
Exploring the Effects of the Interaction of Carbon and MoS2 Catalyst on CO2 Hydrogenation to Methanol

Abstract
:1. Introduction
2. Experiment
2.1. Materials
2.2. Catalyst Preparation
2.2.1. Synthesis of MoS2
2.2.2. Synthesis of MoS2@C
2.2.3. Synthesis of MoS2/C
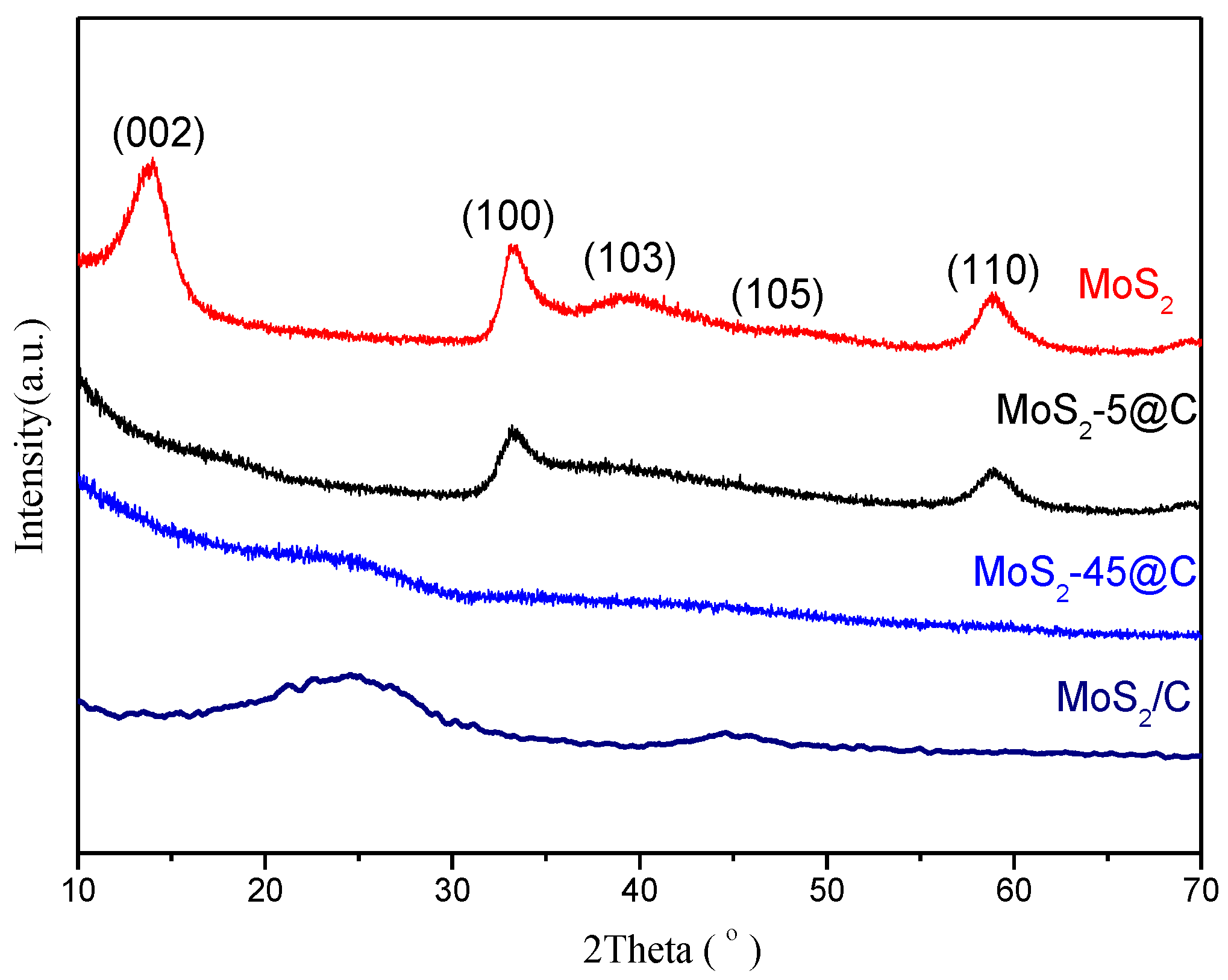
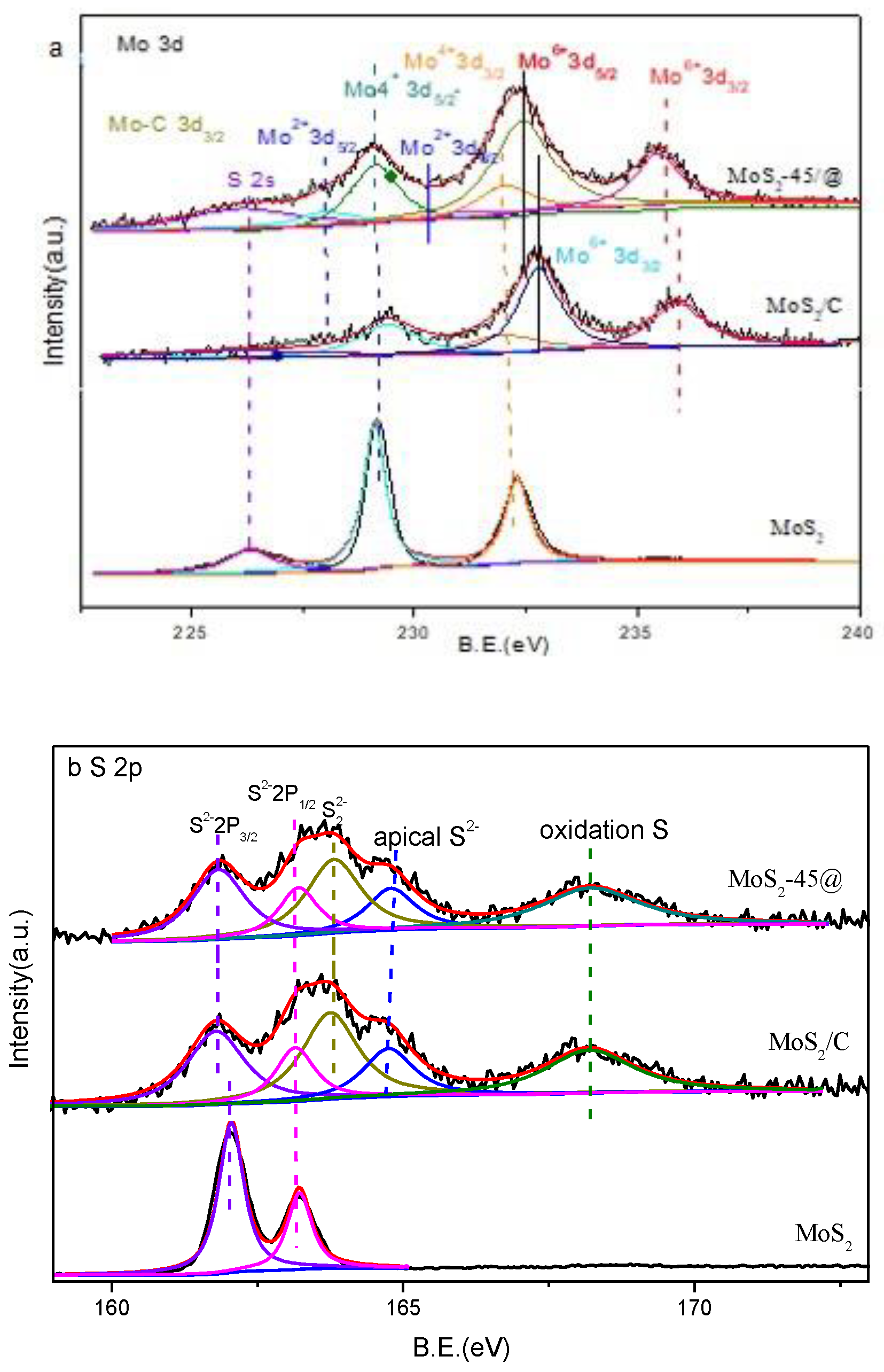
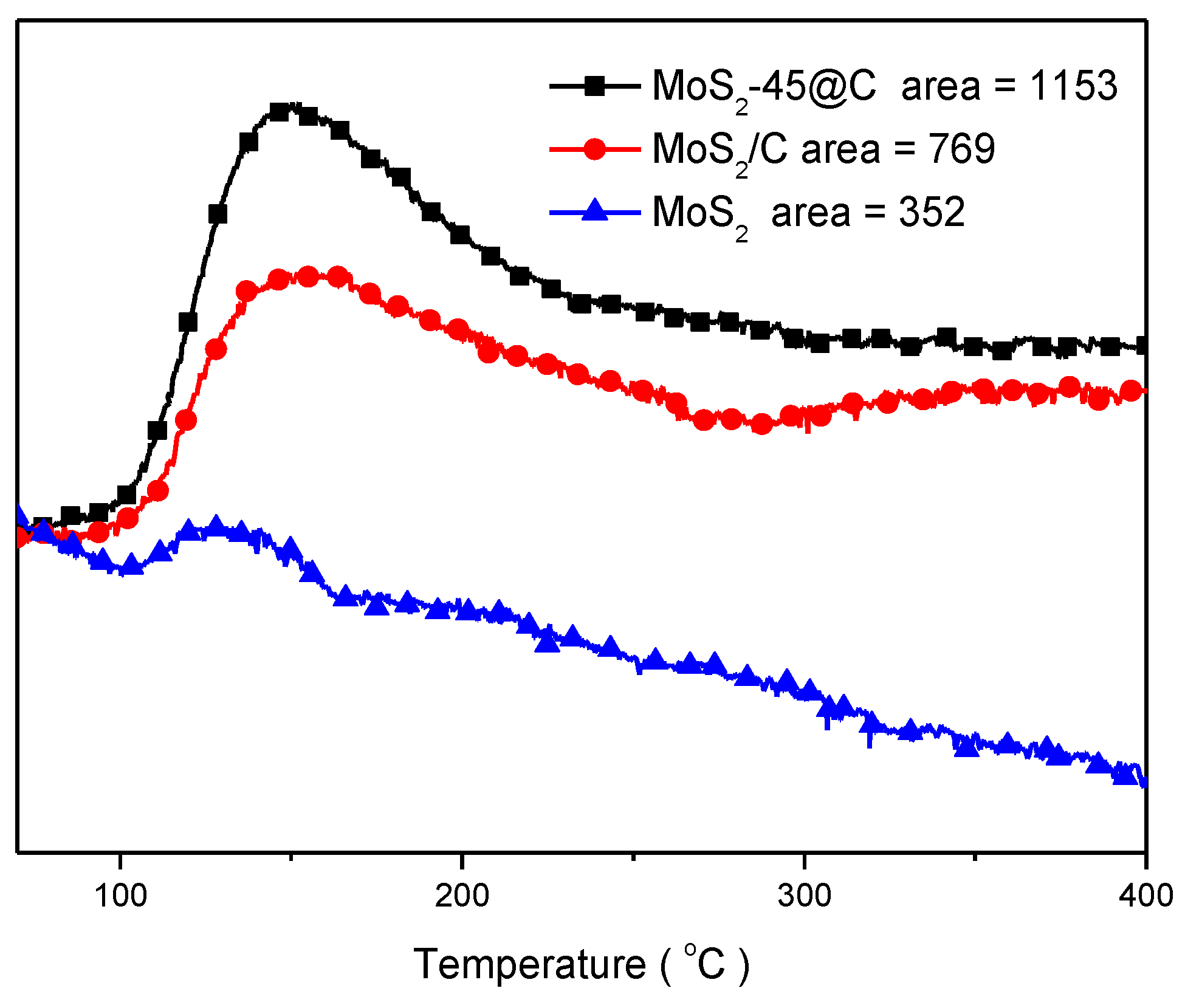
2.3. Catalyst Characterization
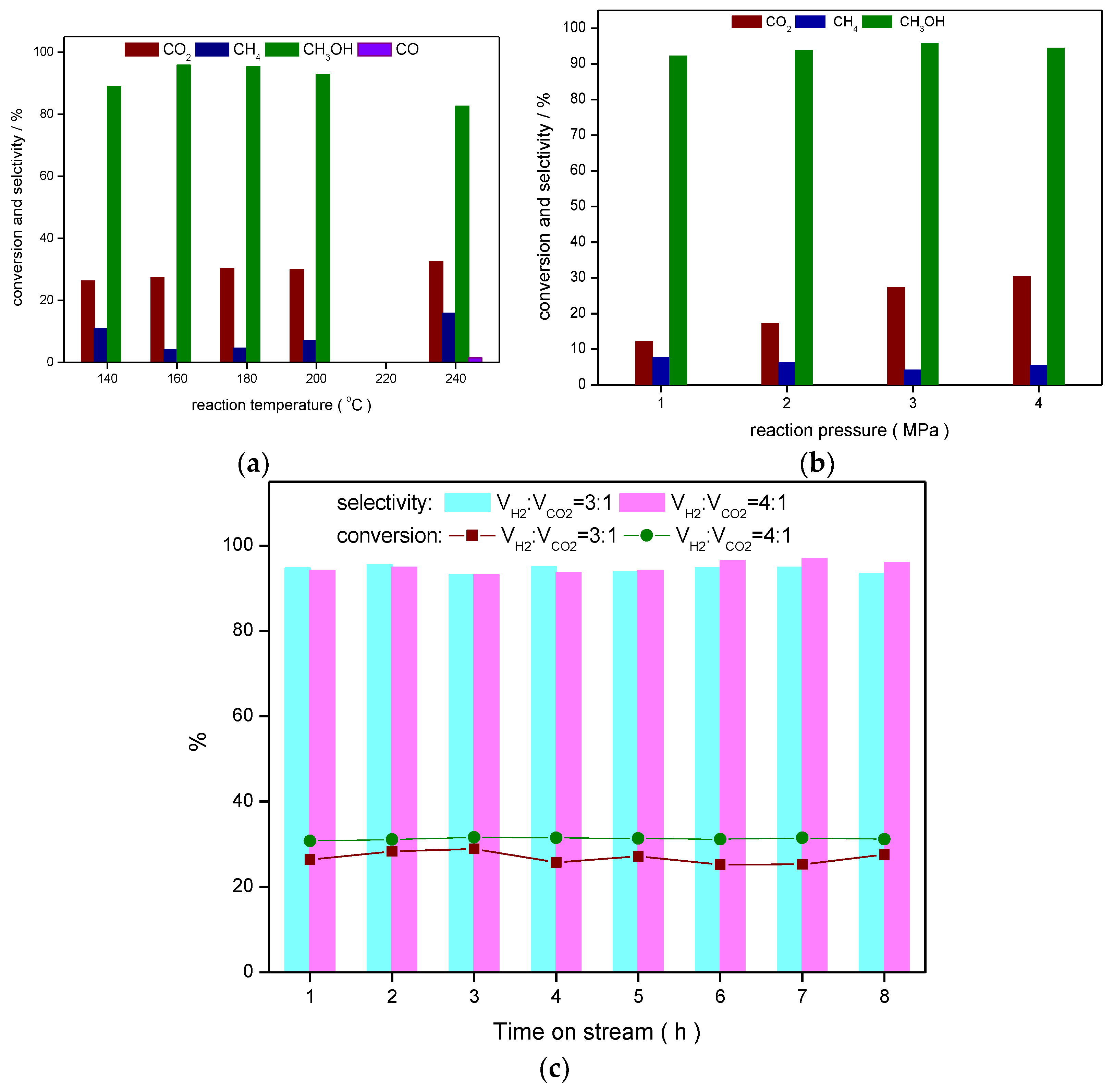
2.4. Catalytic Performance Test
2.5. Calculation of CO2 Conversion and Product Selectivity
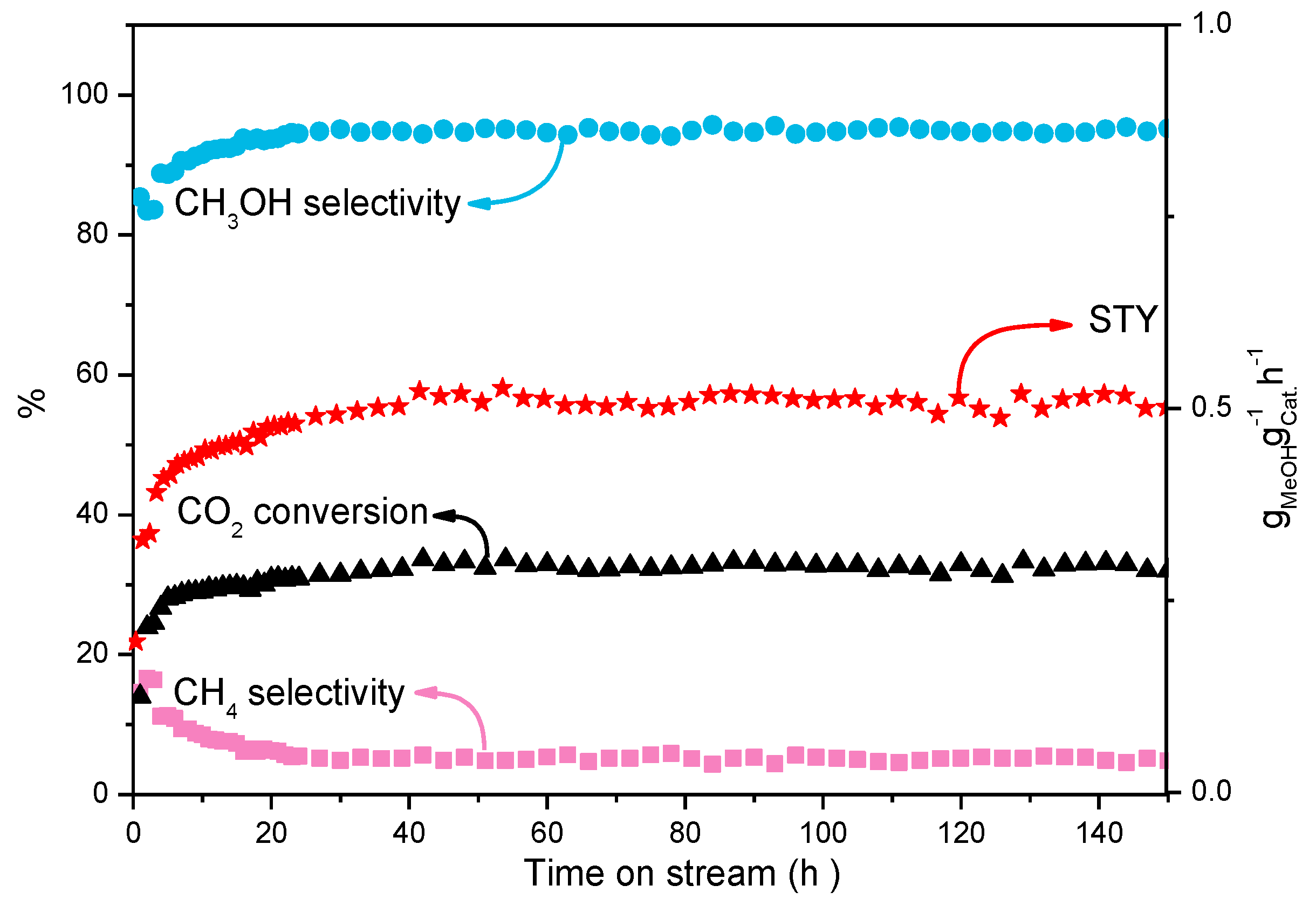
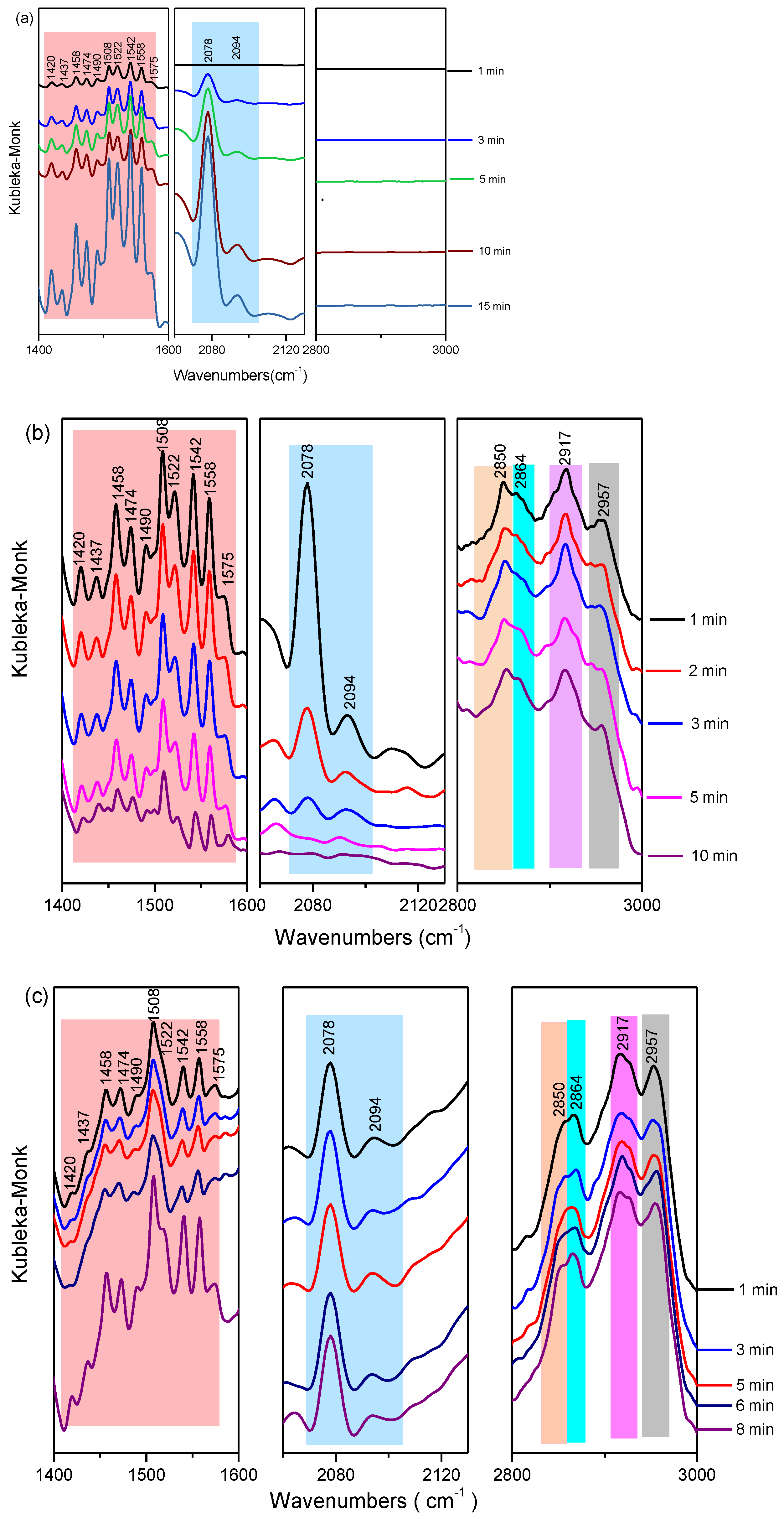
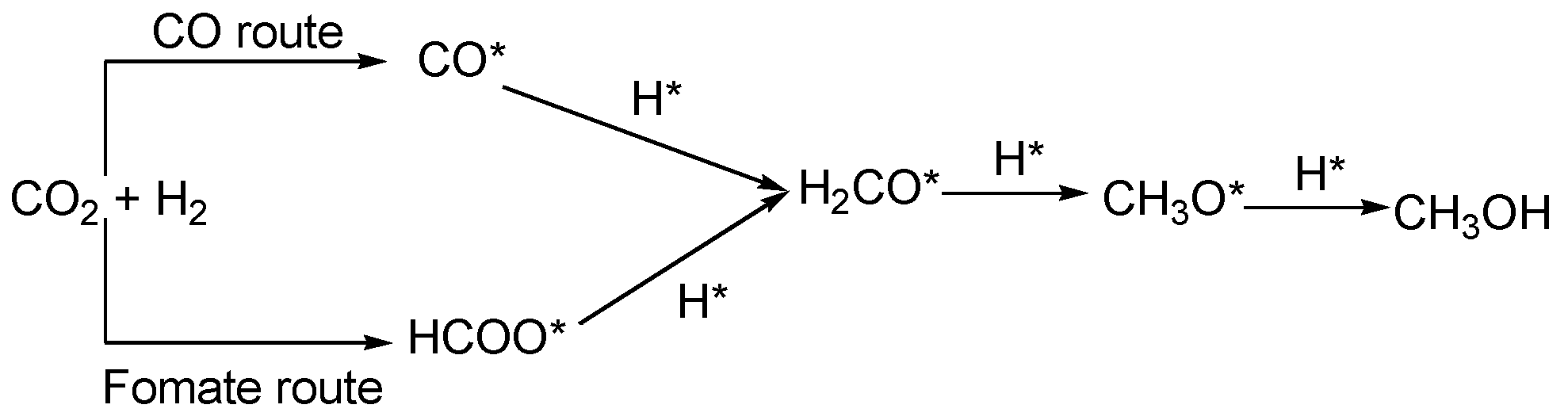
3. Results and Discussions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wei, J.; Yao, R.; Han, Y.; Ge, Q.; Sun, J. Towards the development of the emerging process of CO2 heterogenous hydrogenation into high-value unsaturated heavy hydrocarbons. Chem. Soc. Rev. 2021, 50, 10764–10805. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Nie, X.; Guo, X.; Song, C.; Chen, J.G. Recent advances in carbon dioxide hydrogenation to methanol via heterogeneous catalysis. Chem. Rev. 2020, 120, 7984–8034. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wang, Y.Q.; Ding, M.Y.; Hong, X.L.; Liu, G.L.; Tsang, S.C.E. Advances in higher alcohol synthesis from CO2 hydrogenation. Chem 2021, 7, 849–881. [Google Scholar] [CrossRef]
- Bediako, B.B.A.; Qian, Q.; Han, B.X. Synthesis of C2+ Chemicals from CO2 and H2 via C-C Bond Formation. Acc. Chem. Res. 2021, 54, 2467–2476. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Sanz, I.; Korili, S.A.; Gil, A. Catalytic valorization of CO2 by hydrogenation: Current status and future trends. Catal. Rev. 2021. [Google Scholar] [CrossRef]
- Wang, D.; Xie, Z.H.; Porosoff, M.D.; Chen, J.G. Recent advances in carbon dioxide hydrogenation to produce olefins and aromatics. Chem 2021, 7, 2277–2311. [Google Scholar] [CrossRef]
- Navarro-Jaén, S.; Virginie, M.; Bonin, J.; Robert, M.; Wojcieszak, R.; Khodakov, A.Y. Highlights and challenges in the selective reduction of carbon dioxide to methanol. Nat. Rev. Chem. 2021, 5, 564–579. [Google Scholar] [CrossRef]
- Bai, S.T.; De Smet, G.; Liao, Y.H.; Sun, R.Y.; Zhou, C.; Beller, M.; Maes, B.U.W.; Sels, B.F. Homogeneous and heterogeneous catalysts for hydrogenation of CO2 to methanol under mild conditions. Chem. Soc. Rev. 2021, 50, 4259–4298. [Google Scholar] [CrossRef]
- Zhang, X.B.; Zhang, G.H.; Song, C.S.; Guo, X.W. Catalytic conversion of carbon dioxide to methanol: Current status and future perspective. Front. Energy Res. 2021, 8, 621119. [Google Scholar] [CrossRef]
- Sharma, P.; Sebastian, J.; Ghosh, S.; Creaser, D.; Olsson, L. Recent advances in hydrogenation of CO2 into hydrocarbons via methanol intermediate over heterogeneous catalysts. Catal. Sci. Technol. 2021, 11, 1665–1697. [Google Scholar] [CrossRef]
- Murthy, P.S.; Liang, W.B.; Jiang, Y.J.; Huang, J. Cu-Based Nanocatalysts for CO2 hydrogenation to methanol. Energy Fuels 2021, 35, 8558–8584. [Google Scholar] [CrossRef]
- Shi, Z.; Tan, Q.; Wu, D. Enhanced CO2 hydrogenation to methanol over TiO2 nanotubes-supported CuO-ZnO-CeO2 catalyst. Appl. Catal. A Gen. 2019, 581, 58–66. [Google Scholar] [CrossRef]
- Zhang, G.; Fan, G.; Yang, L.; Li, F. Tuning surface-interface structures of ZrO2 supported copper catalysts by in situ introduction of indium to promote CO2 hydrogenation to methanol. Appl. Catal. A Gen. 2020, 605, 117805. [Google Scholar] [CrossRef]
- Snider, J.L.; Streibel, V.; Hubert, M.A.; Choksi, T.S.; Valle, E.; Upham, D.C.; Schumann, J.; Duyar, M.S.; Gallo, A.; Abild-Pedersen, F.; et al. Revealing the synergy between oxide and alloy phases on the performance of bimetallic In-Pd catalysts for CO2 hydrogenation to methanol. ACS Catal. 2019, 9, 3399–3412. [Google Scholar] [CrossRef]
- Gholinejad, M.; Khosravi, F.; Afrasi, M.; Sansano, J.M.; Nájera, C. Applications of bimetallic PdCu catalysts. Catal. Sci. Technol. 2021, 11, 2652–2702. [Google Scholar] [CrossRef]
- Sha, F.; Han, Z.; Tang, S.; Wang, J.; Li, C. Hydrogenation of carbon dioxide to methanol over non-Cu-based heterogeneous catalysts. ChemSusChem 2020, 13, 6160–6181. [Google Scholar] [CrossRef] [PubMed]
- Khobragade, R.; Roškarič, M.; Žerjav, G.; Košiček, M.; Zavašnik, J.; Van de Velde, N.; Jerman, I.; Tušar, N.N.; Pintar, A. Exploring the effect of morphology and surface properties of nanoshaped Pd/CeO2 catalysts on CO2 hydrogenation to methanol. Appl. Catal. A Gen. 2021, 667, 118394. [Google Scholar] [CrossRef]
- Lin, F.; Jiang, X.; Boreriboon, N.; Wang, Z.; Song, C.; Cen, K. Effects of supports on bimetallic Pd-Cu catalysts for CO2 hydrogenation to methanol. Appl. Catal. A Gen. 2019, 585, 117210. [Google Scholar] [CrossRef]
- Nie, X.W.; Li, W.H.; Jiang, X.; Guo, X.W.; Song, C.S. Recent advances in catalytic CO2 hydrogenation to alcohols and hydrocarbons. Adv. Catal. 2020, 65, 121–233. [Google Scholar]
- Shen, C.Y.; Bao, Q.Q.; Xue, W.J.; Sun, K.H.; Zhang, Z.T.; Jia, X.Y.; Mei, D.H.; Liu, C.J. Synergistic effect of the metal-support interaction and interfacial oxygen vacancy for CO2 hydrogenation to methanol over Ni/In2O3 catalyst: A theoretical study. J. Energy Chem. 2022, 65, 623–629. [Google Scholar] [CrossRef]
- Wang, J.Y.; Zhang, G.H.; Zhu, J.; Zhang, X.B.; Ding, F.S.; Zhang, A.F.; Guo, X.W.; Song, C.S. CO2 hydrogenation to methanol over In2O3-based catalysts: From mechanism to catalyst development. ACS Catal. 2021, 11, 1406–1423. [Google Scholar] [CrossRef]
- Wang, J.J.; Tang, C.Z.; Li, G.N.; Han, Z.; Li, Z.L.; Liu, H.; Cheng, F.; Li, C. High-performance MaZrOx (Ma = Cd, Ga) solid-solution catalysts for CO2 hydrogenation to methanol. ACS Catal. 2019, 9, 10253–10259. [Google Scholar] [CrossRef]
- Wang, J.J.; Li, G.N.; Li, Z.L.; Tang, C.Z.; Feng, Z.C.; An, H.Y.; Liu, H.L.; Liu, T.F.; Li, C. A highly selective and stable ZnO-ZrO2 solid solution catalyst for CO2 hydrogenation to methanol. Sci. Adv. 2017, 3, e1701290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Hong, X.L.; Liu, G.L. Highly dispersed metal doping to ZnZr oxide catalyst for CO2 hydrogenation to methanol: Insight into hydrogen spillover. J. Catal. 2021, 393, 207–214. [Google Scholar] [CrossRef]
- Qiao, S.C.; Liu, X.Y.; Zhu, R.R.; Xue, D.M.; Liu, X.Q.; Sun, L.B. Causation of catalytic activity of Cu-ZnO for CO2 hydrogenation to methanol. Chem. Eng. J. 2022, 430, 132784. [Google Scholar] [CrossRef]
- Saito, M.; Anderson, R.B. The activity of several molybdenum compounds for the methanation of CO2. J. Catal. 1981, 67, 296–302. [Google Scholar] [CrossRef]
- Primo, A.; He, J.; Jurca, B.; Cojocaru, B.; Bucur, C.; Parvulescu, V.I.; Garcia, H. CO2 methanation catalyzed by oriented MoS2 nanoplatelets supported on few layers graphene. Appl. Catal. B Environ. 2019, 245, 351–359. [Google Scholar] [CrossRef]
- Liu, P.; Choi, Y.M.; Yang, Y.; White, M.G. Methanol synthesis from H2 and CO2 on a Mo6S8 cluster: A density functional study. J. Phys. Chem. A 2010, 114, 3888–3895. [Google Scholar] [CrossRef]
- Lu, Z.; Cheng, Y.; Li, S.; Yang, Z.; Wu, R. CO2 thermoreduction to methanol on the MoS2 supported single Co atom catalyst: A DFT study. Appl. Sur. Sci. 2020, 528, 147047. [Google Scholar] [CrossRef]
- Li, H.; Wang, L.; Dai, Y.; Pu, Z.; Lao, Z.; Chen, Y.; Wang, M.; Zheng, X.; Zhu, J.; Zhang, W.; et al. Synergetic interaction between neighbouring platinum monomers in CO2 hydrogenation. Nat. Nanotechnol. 2018, 13, 411–417. [Google Scholar] [CrossRef]
- Hu, J.; Yu, L.; Deng, J.; Wang, Y.; Cheng, K.; Ma, C.; Zhang, Q.; Wen, W.; Yu, S.; Pan, Y.; et al. Sulfur vacancy-rich MoS2 as a catalyst for the hydrogenation of CO2 to methanol. Nat. Catal. 2021, 4, 242–250. [Google Scholar] [CrossRef]
- Han, H.; Cui, P.P.; Xiao, L.F.; Wu, W. MoCS@NSC with interfacial heterojunction nanostructure: A highly selective catalyst for synthesizing methanol from CO2 at low temperature. J. Environ. Chem. Eng. 2021, 9, 106354. [Google Scholar] [CrossRef]
- Panigrahi, P.K.; Pathak, A. Aqueous medium synthesis route for randomly stacked molybdenum disulfide. J. Nanoparticl. 2013, 2013, 671214. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Zhu, Y.; Tang, Y. Preparation of sulfur-doped microporous carbons for the storage of hydrogen and carbon dioxide. Carbon 2012, 50, 5543–5553. [Google Scholar] [CrossRef]
- Djamil, J.; Segler, S.A.; Dabrowski, A.; Bensch, W.; Lotnyk, A.; Schürmann, U.; Kienle, L.; Hansen, S.; Bewerie, T. The influence of carbon content on the structure and properties of MoSxCy photocatalysts for light-driven hydrogen generation. Dalton Trans. 2013, 42, 1287–1292. [Google Scholar] [CrossRef]
- Tsai, C.; Abild-Pedersen, F.; Norskov, J.K. Tuning the MoS2 edge-site activity for hydrogen evolution via support interactions. Nano Lett. 2014, 14, 1381–1387. [Google Scholar] [CrossRef]
- Hinnemann, B.; Moses, P.G.; Bonde, J.; Jørgensen, K.P.; Nielsen, J.H.; Horch, S.; Chorkendorff, I.; Nørskov, J.K. Biomimetic hydrogen evolution: MoS2 nanoparticles as catalyst for hydrogen evolution. J. Am. Chem. Soc. 2005, 127, 5308–5309. [Google Scholar] [CrossRef]
- Prins, R.; De Beer, V.H.J.; Somorjai, G.A. Structure and function of the catalyst and the promoter in Co-Mo Hydrodesulfurization catalysts. Catal. Rev. 1989, 31, 1–41. [Google Scholar] [CrossRef]
- Gao, S.; Yang, L.; Shao, J.; Qu, Q.; Wu, Y.; Holze, R. Construction of hierarchical hollow MoS2/carbon microspheres for enhanced lithium storage performance. J. Electrochem. Soc. 2020, 167, 100525. [Google Scholar] [CrossRef]
- Cao, H.; Wang, H.; Huang, Y.; Sun, Y.; Shi, S.; Tang, M. Quantification of gold(III) in solution and with a test stripe via the quenching of the fluorescence of molybdenum disulfide quantum dots. Microchimi. Acta 2016, 184, 91–100. [Google Scholar] [CrossRef]
- Zhang, J.; Cui, P.; Gu, Y.; Wu, D.; Tao, S.; Qian, B.; Chu, W.; Song, L. Encapsulating carbon-coated MoS2 nanosheets within a nitrogen-doped graphene network for high-performance potassium-ion storage. Adv. Mater. Interfaces 2019, 6, 1901066. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, T.; Fang, T.; Gao, Y.; Gao, S.; Wang, W.; Liao, L. A novel MoS2@C framework architecture composites with three-dimensional cross-linked porous carbon supporting MoS2 nanosheets for sodium storage. J. Alloy. Compd. 2020, 818, 152821. [Google Scholar] [CrossRef]
- Cai, Y.; Yang, H.; Zhou, J.; Luo, Z.; Fang, G.; Liu, S.; Pan, A.; Liang, S. Nitrogen-doped hollow MoS2/C nanospheres as anode for long-life sodium-ion batteries. Chem. Eng. J. 2017, 327, 522–529. [Google Scholar] [CrossRef]
- Hu, R.; Fang, Y.; Zhu, K.; Yang, X.; Yin, J.; Ye, K.; Yan, J.; Cao, D.; Wang, G. Carbon coated MoS2 Hierarchical Microspheres enabling fast and durable potassium ion storage. Appl. Sur. Sci. 2021, 564, 150387. [Google Scholar] [CrossRef]
- Zhao, Z.; Qin, F.; Kasiraju, S.; Xie, L.; Alam, M.K.; Chen, S.; Wang, D.; Ren, Z.; Wang, Z.; Grabow, L.C.; et al. Vertically aligned MoS2/Mo2C hybrid nanosheets grown on carbon paper for efficient electrocatalytic hydrogen evolution. ACS Catal. 2017, 7, 7312–7318. [Google Scholar] [CrossRef]
- Lin, Q.; Dong, X.; Wang, Y.; Zheng, N.; Zhao, Y.; Xu, W.; Ding, T. Molybdenum disulfide with enlarged interlayer spacing decorated on reduced graphene oxide for efficient electrocatalytic hydrogen evolution. J. Mater. Sci. 2020, 55, 6637–6647. [Google Scholar] [CrossRef]
- Yin, X.; Sun, G.; Song, A.; Wang, L.; Wang, Y.; Dong, H.; Shao, G. A novel structure of Ni-(MoS2/GO) composite coatings deposited on Ni foam under supergravity field as efficient hydrogen evolution reaction catalysts in alkaline solution. Electrochimi. Acta 2017, 249, 52–63. [Google Scholar] [CrossRef]
- Yang, S.; Wang, Y.; Zhang, H.; Zhang, Y.; Liu, L.; Fang, L.; Yang, X.; Gu, X.; Wang, Y. Unique three-dimensional Mo2C@MoS2 heterojunction nanostructure with S vacancies as outstanding all-pH range electrocatalyst for hydrogen evolution. J. Catal. 2019, 371, 20–26. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, Y.; Wang, X.; Li, S.; Huang, W.; Dong, L.; Liu, C.; Li, Y.; Lan, Y. Molybdenum disulfide/nitrogen-doped reduced graphene oxide nanocomposite with enlarged interlayer spacing for electrocatalytic hydrogen evolution. Adv. Energy Mater. 2016, 6, 1600116. [Google Scholar] [CrossRef]
- Liu, H.; Hu, H.; Wang, J.; Niehoff, P.; He, X.; Paillard, E.; Eder, D.; Winter, M.; Li, J. Hierarchical ternary MoO2/MoS2/heteroatom-doped carbon hybrid materials for high-performance lithium-ion storage. ChemElectroChem 2016, 3, 922–932. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, J.; Goodman, K.R.; Head, A.R.; Tong, X.; Stacchiola, D.J.; White, M.G. Reactivity of a zirconia-copper inverse catalyst for CO2 hydrogenation. J. Phys. Chem. C 2020, 124, 22158–22172. [Google Scholar] [CrossRef]
- Gao, P.; Li, F.; Zhan, H.J.; Zhao, N.; Xiao, F.K.; Wei, W.; Zhong, L.S.; Wang, H.; Sun, Y.H. Influence of Zr on the performance of Cu/Zn/Al/Zr catalysts via hydrotalcite-like precursors for CO2 hydrogenation to methanol. J. Catal. 2013, 298, 51–60. [Google Scholar] [CrossRef]
- Xiao, J.; Mao, D.S.; Guo, X.M.; Yu, J. Effect of TiO2, ZrO2, and TiO2-ZrO2 on the performance of CuO-ZnO catalyst for CO2 hydrogenation to methanol. Appl. Surf. Sci. 2015, 338, 146–153. [Google Scholar] [CrossRef]
- Liu, T.; Xu, D.; Wu, D.; Liu, G.; Hong, X. Spinel ZnFe2O4 regulates copper sites for CO2 hydrogenation to methanol. ACS Sustain. Chem. Eng. 2021, 9, 4033–4041. [Google Scholar] [CrossRef]
- Kiciński, W.; Szala, M.; Bystrzejewski, M. Sulfur-doped porous carbons: Synthesis and applications. Carbon 2014, 68, 1–32. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Ramírez, P.J.; Gutiérrez, R.A.; Stacchiola, D.J.; Viñes, F.; Liu, P.; Illas, F.; Rodriguez, J.A. The conversion of CO2 to methanol on orthorhombic β-Mo2C and Cu/β-Mo2C catalysts: Mechanism for admetal induced change in the selectivity and activity. Catal. Sci. Technol. 2016, 6, 6766–6777. [Google Scholar] [CrossRef]
- Posada-Pérez, S.; Viñes, F.; Rodriguez, J.A.; Illas, F. Fundamentals of methanol synthesis on metal carbide based catalysts: Activation of CO2 and H2. Top. Catal. 2015, 58, 159–173. [Google Scholar] [CrossRef]
- Posada-Perez, S.; Politi, J.R.d.S.; Vines, F.; Illas, F. Methane capture at room temperature: Adsorption on cubic δ-MoC and orthorhombic β-Mo2C molybdenum carbide (001) surfaces. RSC Adv. 2015, 5, 33737–33746. [Google Scholar] [CrossRef]
- Alves, L.M.N.C.; Almeida, M.P.; Ayala, M.; Watson, C.D.; Jacobs, G.; Rabelo-Neto, R.C.; Noronha, F.B.; Mattos, L.V. CO2 methanation over metal catalysts supported on ZrO2: Effect of the nature of the metallic phase on catalytic performance. Chem. Eng. Sci. 2021, 239, 116604. [Google Scholar] [CrossRef]
- Yu, J.; Yang, M.; Zhang, J.; Ge, Q.; Zimina, A.; Pruessmann, T.; Zheng, L.; Grunwaldt, J.D.; Sun, J. Stabilizing Cu+ in Cu/SiO2 catalysts with a shattuckite-like structure boosts CO2 hydrogenation into methanol. ACS Catal. 2020, 10, 14694–14706. [Google Scholar] [CrossRef]
- Zhao, K.; Wang, L.; Moioli, E.; Calizzi, M.; Züttel, A. Identifying reaction species by evolutionary fitting and kinetic analysis: An example of CO2 hydrogenation in DRIFTS. J. Phys. Chem. C 2021, 123, 8785–8792. [Google Scholar] [CrossRef]
- Fisher, I.A.; Bell, A.T. In situ infrared study of methanol synthesis from H2/CO over Cu/SiO2 and Cu/ZrO2/SiO2. J. Catal. 1998, 178, 153–173. [Google Scholar] [CrossRef]
- Fisher, I.A.; Bell, A.T. In-situ infrared study of methanol synthesis from H2/CO2 over Cu/SiO2 and Cu/ZrO2/SiO2. J. Catal. 1997, 172, 222–237. [Google Scholar] [CrossRef]
- Xu, M.; Yao, S.; Rao, D.; Niu, Y.; Liu, N.; Peng, M.; Zhai, P.; Man, Y.; Zheng, L.; Wang, B.; et al. Insights into interfacial synergistic catalysis over Ni@TiO2−x catalyst toward Water-Gas shift reaction. J. Am. Chem. Soc. 2018, 140, 11241–11251. [Google Scholar] [CrossRef]
- Cerdá-Moreno, C.; Chica, A.; Keller, S.; Rautenberg, C.; Bentrup, U. Ni-sepiolite and Ni-todorokite as efficient CO2 methanation catalysts: Mechanistic insight by operando DRIFTS. Appl. Catal. B Environ. 2020, 264, 118546. [Google Scholar] [CrossRef]
- Malik, A.S.; Zaman, S.F.; Al-Zahrani, A.A.; Daous, M.A.; Driss, H.; Petrov, L.A. Selective hydrogenation of CO2 to CH3OH and in-depth DRIFT analysis for PdZn/ZrO2 and CaPdZn/ZrO2 catalysts. Catal. Today 2020, 357, 573–582. [Google Scholar] [CrossRef]
- Luo, L.; Wang, M.; Cui, Y.; Chen, Z.; Wu, J.; Cao, Y.; Luo, J.; Dai, Y.; Li, W.; Bao, J.; et al. Surface iron species in palladium-iron intermetallic nanocrystals that promote and stabilize CO2 methanation. Angew. Chem. Int. Ed. 2020, 59, 14434–14442. [Google Scholar] [CrossRef]
- Kattel, S.; Yan, B.; Yang, Y.; Chen, J.G.; Liu, P. Optimizing binding energies of key intermediates for CO2 hydrogenation to methanol over oxide-supported copper. J. Am. Chem. Soc. 2016, 138, 12440–12450. [Google Scholar] [CrossRef]
- Romero-Sarria, F.; Bobadilla, L.F.; Barrera, E.M.J.; Odriozola, J.A. Experimental evidence of HCO species as intermediate in the fischer tropsch reaction using operando techniques. Appl. Catal. B Environ. 2020, 272, 119032. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.; Si, H.; Zhang, Q.; Wu, J.; Gao, L.; Wei, X.; Sun, Y.; Liao, Q.; Zhang, Z.; et al. Single-atom vacancy defect to trigger high-efficiency hydrogen evolution of MoS2. J. Am. Chem. Soc. 2020, 142, 4298–4308. [Google Scholar] [CrossRef]
- Mu, X.; Zhu, Y.; Gu, X.; Dai, S.; Mao, Q.; Bao, L.; Li, W.; Liu, S.; Bao, J.; Mu, S. Awakening the oxygen evolution activity of MoS2 by exophilic-metal induced surface reorganization engineering. J. Energy Chem. 2021, 62, 546–551. [Google Scholar] [CrossRef]
- Gong, F.; Ye, S.; Liu, M.; Zhang, J.; Gong, L.; Zeng, G.; Meng, E.; Su, P.; Xie, K.; Zhang, Y.; et al. Boosting electrochemical oxygen evolution over yolk-shell structured O-MoS2 nanoreactors with sulfur vacancy and decorated Pt nanoparticles. Nano Energy 2020, 78, 105284. [Google Scholar] [CrossRef]
- Liu, G.; Robertson, A.W.; Li, M.M.; Kuo, W.C.H.; Darby, M.T.; Muhieddine, M.H.; Lin, Y.C.; Suenaga, K.; Stamatakis, M.; Warner, J.H.; et al. MoS2 monolayer catalyst doped with isolated Co atoms for the hydrodeoxygenation reaction. Nat. Chem. 2017, 9, 810–816. [Google Scholar] [CrossRef]
- Cai, L.; He, J.; Liu, Q.; Yao, T.; Chen, L.; Yan, W.; Hu, F.; Jiang, Y.; Zhao, Y.; Hu, T.; et al. Vacancy-induced ferromagnetism of MoS2 nanosheets. J. Am. Chem. Soc. 2015, 137, 2622–2627. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Conversion/% | Selectivity/% | STY | ||
|---|---|---|---|---|---|
| CH3OH | CH4 | CH3OCH3 | /gMeOH gcat.−1 h−1 | ||
| NSC | - | - | - | - | - |
| MoS2 | 18.3 | 66.9 | 32.7 | 0.4 | 0.252 |
| MoS2-45@C | 27.3 | 95.8 | 4.2 | 0 | 0.538 |
| MoS2-5@C | 18.4 | 79.9 | 20.1 | 0 | 0.302 |
| MoS2/C | 4.2 | 78.5 | 21.5 | 0 | 0.068 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, P.; Sun, R.; Xiao, L.; Wu, W. Exploring the Effects of the Interaction of Carbon and MoS2 Catalyst on CO2 Hydrogenation to Methanol. Int. J. Mol. Sci. 2022, 23, 5220. https://doi.org/10.3390/ijms23095220
Cui P, Sun R, Xiao L, Wu W. Exploring the Effects of the Interaction of Carbon and MoS2 Catalyst on CO2 Hydrogenation to Methanol. International Journal of Molecular Sciences. 2022; 23(9):5220. https://doi.org/10.3390/ijms23095220
Chicago/Turabian StyleCui, Pingping, Ruyu Sun, Linfei Xiao, and Wei Wu. 2022. "Exploring the Effects of the Interaction of Carbon and MoS2 Catalyst on CO2 Hydrogenation to Methanol" International Journal of Molecular Sciences 23, no. 9: 5220. https://doi.org/10.3390/ijms23095220
APA StyleCui, P., Sun, R., Xiao, L., & Wu, W. (2022). Exploring the Effects of the Interaction of Carbon and MoS2 Catalyst on CO2 Hydrogenation to Methanol. International Journal of Molecular Sciences, 23(9), 5220. https://doi.org/10.3390/ijms23095220