
Deciphering Pleiotropic Signatures of Regulatory SNPs in Zea mays L. Using Multi-Omics Data and Machine Learning Algorithms



Abstract
:1. Introduction
2. Materials and Methods
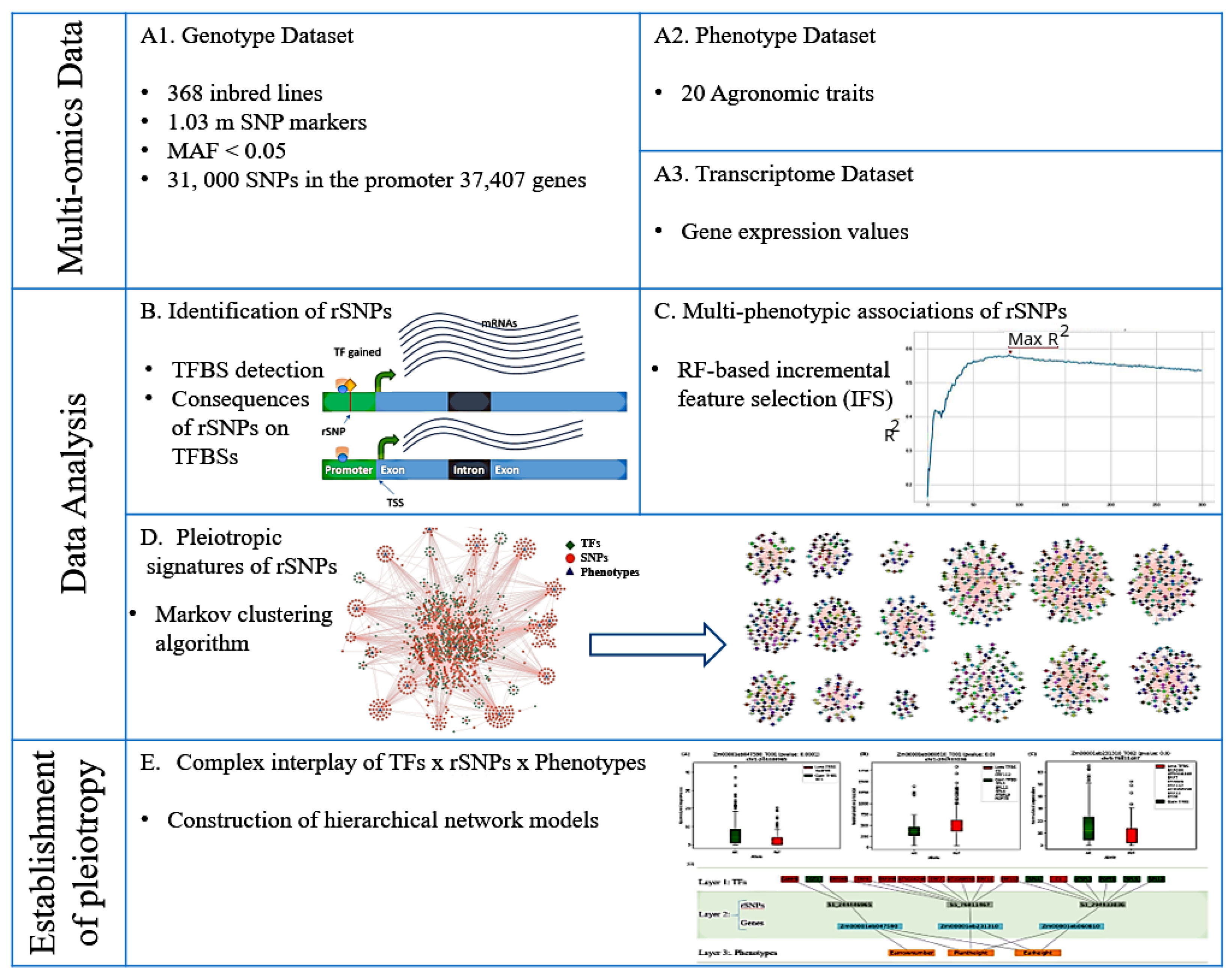
2.1. Multi-Omics Data
2.1.1. Genotype Dataset
2.1.2. Phenotype Dataset
2.1.3. Transcriptome Dataset
2.2. Data Analysis
3. Results and Discussion
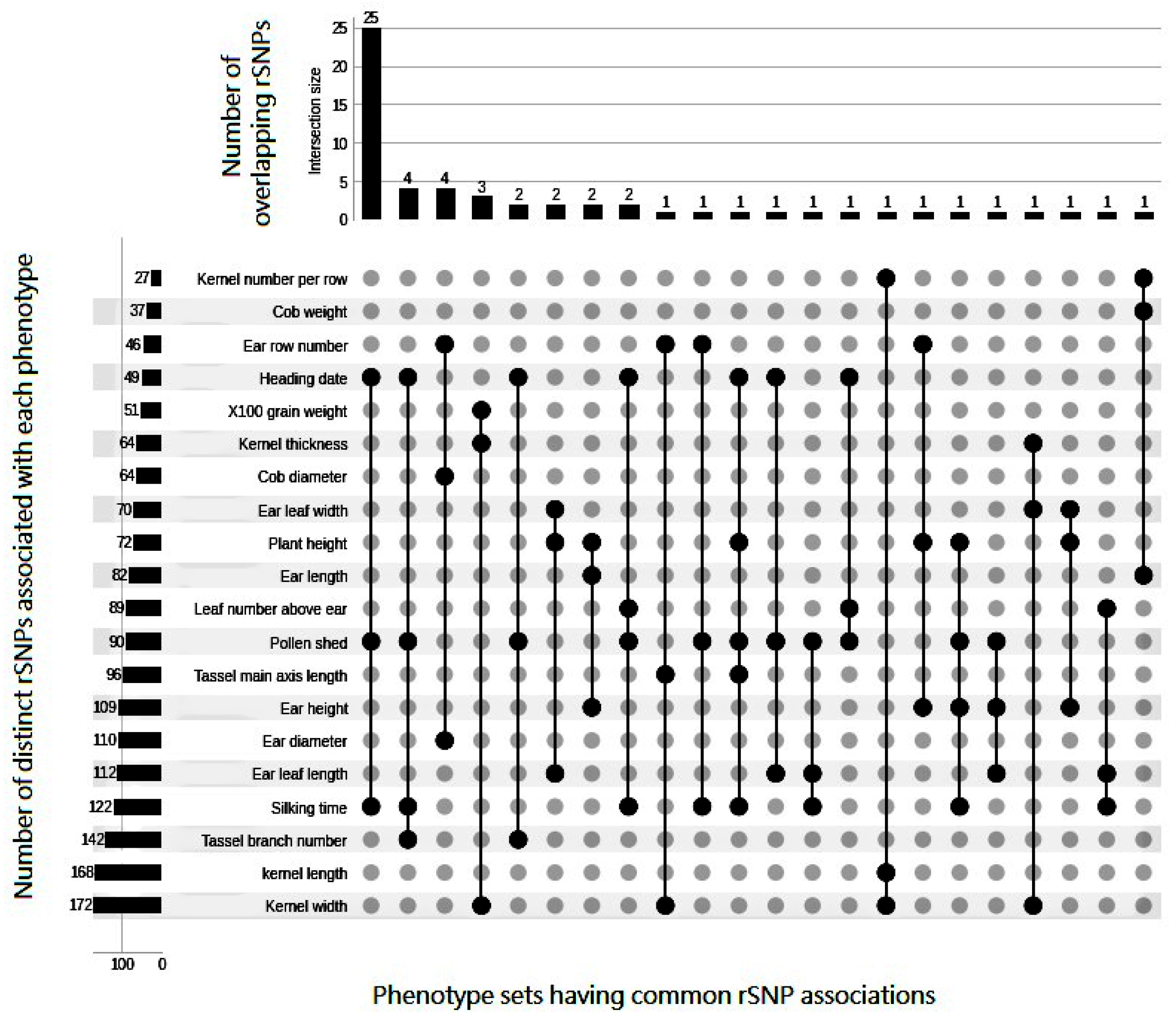
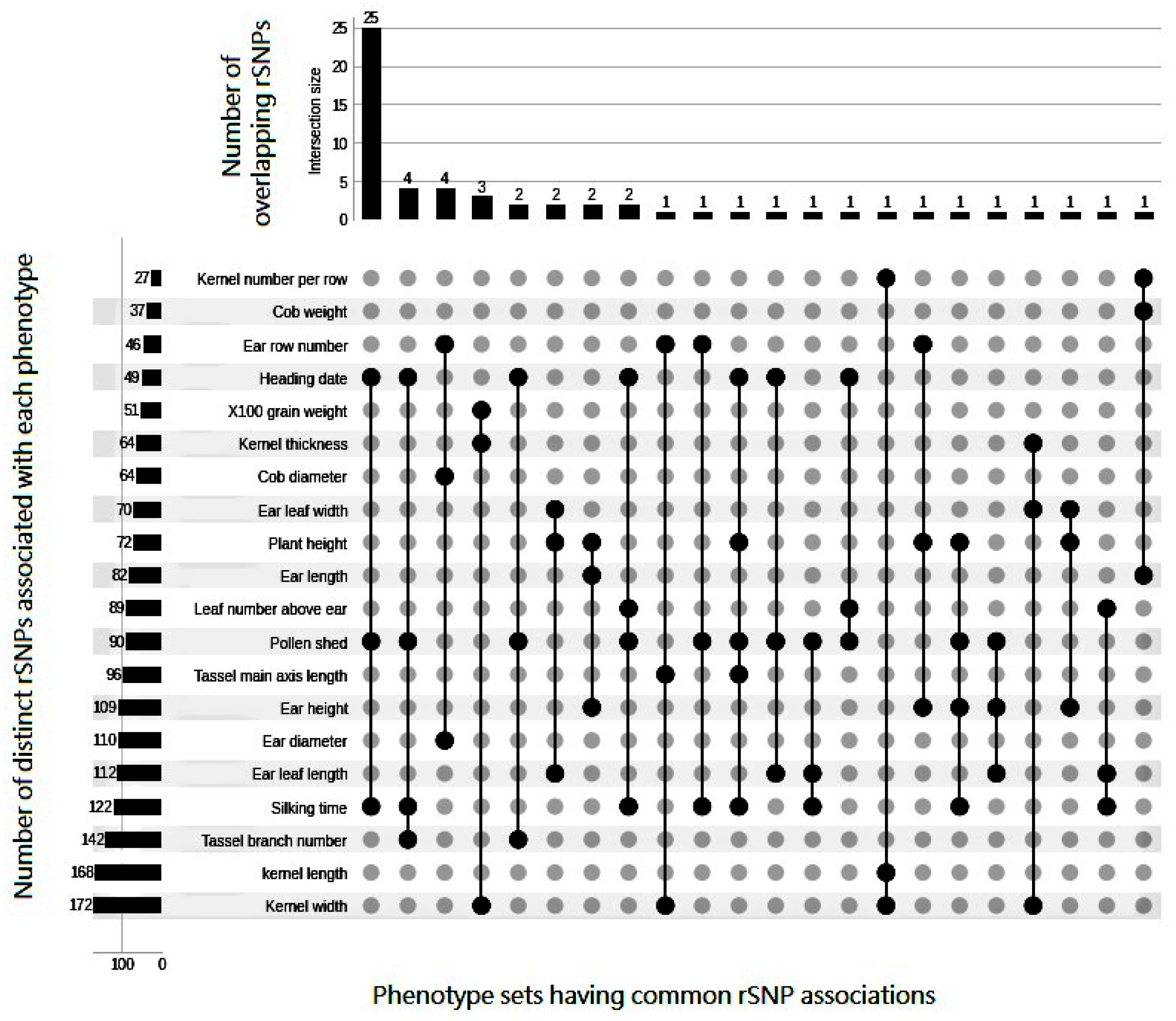
3.1. Pleiotropic Association Signatures of rSNPs
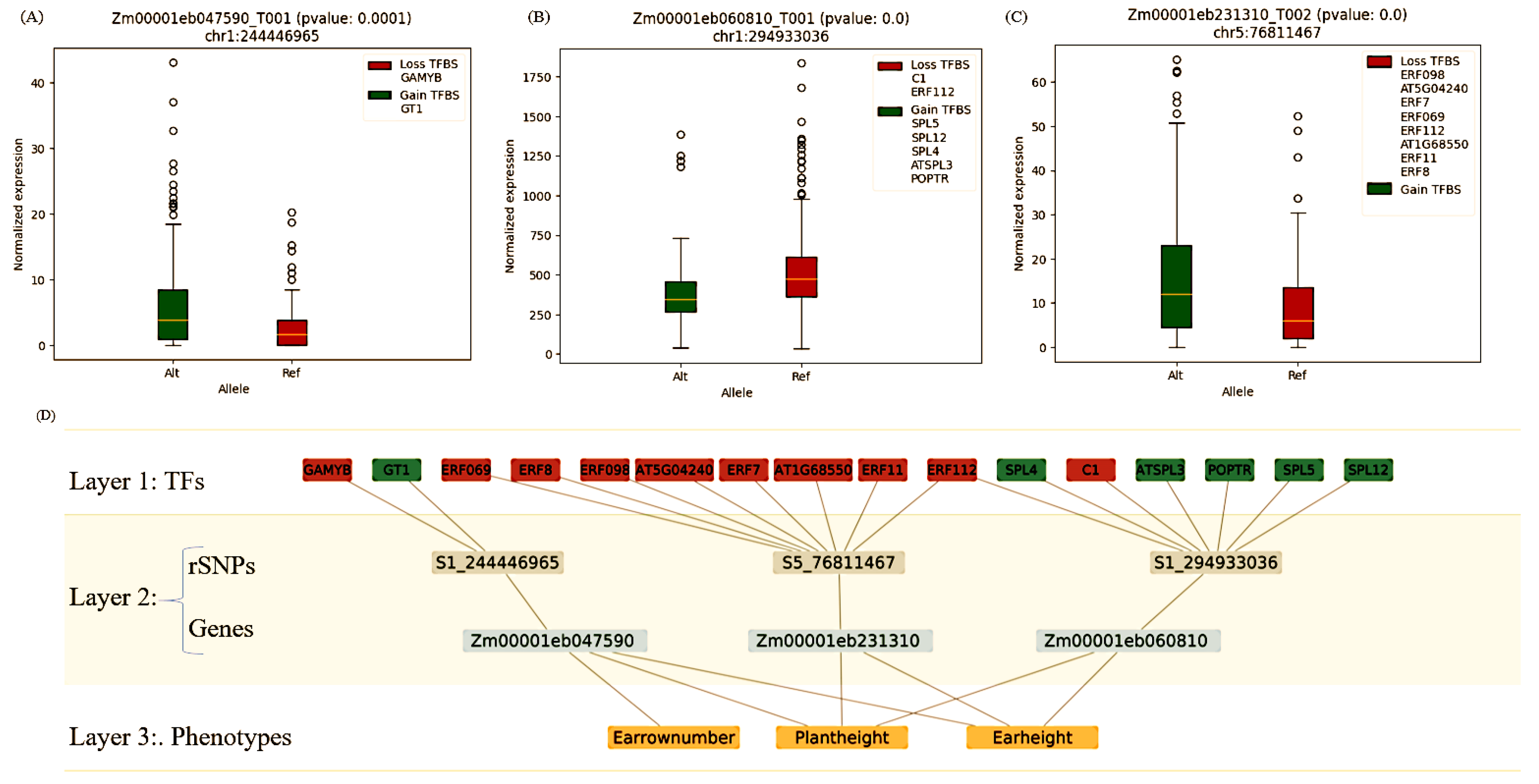
3.2. Construction of Hierarchical Network Models
4. Conclusions
5. Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Shiferaw, B.; Prasanna, B.M.; Hellin, J.; Bänziger, M. Crops that feed the world 6. Past successes and future challenges to the role played by maize in global food security. Food Secur. 2011, 3, 307–327. [Google Scholar] [CrossRef] [Green Version]
- Prasanna, B.M.; Palacios-Rojas, N.; Hossain, F.; Muthusamy, V.; Menkir, A.; Dhliwayo, T.; Ndhlela, T.; San Vicente, F.; Nair, S.K.; Vivek, B.S.; et al. Molecular breeding for nutritionally enriched maize: Status and prospects. Front. Genet. 2020, 10, 1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz-Monasterio, J.I.; Palacios-Rojas, N.; Meng, E.; Pixley, K.; Trethowan, R.; Pena, R. Enhancing the mineral and vitamin content of wheat and maize through plant breeding. J. Cereal Sci. 2007, 46, 293–307. [Google Scholar] [CrossRef]
- Bänziger, M.; Betrán, F.; Lafitte, H. Efficiency of high-nitrogen selection environments for improving maize for low-nitrogen target environments. Crop. Sci. 1997, 37, 1103–1109. [Google Scholar] [CrossRef]
- Suwarno, W.B.; Pixley, K.V.; Palacios-Rojas, N.; Kaeppler, S.M.; Babu, R. Genome-wide association analysis reveals new targets for carotenoid biofortification in maize. Theor. Appl. Genet. 2015, 128, 851–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Lawit, S.J.; Weers, B.; Sun, J.; Mongar, N.; Van Hemert, J.; Melo, R.; Meng, X.; Rupe, M.; Clapp, J.; et al. Overexpression of zmm28 increases maize grain yield in the field. Proc. Natl. Acad. Sci. USA 2019, 116, 23850–23858. [Google Scholar] [CrossRef] [Green Version]
- Boćanski, J.; Srećkov, Z.; Nastasić, A. Genetic and phenotypic relationship between grain yield and components of grain yield of maize (Zea mays L.). Genetika 2009, 41, 145–154. [Google Scholar] [CrossRef]
- Veldboom, L.R.; Lee, M. Genetic mapping of quantitative trait loci in maize in stress and nonstress environments: I. Grain yield and yield components. Crop. Sci. 1996, 36, 1310–1319. [Google Scholar] [CrossRef]
- Betran, F.; Beck, D.; Bänziger, M.; Edmeades, G. Genetic analysis of inbred and hybrid grain yield under stress and nonstress environments in tropical maize. Crop. Sci. 2003, 43, 807–817. [Google Scholar] [CrossRef]
- Dhugga, K.S. Maize biomass yield and composition for biofuels. Crop. Sci. 2007, 47, 2211–2227. [Google Scholar] [CrossRef]
- Fernandez, M.G.S.; Becraft, P.W.; Yin, Y.; Lübberstedt, T. From dwarves to giants? Plant height manipulation for biomass yield. Trends Plant Sci. 2009, 14, 454–461. [Google Scholar] [CrossRef]
- Xue, J.; Gao, S.; Fan, Y.; Li, L.; Ming, B.; Wang, K.; Xie, R.; Hou, P.; Li, S. Traits of plant morphology, stalk mechanical strength, and biomass accumulation in the selection of lodging-resistant maize cultivars. Eur. J. Agron. 2020, 117, 126073. [Google Scholar] [CrossRef]
- Mazaheri, M.; Heckwolf, M.; Vaillancourt, B.; Gage, J.L.; Burdo, B.; Heckwolf, S.; Barry, K.; Lipzen, A.; Ribeiro, C.B.; Kono, T.J.; et al. Genome-wide association analysis of stalk biomass and anatomical traits in maize. BMC Plant Biol. 2019, 19, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, F.; Wutke, M.; Das, P.P.; Kamp, M.; Gültas, M.; Link, W.; Schmitt, A.O. Identification of regulatory SNPs associated with vicine and convicine content of Vicia faba based on genotyping by sequencing data using deep learning. Genes 2020, 11, 614. [Google Scholar] [CrossRef]
- Pearson, T.A.; Manolio, T.A. How to interpret a genome-wide association study. JAMA 2008, 299, 1335–1344. [Google Scholar] [CrossRef]
- Ramzan, F.; Gültas, M.; Bertram, H.; Cavero, D.; Schmitt, A.O. Combining Random Forests and a Signal Detection Method Leads to the Robust Detection of Genotype-Phenotype Associations. Genes 2020, 11, 892. [Google Scholar] [CrossRef] [PubMed]
- Ramzan, F.; Klees, S.; Schmitt, A.O.; Cavero, D.; Gültas, M. Identification of Age-Specific and Common Key Regulatory Mechanisms Governing Eggshell Strength in Chicken Using Random Forests. Genes 2020, 11, 464. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Patron, J.; Serra-Cayuela, A.; Han, B.; Li, C.; Wishart, D.S. Assessing the performance of genome-wide association studies for predicting disease risk. PLoS ONE 2019, 14, e0220215. [Google Scholar] [CrossRef] [Green Version]
- Klees, S.; Lange, T.M.; Bertram, H.; Rajavel, A.; Schlüter, J.S.; Lu, K.; Schmitt, A.O.; Gültas, M. In Silico Identification of the Complex Interplay between Regulatory SNPs, Transcription Factors, and Their Related Genes in Brassica napus L. Using Multi-Omics Data. Int. J. Mol. Sci. 2021, 22, 789. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, D.; He, F.; Wang, J.; Joshi, T.; Xu, D. Phenotype prediction and genome-wide association study using deep convolutional neural network of soybean. Front. Genet. 2019, 10, 1091. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Huang, J.Z.; Wu, Q.; Nguyen, T.T.; Li, M.J. Genome-wide association data classification and SNPs selection using two-stage quality-based Random Forests. BMC Genom. 2015, 16, S5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Chen, F.; Zhai, R.; Lin, X.; Wang, Z.; Su, L.; Christiani, D.C. Correction for population stratification in random forest analysis. Int. J. Epidemiol. 2012, 41, 1798–1806. [Google Scholar] [CrossRef] [Green Version]
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Schrider, D.R.; Kern, A.D. Supervised machine learning for population genetics: A new paradigm. Trends Genet. 2018, 34, 301–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés, A.J.; López-Hernández, F.; Osorio-Rodriguez, D. Predicting thermal adaptation by looking into populations’ genomic past. Front. Genet. 2020, 11, 1093. [Google Scholar] [CrossRef]
- Jansen, S.; Baulain, U.; Habig, C.; Ramzan, F.; Schauer, J.; Schmitt, A.O.; Scholz, A.M.; Sharifi, A.R.; Weigend, A.; Weigend, S. Identification and Functional Annotation of Genes Related to Bone Stability in Laying Hens Using Random Forests. Genes 2021, 12, 702. [Google Scholar] [CrossRef]
- Brieuc, M.S.; Waters, C.D.; Drinan, D.P.; Naish, K.A. A practical introduction to Random Forest for genetic association studies in ecology and evolution. Mol. Ecol. Resour. 2018, 18, 755–766. [Google Scholar] [CrossRef]
- Pendergrass, S.A.; Brown-Gentry, K.; Dudek, S.; Frase, A.; Torstenson, E.S.; Goodloe, R.; Ambite, J.L.; Avery, C.L.; Buyske, S.; Bžková, P.; et al. Phenome-wide association study (PheWAS) for detection of pleiotropy within the Population Architecture using Genomics and Epidemiology (PAGE) Network. PLoS Genet. 2013, 9, e1003087. [Google Scholar] [CrossRef] [Green Version]
- Pendergrass, S.; Brown-Gentry, K.; Dudek, S.; Torstenson, E.; Ambite, J.; Avery, C.; Buyske, S.; Cai, C.; Fesinmeyer, M.; Haiman, C.; et al. The use of phenome-wide association studies (PheWAS) for exploration of novel genotype-phenotype relationships and pleiotropy discovery. Genet. Epidemiol. 2011, 35, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Solovieff, N.; Cotsapas, C.; Lee, P.H.; Purcell, S.M.; Smoller, J.W. Pleiotropy in complex traits: Challenges and strategies. Nat. Rev. Genets. 2013, 14, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Mayfield, S.; Nelson, T.; Taylor, W.; Malkin, R. Carotenoid synthesis and pleiotropic effects in carotenoid-deficient seedlings of maize. Planta 1986, 169, 23–32. [Google Scholar] [CrossRef]
- Pilu, R.; Landoni, M.; Cassani, E.; Doria, E.; Nielsen, E. The maize lpa241 mutation causes a remarkable variability of expression and some pleiotropic effects. Crop. Sci. 2005, 45, 2096–2105. [Google Scholar] [CrossRef]
- Wen, L.; Chase, C.D. Pleiotropic effects of a nuclear restorer-of-fertility locus on mitochondrial transcripts in male-fertile and S male-sterile maize. Curr. Genet. 1999, 35, 521–526. [Google Scholar] [CrossRef]
- Bomblies, K.; Doebley, J.F. Pleiotropic effects of the duplicate maize FLORICAULA/LEAFY genes zfl1 and zfl2 on traits under selection during maize domestication. Genetics 2006, 172, 519–531. [Google Scholar] [CrossRef] [Green Version]
- Asakura, Y.; Hirohashi, T.; Kikuchi, S.; Belcher, S.; Osborne, E.; Yano, S.; Terashima, I.; Barkan, A.; Nakai, M. Maize mutants lacking chloroplast FtsY exhibit pleiotropic defects in the biogenesis of thylakoid membranes. Plant Cell 2004, 16, 201–214. [Google Scholar] [CrossRef] [Green Version]
- Chourey, P.S.; Li, Q.B.; Cevallos-Cevallos, J. Pleiotropy and its dissection through a metabolic gene Miniature1 (Mn1) that encodes a cell wall invertase in developing seeds of maize. Plant Sci. 2012, 184, 45–53. [Google Scholar] [CrossRef]
- Clark, R.M.; Wagler, T.N.; Quijada, P.; Doebley, J. A distant upstream enhancer at the maize domestication gene tb1 has pleiotropic effects on plant and inflorescent architecture. Nat. Genet. 2006, 38, 594–597. [Google Scholar] [CrossRef]
- Wisser, R.J.; Kolkman, J.M.; Patzoldt, M.E.; Holland, J.B.; Yu, J.; Krakowsky, M.; Nelson, R.J.; Balint-Kurti, P.J. Multivariate analysis of maize disease resistances suggests a pleiotropic genetic basis and implicates a GST gene. Proc. Natl. Acad. Sci. USA 2011, 108, 7339–7344. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.J.; Upadyayula, N.; Mahone, G.S.; Tian, F.; Bradbury, P.J.; Myles, S.; Holland, J.B.; Flint-Garcia, S.; McMullen, M.D.; Buckler, E.S.; et al. Distinct genetic architectures for male and female inflorescence traits of maize. PLoS Genet. 2011, 7, e1002383. [Google Scholar] [CrossRef] [Green Version]
- Houle, D.; Govindaraju, D.R.; Omholt, S. Phenomics: The next challenge. Nat. Rev. Genet. 2010, 11, 855–866. [Google Scholar] [CrossRef]
- Rajavel, A.; Klees, S.; Schlüter, J.S.; Bertram, H.; Lu, K.; Schmitt, A.O.; Gültas, M. Unravelling the Complex Interplay of Transcription Factors Orchestrating Seed Oil Content in Brassica napus L. Int. J. Mol. Sci. 2021, 22, 1033. [Google Scholar] [CrossRef]
- Liu, H.; Wang, F.; Xiao, Y.; Tian, Z.; Wen, W.; Zhang, X.; Chen, X.; Liu, N.; Li, W.; Liu, L.; et al. MODEM: Multi-omics data envelopment and mining in maize. Database 2016, 2016, baw117. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Gao, S.; Xu, S.; Zhang, Z.; Prasanna, B.M.; Li, L.; Li, J.; Yan, J. Characterization of a global germplasm collection and its potential utilization for analysis of complex quantitative traits in maize. Mol. Breed. 2011, 28, 511–526. [Google Scholar] [CrossRef]
- Wen, W.; Araus, J.L.; Shah, T.; Cairns, J.; Mahuku, G.; Bänziger, M.; Torres, J.L.; Sánchez, C.; Yan, J. Molecular characterization of a diverse maize inbred line collection and its potential utilization for stress tolerance improvement. Crop. Sci. 2011, 51, 2569–2581. [Google Scholar] [CrossRef]
- Fu, J.; Cheng, Y.; Linghu, J.; Yang, X.; Kang, L.; Zhang, Z.; Zhang, J.; He, C.; Du, X.; Peng, Z.; et al. RNA sequencing reveals the complex regulatory network in the maize kernel. Nat. Commun. 2013, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Peng, Z.; Yang, X.; Wang, W.; Fu, J.; Wang, J.; Han, Y.; Chai, Y.; Guo, T.; Yang, N.; et al. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 2013, 45, 43–50. [Google Scholar] [CrossRef]
- Wen, W.; Li, D.; Li, X.; Gao, Y.; Li, W.; Li, H.; Liu, J.; Liu, H.; Chen, W.; Luo, J.; et al. Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Lu, Y.; Yang, X.; Huang, J.; Zhou, Y.; Ali, F.; Wen, W.; Liu, J.; Li, J.; Yan, J. Genome Wide Association Studies Using a New Nonparametric Model Reveal the Genetic Architecture of 17 Agronomic Traits in an Enlarged Maize Association Panel. PLoS Genet. 2014, 10, e1004573. [Google Scholar] [CrossRef] [Green Version]
- Van Dongen, S. Graph Clustering by Flow Simulation. Ph.D. Thesis, University of Utrecht, Utrecht, The Netherlands, 2000. [Google Scholar]
- Kel, A.E.; Gössling, E.; Reuter, I.; Cheremushkin, E.; Kel-Margoulis, O.V.; Wingender, E. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003, 31, 3576–3579. [Google Scholar] [CrossRef] [Green Version]
- Wingender, E. The TRANSFAC project as an example of framework technology that supports the analysis of genomic regulation. Brief. Bioinform. 2008, 9, 326–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Kursa, M.B.; Rudnicki, W.R. Feature selection with the Boruta package. J. Stat. Softw. 2010, 36, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Li, B.Q.; Hu, L.L.; Chen, L.; Feng, K.Y.; Cai, Y.D.; Chou, K.C. Prediction of Protein Domain with mRMR Feature Selection and Analysis. PLoS ONE 2012, 7, e39308. [Google Scholar] [CrossRef] [Green Version]
- Li, B.Q.; Feng, K.Y.; Chen, L.; Huang, T.; Cai, Y.D. Prediction of Protein-Protein Interaction Sites by Random Forest Algorithm with mRMR and IFS. PLoS ONE 2012, 7, e43927. [Google Scholar] [CrossRef]
- Weighill, D.; Jones, P.; Bleker, C.; Ranjan, P.; Shah, M.; Zhao, N.; Martin, M.; DiFazio, S.; Macaya-Sanz, D.; Schmutz, J.; et al. Multi-phenotype association decomposition: Unraveling complex gene-phenotype relationships. Front. Genet. 2019, 10, 417. [Google Scholar] [CrossRef]
- Ganal, M.W.; Durstewitz, G.; Polley, A.; Bérard, A.; Buckler, E.S.; Charcosset, A.; Clarke, J.D.; Graner, E.M.; Hansen, M.; Joets, J.; et al. A large maize (Zea mays L.) SNP genotyping array: Development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS ONE 2011, 6, e28334. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Chen, G.; Hermanson, P.J.; Xu, Q.; Sun, C.; Chen, W.; Kan, Q.; Li, M.; Crisp, P.A.; Yan, J.; et al. Population-level analysis reveals the widespread occurrence and phenotypic consequence of DNA methylation variation not tagged by genetic variation in maize. Genome Biol. 2019, 20, 1–16. [Google Scholar] [CrossRef]
- Zhao, H.; Sun, Z.; Wang, J.; Huang, H.; Kocher, J.P.; Wang, L. CrossMap: A versatile tool for coordinate conversion between genome assemblies. Bioinformatics 2014, 30, 1006–1007. [Google Scholar] [CrossRef]
- Sun, K. Ktrim: An extra-fast and accurate adapter-and quality-trimmer for sequencing data. Bioinformatics 2020, 36, 3561–3562. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Putri, G.H.; Anders, S.; Pyl, P.T.; Pimanda, J.E.; Zanini, F. Analysing high-throughput sequencing data in Python with HTSeq 2.0. arXiv 2021, arXiv:2112.00939. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klees, S.; Heinrich, F.; Schmitt, A.O.; Gültas, M. agReg-SNPdb: A Database of Regulatory SNPs for Agricultural Animal Species. Biology 2021, 10, 790. [Google Scholar] [CrossRef] [PubMed]
- Bloom, S.A. Similarity indices in community studies: Potential pitfalls. Mar. Ecol. Prog. Ser. 1981, 5, 125–128. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lucia, F.; Crevillen, P.; Jones, A.M.; Greb, T.; Dean, C. A PHD-polycomb repressive complex 2 triggers the epigenetic silencing of FLC during vernalization. Proc. Natl. Acad. Sci. USA 2008, 105, 16831–16836. [Google Scholar] [CrossRef] [Green Version]
- Mylne, J.; Greb, T.; Lister, C.; Dean, C. Epigenetic regulation in the control of flowering. In Proceedings of the Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2004; Volume 69, pp. 457–464. [Google Scholar]
- Berardini, T.Z.; Reiser, L.; Li, D.; Mezheritsky, Y.; Muller, R.; Strait, E.; Huala, E. The Arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 2015, 53, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Sung, S. Role of VIN3-LIKE 2 in facultative photoperiodic flowering response in Arabidopsis. Plant Signal. Behav. 2010, 5, 1672–1673. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Jiang, Z.; Zhang, K.; Yang, S.; He, F.; Zhang, Z. PlaD: A transcriptomics database for plant defense responses to pathogens, providing new insights into plant immune system. Genom. Proteom. Bioinform. 2018, 16, 283–293. [Google Scholar] [CrossRef]
- Stein, O.; Avin-Wittenberg, T.; Krahnert, I.; Zemach, H.; Bogol, V.; Daron, O.; Aloni, R.; Fernie, A.R.; Granot, D. Corrigendum: Arabidopsis fructokinases are important for seed oil accumulation and vascular development. Front. Plant Sci. 2017, 8, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Baudisch, B.; Klösgen, R.B. Dual targeting of a processing peptidase into both endosymbiotic organelles mediated by a transport signal of unusual architecture. Mol. Plant 2012, 5, 494–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Guan, X.; Garlock, R.; Nikolau, B.J. Mitochondrial Fatty Acid Synthase Utilizes Multiple Acyl Carrier Protein Isoforms1[OPEN]. Plant Physiol. 2020, 183, 547–557. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Gügel, I.L.; Giavalisco, P.; Zeisler, V.; Schreiber, L.; Soll, J.; Philippar, K. FAX1, a novel membrane protein mediating plastid fatty acid export. PLoS Biol. 2015, 13, e1002053. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Chandrasekar, B.; Rea, A.C.; Danhof, L.; Zemelis-Durfee, S.; Thrower, N.; Shepard, Z.S.; Pauly, M.; Brandizzi, F.; Keegstra, K. The synthesis of xyloglucan, an abundant plant cell wall polysaccharide, requires CSLC function. Proc. Natl. Acad. Sci. USA 2020, 117, 20316–20324. [Google Scholar] [CrossRef]
- Seebauer, J.R.; Moose, S.P.; Fabbri, B.J.; Crossland, L.D.; Below, F.E. Amino acid metabolism in maize earshoots. Implications for assimilate preconditioning and nitrogen signaling. Plant Physiol. 2004, 136, 4326–4334. [Google Scholar] [CrossRef] [Green Version]
- Gocal, G.F.; Sheldon, C.C.; Gubler, F.; Moritz, T.; Bagnall, D.J.; MacMillan, C.P.; Li, S.F.; Parish, R.W.; Dennis, E.S.; Weigel, D.; et al. GAMYB-like genes, flowering, and gibberellin signaling in Arabidopsis. Plant Physiol. 2001, 127, 1682–1693. [Google Scholar] [CrossRef]
- Woodger, F.J.; Millar, A.; Murray, F.; Jacobsen, J.V.; Gubler, F. The role of GAMYB transcription factors in GA-regulated gene expression. J. Plant Growth Regul. 2003, 22, 176–184. [Google Scholar] [CrossRef]
- Fang, Y.; Xie, K.; Hou, X.; Hu, H.; Xiong, L. Systematic analysis of GT factor family of rice reveals a novel subfamily involved in stress responses. Mol. Genet. Genom. 2010, 283, 157–169. [Google Scholar] [CrossRef]
- Hiratsuka, K.; Wu, X.; Fukuzawa, H.; Chua, N.H. Molecular dissection of GT-1 from Arabidopsis. Plant Cell 1994, 6, 1805–1813. [Google Scholar] [PubMed] [Green Version]
- Green, P.J.; Yong, M.H.; Cuozzo, M.; Kano-Murakami, Y.; Silverstein, P.; Chua, N. Binding site requirements for pea nuclear protein factor GT-1 correlate with sequences required for light-dependent transcriptional activation of the rbcS-3A gene. EMBO J. 1988, 7, 4035–4044. [Google Scholar] [CrossRef] [PubMed]
- Le Gourrierec, J.; Delaporte, V.; Ayadi, M.; Li, Y.F.; Zhou, D.X. Functional analysis of Arabidopsis transcription factor GT-1 in the expression of light-regulated genes. Genome Lett. 2002, 1, 77–82. [Google Scholar] [CrossRef]
- Cheng, H.; Qin, L.; Lee, S.; Fu, X.; Richards, D.E.; Cao, D.; Luo, D.; Harberd, N.P.; Peng, J. Gibberellin regulates Arabidopsis floral development via suppression of DELLA protein function. Development 2004, 131, 1055–1064. [Google Scholar] [CrossRef] [Green Version]
- Cone, K.C.; Cocciolone, S.M.; Burr, F.A.; Burr, B. Maize anthocyanin regulatory gene pl is a duplicate of c1 that functions in the plant. Plant Cell 1993, 5, 1795–1805. [Google Scholar]
- Caarls, L.; Van der Does, D.; Hickman, R.; Jansen, W.; Verk, M.C.V.; Proietti, S.; Lorenzo, O.; Solano, R.; Pieterse, C.M.; Van Wees, S. Assessing the role of ETHYLENE RESPONSE FACTOR transcriptional repressors in salicylic acid-mediated suppression of jasmonic acid-responsive genes. Plant Cell Physiol. 2017, 58, 266–278. [Google Scholar] [CrossRef]
- Yu, N.; Yang, J.C.; Yin, G.T.; Li, R.S.; Zou, W.T. Genome-wide characterization of the SPL gene family involved in the age development of Jatropha curcas. BMC Genom. 2020, 21, 68. [Google Scholar] [CrossRef]
- Jung, J.H.; Seo, P.J.; Kang, S.K.; Park, C.M. miR172 signals are incorporated into the miR156 signaling pathway at the SPL3/4/5 genes in Arabidopsis developmental transitions. Plant Mol. Biol. 2011, 76, 35–45. [Google Scholar] [CrossRef]
- Jung, J.H.; Lee, H.J.; Ryu, J.Y.; Park, C.M. SPL3/4/5 integrate developmental aging and photoperiodic signals into the FT-FD module in Arabidopsis flowering. Mol. Plant 2016, 9, 1647–1659. [Google Scholar] [CrossRef] [Green Version]
- Cardon, G.; Höhmann, S.; Klein, J.; Nettesheim, K.; Saedler, H.; Huijser, P. Molecular characterisation of the Arabidopsis SBP-box genes. Gene 1999, 237, 91–104. [Google Scholar] [CrossRef]
- Chao, L.M.; Liu, Y.Q.; Chen, D.Y.; Xue, X.Y.; Mao, Y.B.; Chen, X.Y. Arabidopsis transcription factors SPL1 and SPL12 confer plant thermotolerance at reproductive stage. Mol. Plant 2017, 10, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Ohta, M.; Matsui, K.; Hiratsu, K.; Shinshi, H.; Ohme-Takagi, M. Repression domains of class II ERF transcriptional repressors share an essential motif for active repression. Plant Cell 2001, 13, 1959–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés, A.J.; López-Hernández, F. Harnessing crop wild diversity for climate change adaptation. Genes 2021, 12, 783. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Escudero, M.; Osorio, A.N.; Cortés, A.J. Integrative pre-breeding for biotic resistance in forest trees. Plants 2021, 10, 2022. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, H.H.; Wang, X. Machine learning for big data analytics in plants. Trends Plant Sci. 2014, 19, 798–808. [Google Scholar] [CrossRef]
- Cortés, A.J.; Restrepo-Montoya, M.; Bedoya-Canas, L.E. Modern strategies to assess and breed forest tree adaptation to changing climate. Front. Plant Sci. 2020, 11, 1606. [Google Scholar] [CrossRef]
- Tong, H.; Nikoloski, Z. Machine learning approaches for crop improvement: Leveraging phenotypic and genotypic big data. J. Plant Physiol. 2021, 257, 153354. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | Max | #rSNPs |
|---|---|---|
| Leaf number above ear | 0.490740 | 89 |
| Ear leaf width | 0.484029 | 70 |
| Cob diameter | 0.445720 | 64 |
| Ear height | 0.509523 | 109 |
| Kernel width | 0.418115 | 172 |
| Ear leaf length | 0.553292 | 112 |
| Tassel main axis length | 0.498562 | 96 |
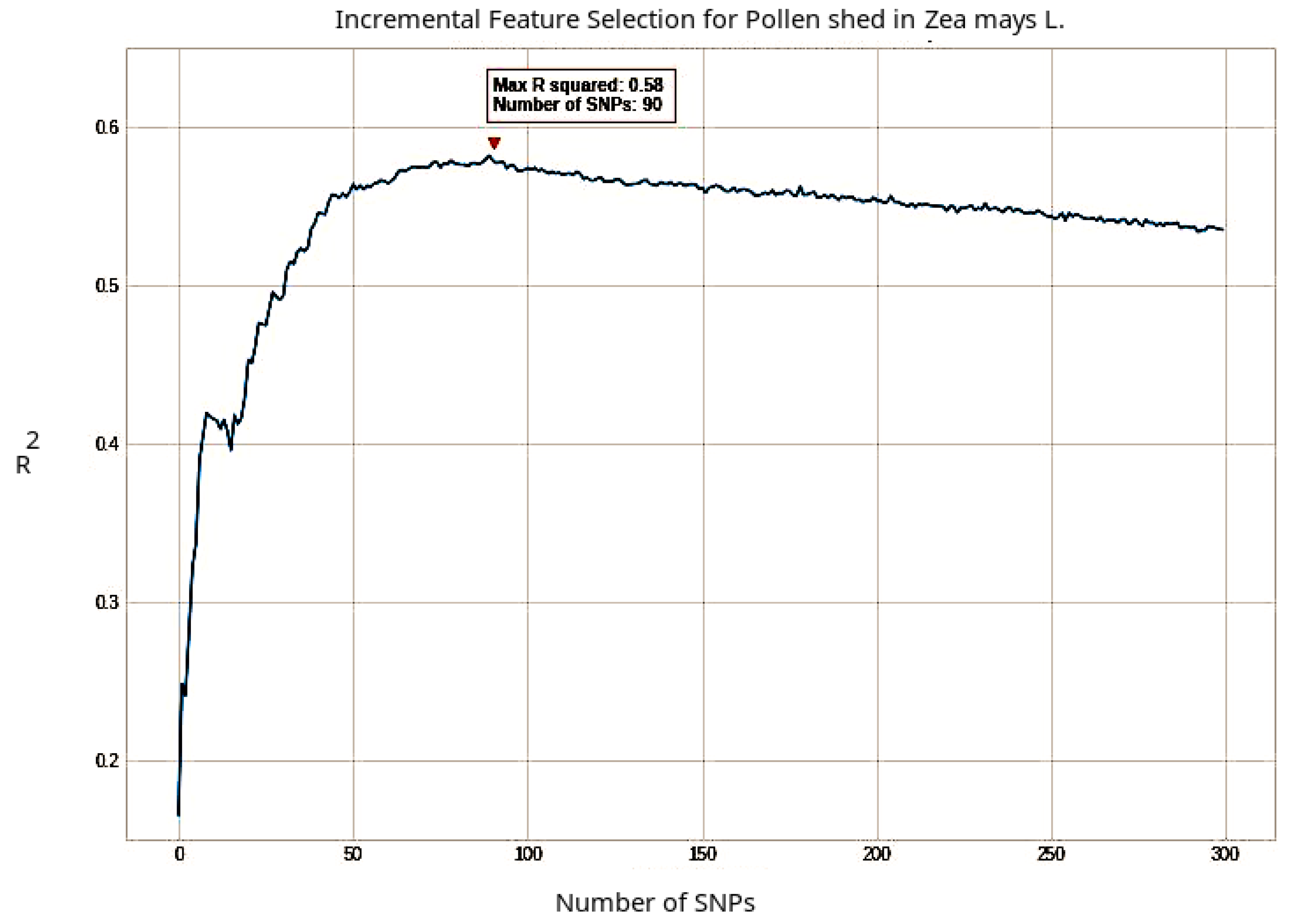
| Pollen shed | 0.581765 | 90 |
| Heading date | 0.537987 | 49 |
| Ear length | 0.434011 | 82 |
| Silking time | 0.506520 | 122 |
| Ear diameter | 0.481445 | 110 |
| Cob weight | 0.460850 | 37 |
| X100 grain weight | 0.389332 | 51 |
| Tassel branch number | 0.507112 | 142 |
| Ear row number | 0.491663 | 46 |
| Kernel number per row | 0.350717 | 27 |
| Plant height | 0.532837 | 72 |
| kernel length | 0.580691 | 168 |
| Kernel thickness | 0.437589 | 64 |
| Cluster | Numbers of Pleiotropic | Phenotypes | |
|---|---|---|---|
| rSNPs | Genes | ||
| Cluster-1 | 15 | 10 | Heading date, Pollen shed, Silking time and Ear height |
| Cluster-2 | 9 | 7 | Cob weight, Heading date, Pollen shed, Tassel main axis length, Ear leaf length, Plant height, Ear leaf width, Ear row number and Ear height |
| Cluster-3 | 7 | 6 | Kernel length, Kernel thickness, Kernel number per row, Ear diameter and X100 grain weight |
| Cluster-4 | 6 | 5 | Ear diameter, Cob diameter and Ear row number |
| Cluster-5 | 6 | 4 | Heading date, Pollen shed, Silking time, Ear height and Tassel branch number |
| Cluster-6 | 5 | 3 | Ear diameter, Cob diameter and Ear row number |
| Cluster-7 | 3 | 3 | Ear height, Plant height and Ear row number |
| Cluster-8 | 3 | 3 | Kernel width, Kernel length, Kernel thickness and X100 grain weight |
| Cluster-9 | 2 | 2 | Ear leaf length, Leaf number above ear, Kernel length |
| Cluster-10 | 2 | 2 | Ear length and Kernel number per row |
| Cluster-11 | 2 | 2 | Ear diameter, Tassel main axis length and Cob weight |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haleem, A.; Klees, S.; Schmitt, A.O.; Gültas, M. Deciphering Pleiotropic Signatures of Regulatory SNPs in Zea mays L. Using Multi-Omics Data and Machine Learning Algorithms. Int. J. Mol. Sci. 2022, 23, 5121. https://doi.org/10.3390/ijms23095121
Haleem A, Klees S, Schmitt AO, Gültas M. Deciphering Pleiotropic Signatures of Regulatory SNPs in Zea mays L. Using Multi-Omics Data and Machine Learning Algorithms. International Journal of Molecular Sciences. 2022; 23(9):5121. https://doi.org/10.3390/ijms23095121
Chicago/Turabian StyleHaleem, Ataul, Selina Klees, Armin Otto Schmitt, and Mehmet Gültas. 2022. "Deciphering Pleiotropic Signatures of Regulatory SNPs in Zea mays L. Using Multi-Omics Data and Machine Learning Algorithms" International Journal of Molecular Sciences 23, no. 9: 5121. https://doi.org/10.3390/ijms23095121
APA StyleHaleem, A., Klees, S., Schmitt, A. O., & Gültas, M. (2022). Deciphering Pleiotropic Signatures of Regulatory SNPs in Zea mays L. Using Multi-Omics Data and Machine Learning Algorithms. International Journal of Molecular Sciences, 23(9), 5121. https://doi.org/10.3390/ijms23095121