- Takasato, M.; Er, P.X.; Chiu, H.S.; Maier, B.; Baillie, G.J.; Ferguson, C.; Parton, R.G.; Wolvetang, E.J.; Roost, M.S.; Chuva de Sousa Lopes, S.M.; et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 2015, 526, 564–568. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Wiegerinck, C.L.; De Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; De Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.; Bijvelds, M.J.; Scholte, B.J.; et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef]
- Gracz, A.; Williamson, I.; Roche, K.C.; Johnston, M.J.; Wang, F.; Wang, Y.; Attayek, P.; Balowski, J.; Liu, X.F.; Laurenza, R.J.; et al. A high-throughput platform for stem cell niche co-cultures and downstream gene expression analysis. Nat. Cell Biol. 2015, 17, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Farach-Carson, M.C.; Jia, X. Three-dimensional in vitro tumor models for cancer research and drug evaluation. Biotechnol. Adv. 2014, 32, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.; Walter, S.; Walzl, A.; Kramer, N.; Unger, C.; Scherzer, M.; Unterleuthner, D.; Hengstschläger, M.; Krupitza, G.; Dolznig, H. Increased complexity in carcinomas: Analyzing and modeling the interaction of human cancer cells with their microenvironment. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 35, pp. 107–124. [Google Scholar]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Weeber, F.; Ooft, S.N.; Dijkstra, K.K.; Voest, E.E. Tumor organoids as a pre-clinical cancer model for drug discovery. Cell Chem. Biol. 2017, 24, 1092–1100. [Google Scholar] [CrossRef]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Shang, M.; Soon, R.H.; Lim, C.T.; Khoo, B.L.; Han, J. Microfluidic modelling of the tumor microenvironment for anti-cancer drug development. Lab Chip 2019, 19, 369–386. [Google Scholar] [CrossRef]
- Alepee, N.; Bahinski, A.; Daneshian, M.; De Wever, B.; Fritsche, E.; Goldberg, A.; Hansmann, J.; Hartung, T.; Haycock, J.; Hogberg, H.T.; et al. t4 workshop report: State-of-the-art of 3D cultures (organs-on-a-chip) in safety testing and pathophysiology. Altex 2014, 31, 441. [Google Scholar] [CrossRef]
- Pamies, D.; Hartung, T.; Hogberg, H.T. Biological and medical applications of a brain-on-a-chip. Exp. Biol. Med. 2014, 239, 1096–1107. [Google Scholar] [CrossRef]
- Abaci, H.; Guo, Z.; Doucet, Y.; Jacków, J.; Christiano, A. Next generation human skin constructs as advanced tools for drug development. Exp. Biol. Med. 2017, 242, 1657–1668. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, W.; Kuss, M.A.; Mirza, S.; Qi, D.; Krasnoslobodtsev, A.; Zeng, J.; Band, H.; Band, V.; Duan, B. 3D bioprinting of breast cancer models for drug resistance study. ACS Biomater. Sci. Eng. 2018, 4, 4401–4411. [Google Scholar] [CrossRef]
- Grolman, J.M.; Zhang, D.; Smith, A.M.; Moore, J.S.; Kilian, K.A. Rapid 3D extrusion of synthetic tumor microenvironments. Adv. Mater. 2015, 27, 5512–5517. [Google Scholar] [CrossRef] [PubMed]
- Bischel, L.L.; Beebe, D.J.; Sung, K.E. Microfluidic model of ductal carcinoma in situ with 3D, organotypic structure. BMC Cancer 2015, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Toh, Y.-C.; Raja, A.; Yu, H.; van Noort, D. A 3D microfluidic model to recapitulate cancer cell migration and invasion. Bioengineering 2018, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Hyun, E.; Seo, J.; Blundell, C.; Kim, H.C.; Lee, E.; Lee, S.H.; Moon, A.; Moon, W.K.; Huh, D. A microengineered pathophysiological model of early-stage breast cancer. Lab Chip 2015, 15, 3350–3357. [Google Scholar] [CrossRef] [PubMed]
- Gioiella, F.; Urciuolo, F.; Imparato, G.; Brancato, V.; Netti, P.A. An Engineered Breast Cancer Model on a Chip to Replicate ECM-Activation in vitro during Tumor Progression. Adv. Healthc. Mater. 2016, 5, 3074–3084. [Google Scholar] [CrossRef]
- Bersini, S.; Jeon, J.S.; Dubini, G.; Arrigoni, C.; Chung, S.; Charest, J.L.; Moretti, M.; Kamm, R.D. A microfluidic 3D in vitro model for specificity of breast cancer metastasis to bone. Biomaterials 2014, 35, 2454–2461. [Google Scholar] [CrossRef]
- Jeon, J.S.; Bersini, S.; Gilardi, M.; Dubini, G.; Charest, J.L.; Moretti, M.; Kamm, R.D. Human 3D vascularized organotypic microfluidic assays to study breast cancer cell extravasation. Proc. Natl. Acad. Sci. USA 2015, 112, 214–219. [Google Scholar] [CrossRef]
- van Marion, D.M.; Domanska, U.M.; Timmer-Bosscha, H.; Walenkamp, A.M. Studying cancer metastasis: Existing models, challenges and future perspectives. Crit. Rev. Oncol./Hematol. 2016, 97, 107–117. [Google Scholar] [CrossRef]
- Bahcecioglu, G.; Basara, G.; Ellis, B.W.; Ren, X.; Zorlutuna, P. Breast cancer models: Engineering the tumor microenvironment. Acta Biomater. 2020, 106, 1–21. [Google Scholar] [CrossRef]
- Pape, J.; Emberton, M.; Cheema, U. 3D Cancer Models: The Need for a Complex Stroma, Compartmentalization and Stiffness. Front. Bioeng. Biotechnol. 2021, 9, 276. [Google Scholar] [CrossRef]
- Datta, P.; Dey, M.; Ataie, Z.; Unutmaz, D.; Ozbolat, I.T. 3D bioprinting for reconstituting the cancer microenvironment. NPJ Precis. Oncol. 2020, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Garakani, M.M.; Ahangar, P.; Watson, S.; Nisol, B.; Wertheimer, M.R.; Rosenzweig, D.H.; Ajji, A. A novel 3D co-culture platform for integrating tissue interfaces for tumor growth, migration and therapeutic sensitivity: “PP-3D-S”. Mater. Sci. Eng. C 2021, 134, 112566. [Google Scholar]
- Garakani, M.M.; Cooke, M.E.; Wertheimer, M.R.; Rosenzweig, D.H.; Ajji, A. A novel 3D in vitro tissue model for bone-metastasized breast cancer: A preclinical tool in drug discovery and testing. Plasma Process. Polym. 2022, 19, 2100206. [Google Scholar] [CrossRef]
- Chang, H.-C.; Lin, C.-H.; Juang, D.S.; Wu, H.-W.; Lee, C.-Y.; Chen, C.; Hsu, C.-H. Multilayer architecture microfluidic network array for combinatorial drug testing on 3D-cultured cells. Biofabrication 2019, 11, 035024. [Google Scholar] [CrossRef] [PubMed]
- Eduati, F.; Utharala, R.; Madhavan, D.; Neumann, U.P.; Longerich, T.; Cramer, T.; Saez-Rodriguez, J.; Merten, C.A. A microfluidics platform for combinatorial drug screening on cancer biopsies. Nat. Commun. 2018, 9, 2434. [Google Scholar] [CrossRef]
- Dhiman, N.; Kingshott, P.; Sumer, H.; Sharma, C.S.; Rath, S.N. On-chip anticancer drug screening–recent progress in microfluidic platforms to address challenges in chemotherapy. Biosens. Bioelectron. 2019, 137, 236–254. [Google Scholar] [CrossRef]
- Niepel, M.; Hafner, M.; Duan, Q.; Wang, Z.; Paull, E.O.; Chung, M.; Lu, X.; Stuart, J.M.; Golub, T.R.; Subramanian, A.; et al. Common and cell-type specific responses to anti-cancer drugs revealed by high throughput transcript profiling. Nat. Commun. 2017, 8, 1186. [Google Scholar] [CrossRef]
- Ramani, V.C.; Vlodavsky, I.; Ng, M.; Zhang, Y.; Barbieri, P.; Noseda, A.; Sanderson, R.D. Chemotherapy induces expression and release of heparanase leading to changes associated with an aggressive tumor phenotype. Matrix Biol. 2016, 55, 22–34. [Google Scholar] [CrossRef]
- Sun, L.; Yao, Y.; Liu, B.; Lin, Z.; Lin, L.; Yang, M.; Zhang, W.; Chen, W.; Pan, C.; Liu, Q.; et al. MiR-200b and miR-15b regulate chemotherapy-induced epithelial-mesenchymal transition in human tongue cancer cells by targeting BMI1. Oncogene 2012, 31, 432–445. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Condeelis, J.S.; Oktay, M.H. Chemotherapy-induced metastasis: Molecular mechanisms, clinical manifestations, therapeutic interventions. Cancer Res. 2019, 79, 4567–4576. [Google Scholar] [CrossRef]
- Lu, H.; Samanta, D.; Xiang, L.; Zhang, H.; Hu, H.; Chen, I.; Bullen, J.W.; Semenza, G.L. Chemotherapy triggers HIF-1–dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc. Natl. Acad. Sci. USA 2015, 112, E4600–E4609. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Chen, I.; Shimoda, L.A.; Park, Y.; Zhang, C.; Tran, L.; Zhang, H.; Semenza, G.L. Chemotherapy-induced Ca2+ release stimulates breast cancer stem cell enrichment. Cell Rep. 2017, 18, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Tonnessen-Murray, C.A.; Frey, W.D.; Rao, S.G.; Shahbandi, A.; Ungerleider, N.A.; Olayiwola, J.O.; Murray, L.B.; Vinson, B.T.; Chrisey, D.B.; Lord, C.J.; et al. Chemotherapy-induced senescent cancer cells engulf other cells to enhance their survival. J. Cell Biol. 2019, 218, 3827–3844. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-C.; Xu, X.; Lim, Y.-W.; Iau, P.; Sukri, N.; Lim, S.-E.; Yap, H.L.; Yeo, W.-L.; Tan, P.; Tan, S.-H.; et al. Chemotherapy-induced tumor gene expression changes in human breast cancers. Pharm. Genom. 2009, 19, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Q.; Chen, Z.Q.; Cao, X.X.; Xu, J.D.; Xu, J.W.; Chen, Y.Y.; Wang, W.J.; Chen, Q.; Tang, F.; Liu, X.P.; et al. Involvement of NF-κB/miR-448 regulatory feedback loop in chemotherapy-induced epithelial–mesenchymal transition of breast cancer cells. Cell Death Differ. 2011, 18, 16–25. [Google Scholar] [CrossRef]
- Ahangar, P.; Akoury, E.; Luna, A.S.R.G.; Nour, A.; Weber, M.H.; Rosenzweig, D.H. Nanoporous 3D-printed scaffolds for local doxorubicin delivery in bone metastases secondary to prostate cancer. Materials 2018, 11, 1485. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef]
- Nguyen, P.T.; Kudo, Y.; Yoshida, M.; Kamata, N.; Ogawa, I.; Takata, T. N-cadherin expression is involved in malignant behavior of head and neck cancer in relation to epithelial-mesenchymal transition. Histol. Histopathol. 2011, 26, 147. [Google Scholar]
- Ye, Z.; Zhou, M.; Tian, B.; Wu, B.; Li, J. Expression of lncRNA-CCAT1, E-cadherin and N-cadherin in colorectal cancer and its clinical significance. Int. J. Clin. Exp. Med. 2015, 8, 3707. [Google Scholar] [PubMed]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-cadherin and N-cadherin switch in epithelial-to-mesenchymal transition: Signaling, therapeutic implications, and challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Y.; Lin, H.-H.; Tang, M.-J.; Wang, Y.-K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Shen, W.; Peng, H.; Li, Y.; Chen, F.; Zheng, L.; Xu, J.; Jia, L. Fibronectin 1 promotes melanoma proliferation and metastasis by inhibiting apoptosis and regulating EMT. OncoTargets Ther. 2019, 12, 3207. [Google Scholar] [CrossRef]
- Yang, N.; Hui, L.; Wang, Y.; Yang, H.; Jiang, X. Overexpression of SOX2 promotes migration, invasion, and epithelial-mesenchymal transition through the Wnt/β-catenin pathway in laryngeal cancer Hep-2 cells. Tumor Biol. 2014, 35, 7965–7973. [Google Scholar] [CrossRef]
- Zhu, Q.-Q.; Ma, C.; Wang, Q.; Song, Y.; Lv, T. The role of TWIST1 in epithelial-mesenchymal transition and cancers. Tumor Biol. 2016, 37, 185–197. [Google Scholar] [CrossRef]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.; Weberpals, J.; et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012, 12, 91. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef]
- Chao, Y.L.; Shepard, C.R.; Wells, A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol. Cancer 2010, 9, 179. [Google Scholar] [CrossRef]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef]
- Savoji, H.; Hadjizadeh, A.; Maire, M.; Ajji, A.; Wertheimer, M.R.; Lerouge, S. Electrospun nanofiber scaffolds and plasma polymerization: A promising combination towards complete, stable endothelial lining for vascular grafts. Macromol. Biosci. 2014, 14, 1084–1095. [Google Scholar] [CrossRef] [PubMed]
- Savoji, H.; Lerouge, S.; Ajji, A.; Wertheimer, M.R. Plasma-etching for controlled modification of structural and mechanical properties of electrospun PET scaffolds. Plasma Process. Polym. 2015, 12, 314–327. [Google Scholar] [CrossRef]
- Savoji, H.; Maire, M.; Lequoy, P.; Liberelle, B.; De Crescenzo, G.; Ajji, A.; Wertheimer, M.R.; Lerouge, S. Combining electrospun fiber mats and bioactive coatings for vascular graft prostheses. Biomacromolecules 2017, 18, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Ganesh, K.; Massagué, J. Targeting metastatic cancer. Nat. Med. 2021, 27, 34–44. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev.™ Oncog. 2013, 18, 43. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Full Name | Forward Primer 5′ > 3′ | Reverse Primer 5′ > 3′ | Template Size (Base Pairs) |
|---|---|---|---|
| E-cadherin | CAAATCCAACAAAGACAAAGAAGGC | ACACAGCGTGAGAGAAGAGAGT | 1 ea @ 25 (FWD), 22 (REV) bases |
| N-cadherin | CATCATCATCCTGCTTATCCTTGT | GGTCTTCTTCTCCTCCACCTTCT | 1 ea @ 24 (FWD), 23 (REV) bases |
| TWIST1 | ATGGCAAGCTGCAGCTATG | AGTTATCCAGCTCCAGAGTC | 1 ea @ 19 (FWD), 20 (REV) bases |
| SLUG | AGCATTTCAACGCCTCCAA | ACACAGTGATGGGGCTGTAT | 1 ea @ 19 (FWD), 20 (REV) bases |
| SNAIL1 | GAAAGGCCTTCAACTGCAAA | TGACATCTGAGTGGGTCTGG | 1 ea @ 20 (FWD), 20 (REV) bases |
| SOX2 | CATCACCCACAGCAAATGAC | CAAAGCTCCTACCGTACCACT | 1 ea @ 20 (FWD), 21 (REV) bases |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohseni Garakani, M.; Cooke, M.E.; Weber, M.H.; Wertheimer, M.R.; Ajji, A.; Rosenzweig, D.H. A 3D, Compartmental Tumor-Stromal Microenvironment Model of Patient-Derived Bone Metastasis. Int. J. Mol. Sci. 2023, 24, 160. https://doi.org/10.3390/ijms24010160
Mohseni Garakani M, Cooke ME, Weber MH, Wertheimer MR, Ajji A, Rosenzweig DH. A 3D, Compartmental Tumor-Stromal Microenvironment Model of Patient-Derived Bone Metastasis. International Journal of Molecular Sciences. 2023; 24(1):160. https://doi.org/10.3390/ijms24010160
Chicago/Turabian StyleMohseni Garakani, Mansoureh, Megan E. Cooke, Michael H. Weber, Michael R. Wertheimer, Abdellah Ajji, and Derek H. Rosenzweig. 2023. "A 3D, Compartmental Tumor-Stromal Microenvironment Model of Patient-Derived Bone Metastasis" International Journal of Molecular Sciences 24, no. 1: 160. https://doi.org/10.3390/ijms24010160
APA StyleMohseni Garakani, M., Cooke, M. E., Weber, M. H., Wertheimer, M. R., Ajji, A., & Rosenzweig, D. H. (2023). A 3D, Compartmental Tumor-Stromal Microenvironment Model of Patient-Derived Bone Metastasis. International Journal of Molecular Sciences, 24(1), 160. https://doi.org/10.3390/ijms24010160