
Angiotensin II Promotes SARS-CoV-2 Infection via Upregulation of ACE2 in Human Bronchial Cells

 , , , , ,
, , , , ,  , ,
, ,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. ACE2 and AGTR1 Expression in HUVEC and Calu-3 Cells
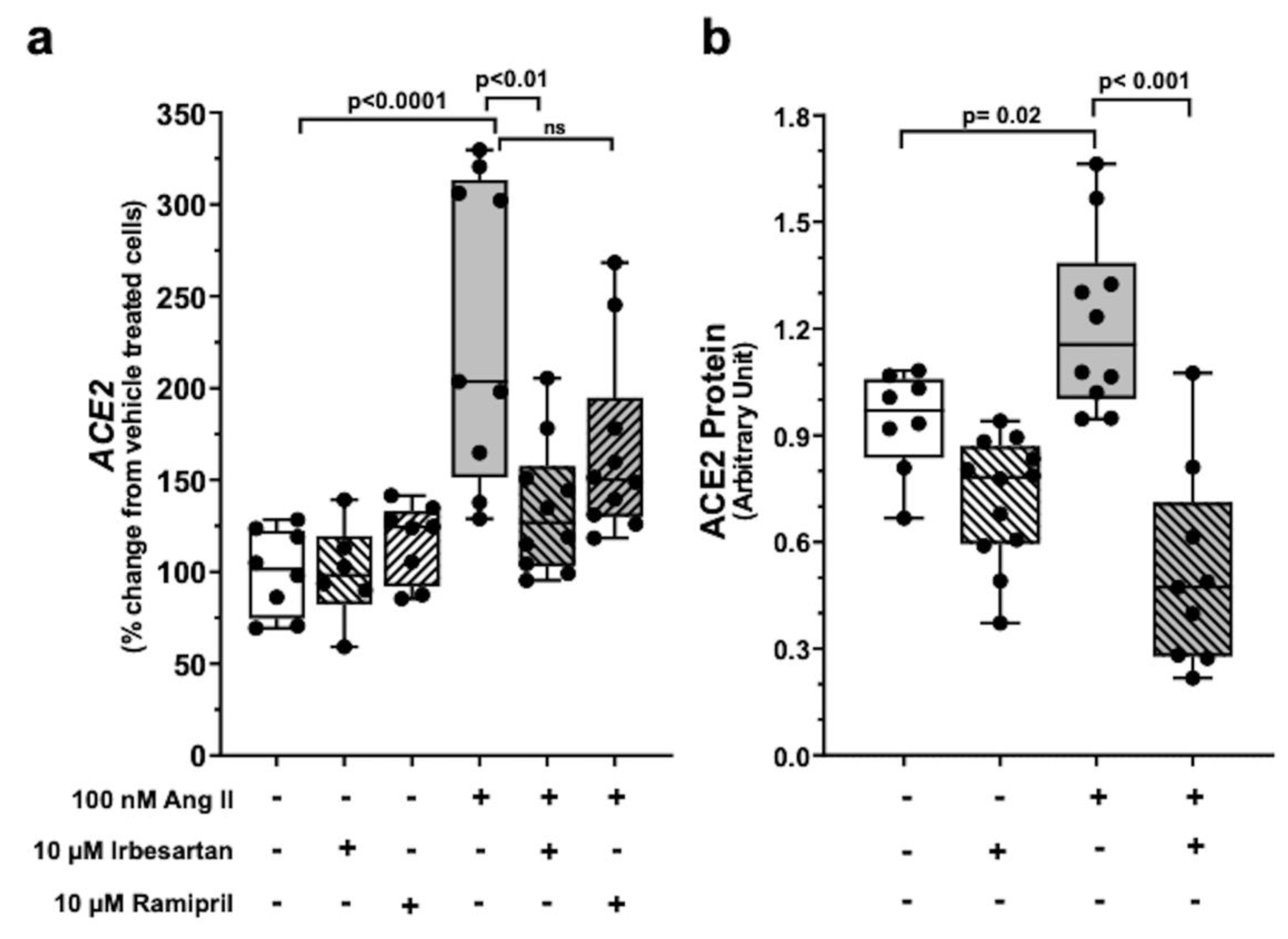
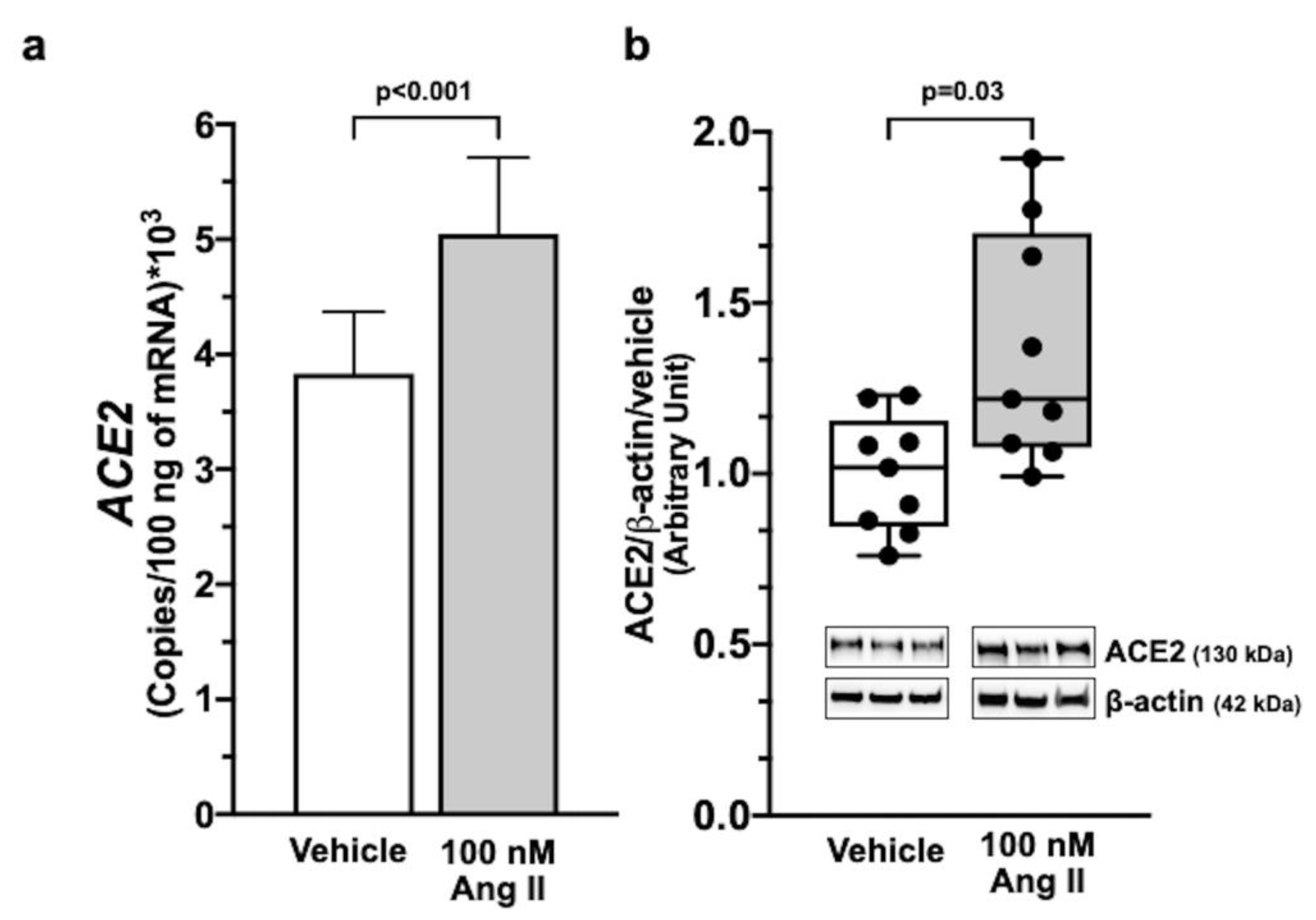
2.2. Ang II Upregulates ACE2 Gene Expression via AT1R Signalling
2.3. Exclusion of Ang 1-7-Mediated Effect on ACE2 in Calu-3 cells
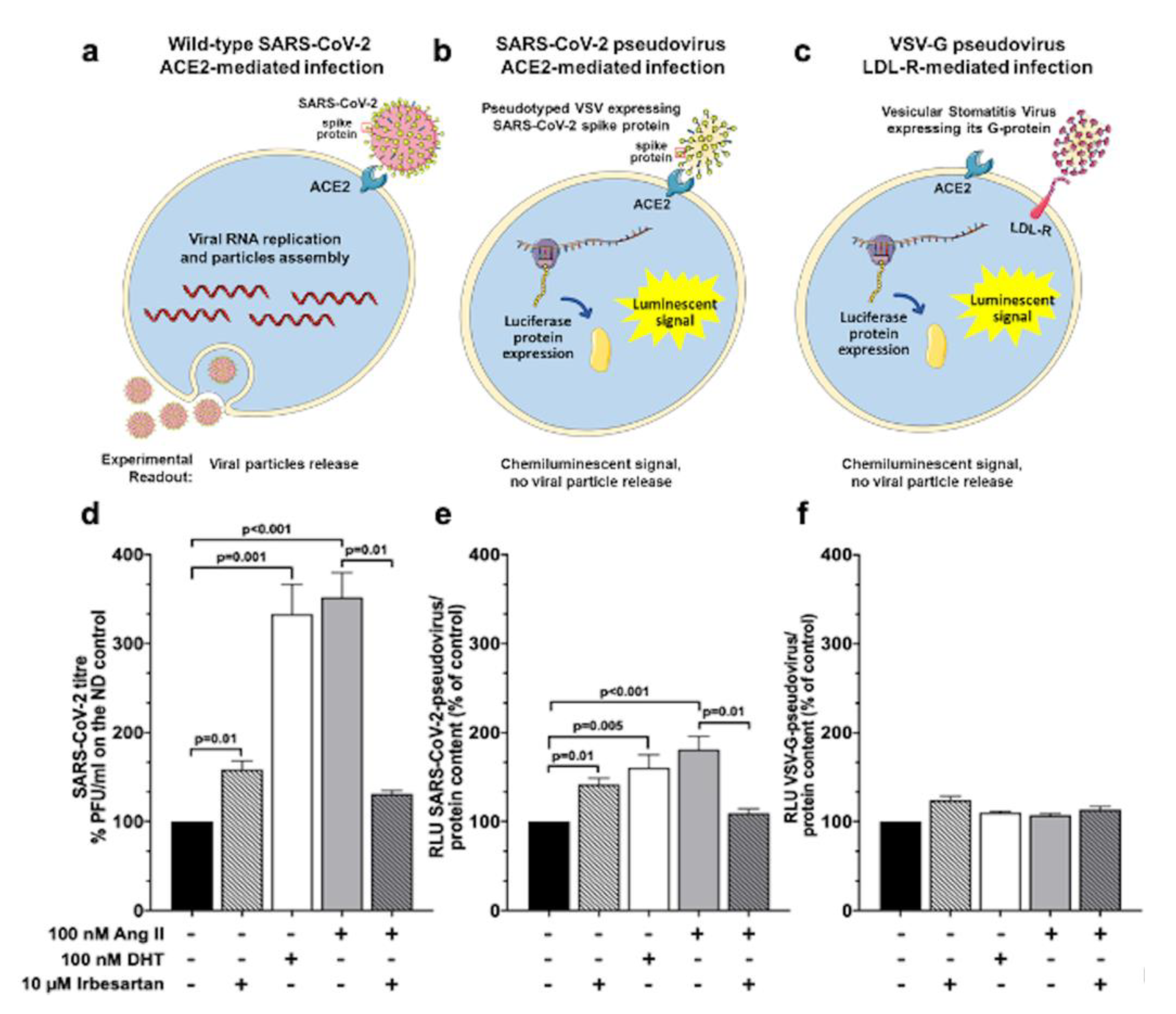
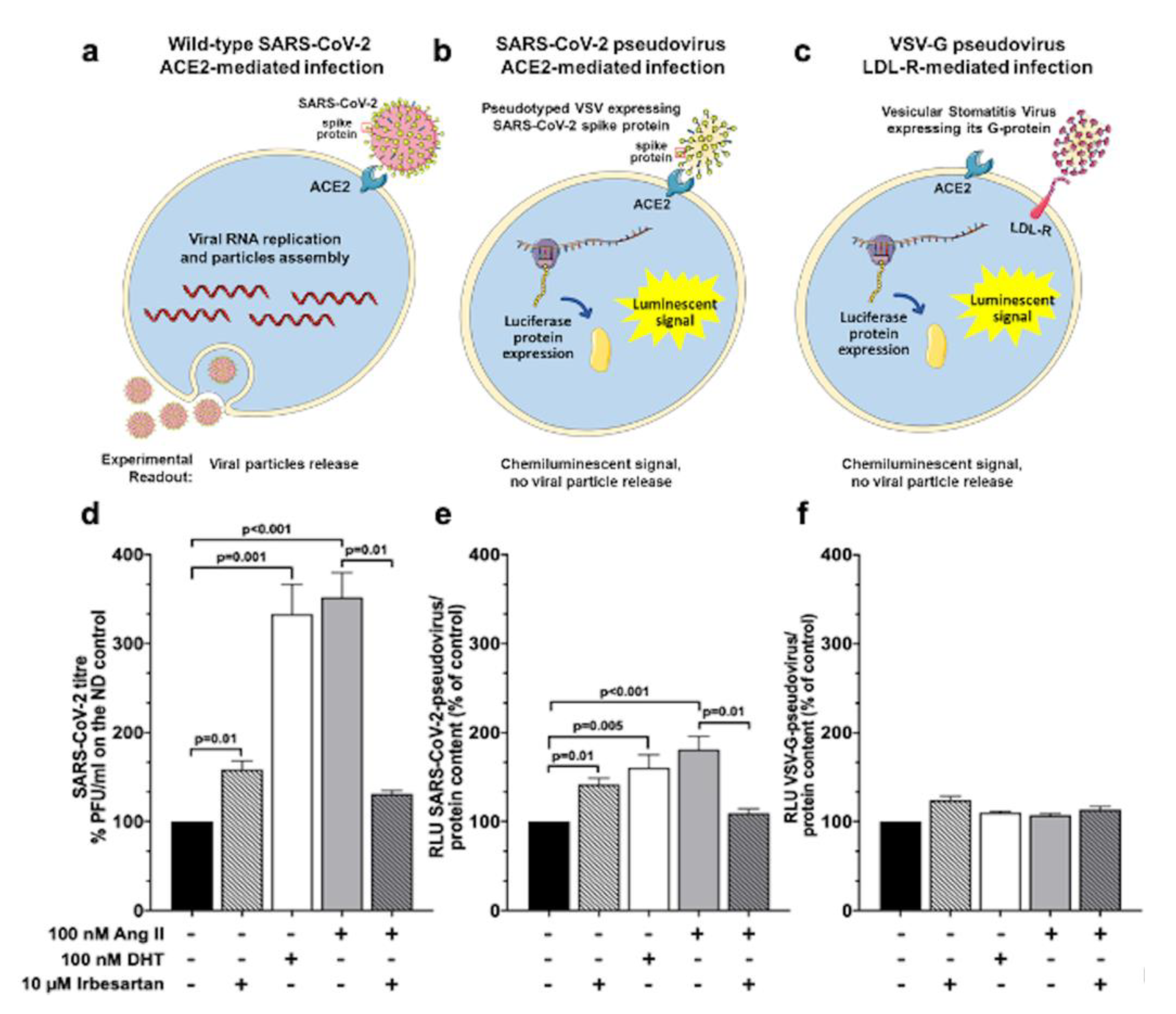
2.4. Ang II and Irbesartan Modulate SARS-CoV-2 Infection in Human Lung Cells
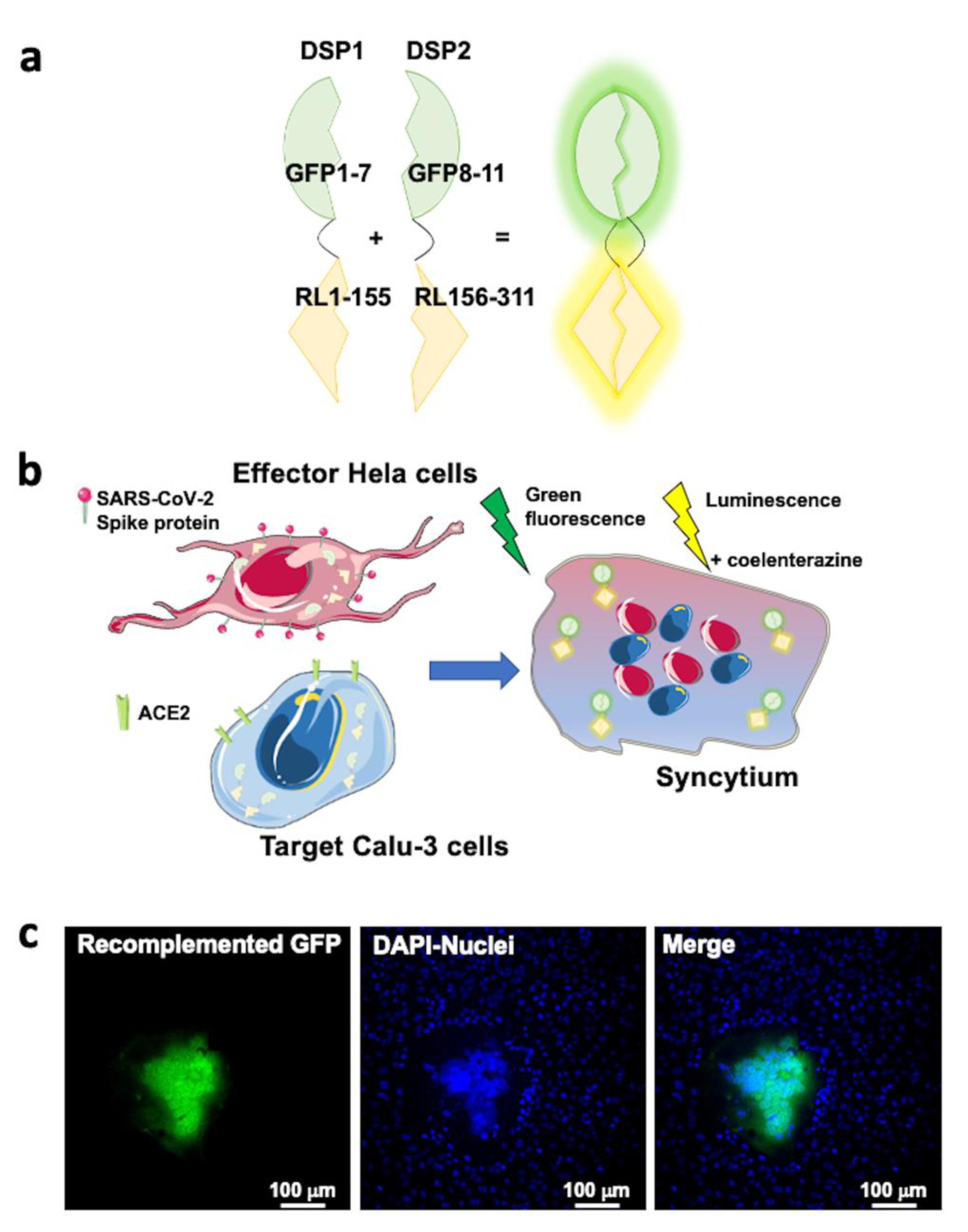
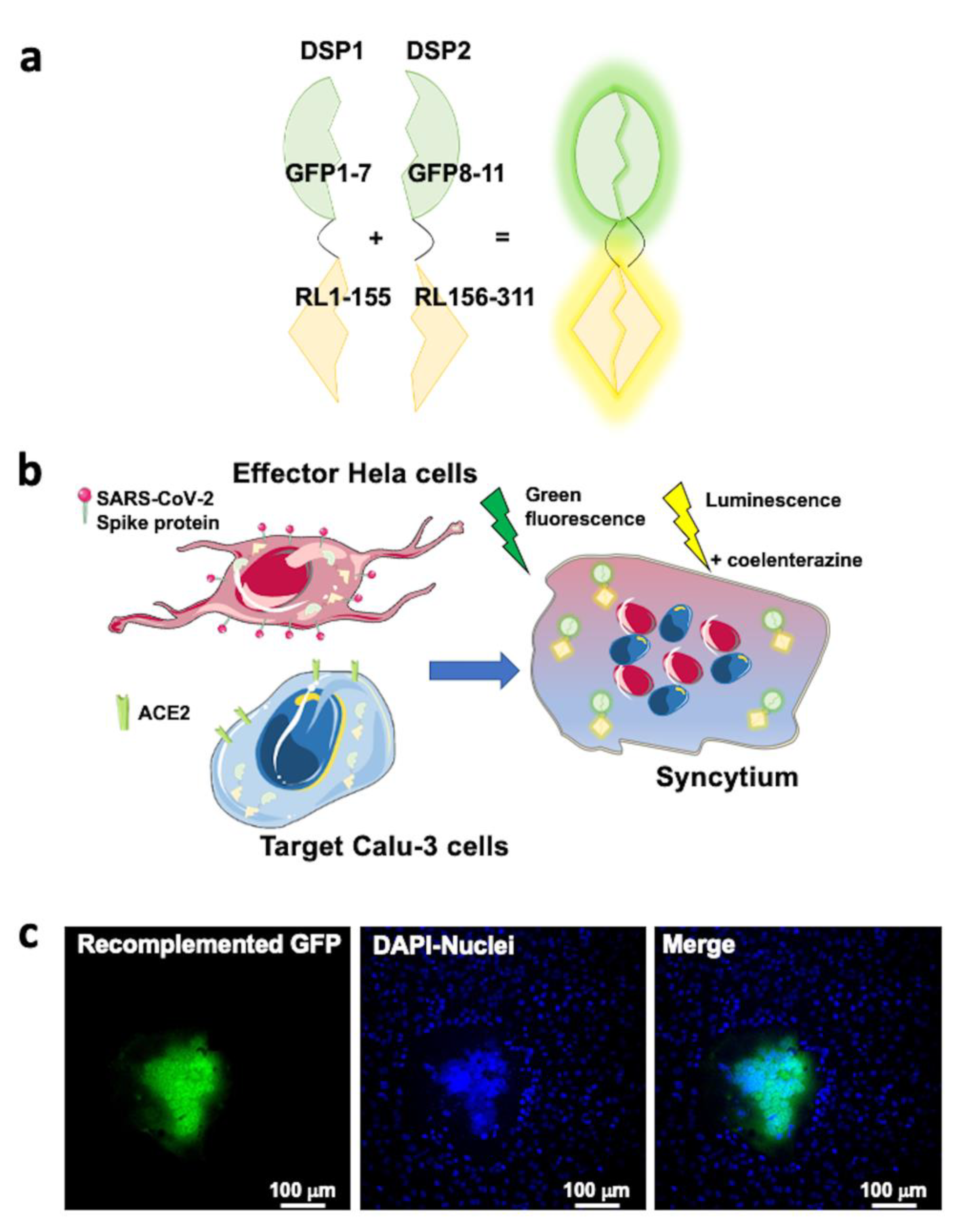
2.5. SARS-CoV-2 Entry in Human Lung Cells Involves ACE2, TMPRSS2, and Spike Protein
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Cell Treatments
4.3. Gene Expression Analysis
4.4. Immunoblotting
4.5. Pseudotyping of Vesicular Stomatitis Virus
4.6. Infection with SARS-CoV-2 Pseudovirus
4.7. Generation of SARS-CoV-2 Viral Stock
4.8. Infection with SARS-CoV-2 Virus
4.9. DSP1 and DSP2 Expression Plasmids
4.10. Generation of DSP1 and DSP2 Expressing Cells
4.11. Cell–Cell Fusion Assay
4.12. Statitistical Analysis and Softwares
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Mü, M.A.; Drosten, C.; Pö, S.; Krü, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Burrell, L.M.; Risvanis, J.; Kubota, E.; Dean, R.G.; MacDonald, P.S.; Lu, S.; Tikellis, C.; Grant, S.L.; Lew, R.A.; Smith, A.I.; et al. Myocardial Infarction Increases ACE2 Expression in Rat and Humans. Eur. Heart J. 2005, 26, 369–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiyama, Y.; Gallagher, P.E.; Averill, D.B.; Tallant, E.A.; Brosnihan, K.B.; Ferrario, C.M. Upregulation of Angiotensin-Converting Enzyme 2 after Myocardial Infarction by Blockade of Angiotensin II Receptors. Hypertension 2004, 43, 970–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, Y.; Zhu, A.; Yoneda, T.; Usukura, M.; Takata, H.; Yamagishi, M. Effects of Aldosterone and Angiotensin II Receptor Blockade on Cardiac Angiotensinogen and Angiotensin-Converting Enzyme 2 Expression in Dahl Salt-Sensitive Hypertensive Rats. Am. J. Hypertens. 2007, 20, 1119–1124. [Google Scholar] [CrossRef]
- Igase, M.; Strawn, W.B.; Gallagher, P.E.; Geary, R.L.; Ferrario, C.M. Angiotensin II At1 Receptors Regulate ACE2 and Angiotensin-(1-7) Expression in the Aorta of Spontaneously Hypertensive Rats. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1013–H1019. [Google Scholar] [CrossRef]
- Fang, L.; Karakiulakis, G.; Roth, M. Are Patients with Hypertension and Diabetes Mellitus at Increased Risk for COVID-19 Infection? Lancet. Respir. Med. 2020, 2600, 30116. [Google Scholar] [CrossRef]
- Zheng, Y.-Y.; Ma, Y.-T.; Zhang, J.-Y.; Xie, X. COVID-19 and the Cardiovascular System. Nat. Rev. Cardiol. 2020, 17, 259–260. [Google Scholar] [CrossRef] [Green Version]
- Koka, V.; Xiao, R.H.; Chung, A.C.K.; Wang, W.; Truong, L.D.; Lan, H.Y. Angiotensin II Up-Regulates Angiotensin I-Converting Enzyme (ACE), but down-Regulates ACE2 via the AT1-ERK/P38 MAP Kinase Pathway. Am. J. Pathol. 2008, 172, 1174–1183. [Google Scholar] [CrossRef] [Green Version]
- Caldeira, D.; Alarcão, J.; Vaz-Carneiro, A.; Costa, J. Risk of Pneumonia Associated with Use of Angiotensin Converting Enzyme Inhibitors and Angiotensin Receptor Blockers: Systematic Review and Meta-Analysis. BMJ 2012, 345, e4260. [Google Scholar] [CrossRef] [Green Version]
- Mentz, R.J.; Bakris, G.L.; Waeber, B.; McMurray, J.J.V.; Gheorghiade, M.; Ruilope, L.M.; Maggioni, A.P.; Swedberg, K.; Piña, I.L.; Fiuzat, M.; et al. The Past, Present and Future of Renin-Angiotensin Aldosterone System Inhibition. Int. J. Cardiol. 2013, 167, 1677–1687. [Google Scholar] [CrossRef] [Green Version]
- Mancia, G.; Rea, F.; Ph, D.; Ludergnani, M.; Sc, M.; Apolone, G.; Corrao, G.; Ph, D. Renin–Angiotensin–Aldosterone System Blockers and the Risk of Covid-19. N. Engl. J. Med. 2020, 382, 2431–2440. [Google Scholar] [CrossRef]
- Guo, X.; Zhu, Y.; Hong, Y. Decreased Mortality of COVID-19 With Renin-Angiotensin- Aldosterone System Inhibitors Therapy in Patients with Hypertension A Meta-Analysis. Hypertension 2020, 76, e13–e14. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Lin, C.C.; Lau, C.I.; Chang, A.; Kao, C.H. Angiotensin-Converting Enzyme Inhibitors and Bacterial Pneumonia in Patients with Parkinson Disease. Mov. Disord. 2015, 30, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, E.M.; Pugh, M.J.; Copeland, L.A.; Restrepo, M.I.; Cornell, J.E.; Anzueto, A.; Pugh, J.A. Impact of Statins and Angiotensin-Converting Enzyme Inhibitors on Mortality of Subjects Hospitalised with Pneumonia. Eur. Respir. J. 2008, 31, 611–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.L.; Shau, W.Y.; Wu, C.S.; Lai, M.S. Angiotensin-Converting Enzyme Inhibitor/Angiotensin II Receptor Blockers and Pneumonia Risk among Stroke Patients. J. Hypertens. 2012, 30, 2223–2229. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-Converting Enzyme 2 Protects from Severe Acute Lung Failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Unger, T.U.; Muscha, S.; Souza dos Santos, R.A. The Protective Arm of the Renin Angiotensin System (RAS); Academic Press: Cambridge, MA, USA, 2015; ISBN 9780128013649. [Google Scholar]
- Rossi, G.P.; Sanga, V.; Barton, M. Potential Harmful Effects of Discontinuing ACE-Inhibitors and ARBs in COVID-19 Patients. eLife 2020, 9, e57278. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.B.; Hanff, T.C.; William, P.; Sweitzer, N.; Rosado-Santander, N.R.; Medina, C.; Rodriguez-Mori, J.E.; Renna, N.; Chang, T.I.; Corrales-Medina, V.; et al. Continuation versus Discontinuation of Renin–Angiotensin System Inhibitors in Patients Admitted to Hospital with COVID-19: A Prospective, Randomised, Open-Label Trial. Lancet Respir. Med. 2021, 9, 275–284. [Google Scholar] [CrossRef]
- Lopes, R.D.; Macedo, A.V.S.; De Barros, E.; Silva, P.G.M.; Moll-Bernardes, R.J.; Dos Santos, T.M.; Mazza, L.; Feldman, A.; D’Andréa Saba Arruda, G.; De Albuquerque, D.C.; et al. Effect of Discontinuing vs Continuing Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers on Days Alive and out of the Hospital in Patients Admitted with COVID-19: A Randomized Clinical Trial. JAMA 2021, 325, 254–264. [Google Scholar] [CrossRef]
- Leach, D.A.; Mohr, A.; Giotis, E.S.; Cil, E.; Isac, A.M.; Yates, L.L.; Barclay, W.S.; Zwacka, R.M.; Bevan, C.L.; Brooke, G.N. The Antiandrogen Enzalutamide Downregulates TMPRSS2 and Reduces Cellular Entry of SARS-CoV-2 in Human Lung Cells. Nat. Commun. 2021, 12, 4068. [Google Scholar] [CrossRef]
- Kim, I.S.; Jenni, S.; Stanifer, M.L.; Roth, E.; Whelan, S.P.J.; Van Oijen, A.M.; Harrison, S.C. Mechanism of Membrane Fusion Induced by Vesicular Stomatitis Virus G Protein. Proc. Natl. Acad. Sci. USA 2017, 114, E28–E36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolic, J.; Belot, L.; Raux, H.; Legrand, P.; Gaudin, Y.; Albertini, A.A. Structural Basis for the Recognition of LDL-Receptor Family Members by VSV Glycoprotein. Nat. Commun. 2018, 9, 1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, J.D.; McCready, T.; Lopez-Jaramillo, P.; Yusoff, K.; Attaran, A.; Lamelas, P.; Camacho, P.A.; Majid, F.; Bangdiwala, S.I.; Thabane, L.; et al. A Community-Based Comprehensive Intervention to Reduce Cardiovascular Risk in Hypertension (HOPE 4): A Cluster-Randomised Controlled Trial. Lancet 2019, 394, 1231–1242. [Google Scholar] [CrossRef]
- Wyler, E.; Mösbauer, K.; Franke, V.; Diag, A.; Gottula, L.T.; Arsiè, R.; Klironomos, F.; Koppstein, D.; Hönzke, K.; Ayoub, S.; et al. Transcriptomic Profiling of SARS-CoV-2 Infected Human Cell Lines Identifies HSP90 as Target for COVID-19 Therapy. iScience 2021, 24, 102151. [Google Scholar] [CrossRef]
- Lamers, M.; Mykytyn, A.; Breugem, T.; Wang, Y.; Wu, D.; Riesebosch, S.; van den Doel, P.; Schipper, D.; Bestebroer, T.; Wu, N.; et al. Human Airway Cells Prevent SARS-CoV-2 Multibasic Cleavage Site Cell Culture Adaptation. eLife 2021, 10, e66815. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, J.; Schiavon, C.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef]
- Wu, Z.; Hu, R.; Zhang, C.; Ren, W.; Yu, A.; Zhou, X. Elevation of Plasma Angiotensin II Level Is a Potential Pathogenesis for the Critically Ill COVID-19 Patients. Crit. Care 2020, 24, 290. [Google Scholar] [CrossRef]
- Bauer, A.; Schreinlechner, M.; Sappler, N.; Dolejsi, T.; Tilg, H.; Aulinger, B.A.; Weiss, G.; Bellmann-Weiler, R.; Adolf, C.; Wolf, D.; et al. Discontinuation versus Continuation of Renin-Angiotensin-System Inhibitors in COVID-19 (ACEI-COVID): A Prospective, Parallel Group, Randomised, Controlled, Open-Label Trial. Lancet Respir. Med. 2021, 9, 863–872. [Google Scholar] [CrossRef]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury with Mortality in Hospitalized Patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020, 5, 802–810. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotfis, K.; Lechowicz, K.; Drożdżal, S.; Niedźwiedzka-Rystwej, P.; Wojdacz, T.K.; Grywalska, E.; Biernawska, J.; Wiśniewska, M.; Parczewski, M. COVID-19—The Potential Beneficial Therapeutic Effects of Spironolactone during SARS-CoV-2 Infection. Pharmaceuticals 2021, 14, 71. [Google Scholar] [CrossRef] [PubMed]
- Tai, C.J.; Li, C.L.; Tai, C.J.; Wang, C.K.; Lin, L.T. Early Viral Entry Assays for the Identification and Evaluation of Antiviral Compounds. J. Vis. Exp. 2015, 105, e53124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, N.; Gullberg, M.; Israelsson, S.; Lindberg, A.M. A Rapid and Efficient Method for Studies of Virus Interaction at the Host Cell Surface Using Enteroviruses and Real-Time PCR. Virol. J. 2009, 6, 217. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Meyerholz, D.K.; Bartlett, J.A.; McCray, P.B. The Tmprss2 Inhibitor Nafamostat Reduces Sars-Cov-2 Pulmonary Infection in Mouse Models of Covid-19. MBio 2021, 12, e0097021. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kiso, M.; Sakai-Tagawa, Y.; Iwatsuki-Horimoto, K.; Imai, M.; Takeda, M.; Kinoshita, N.; Ohmagari, N.; Gohda, J.; Semba, K.; et al. The Anticoagulant Nafamostat Potently Inhibits SARS-CoV-2 S Protein-Mediated Fusion in a Cell Fusion Assay System and Viral Infection in Vitro in a Cell-Type-Dependent Manner. Viruses 2020, 12, 629. [Google Scholar] [CrossRef]
- Hoffmann, M.; Schoreder, S.; Kleine-Weber, H.; Muller, M.A.; Drosten, C.; Pohlmann, S. Nafamostat Mesylate Blocks Activation of SARS-CoV-2: New Treatment Option for COVID-19. Antimicrob. Agents Chemother. 2020, 64, e00754-20. [Google Scholar] [CrossRef] [Green Version]
- Cegolon, L.; Mirandola, M.; Salaris, C.; Salvati, M.V.; Mastrangelo, G.; Salata, C. Hypothiocyanite and Hypothiocyanite/Lactoferrin Mixture Exhibit Virucidal Activity in Vitro against Sars-Cov-2. Pathogens 2021, 10, 233. [Google Scholar] [CrossRef]
- Whitt, M.A. Generation of VSV Pseudotypes Using Recombinant ΔG-VSV for Studies on Virus Entry, Identification of Entry Inhibitors, and Immune Responses to Vaccines. J. Virol. Methods 2011, 169, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Rentsch, M.B.; Zimmer, G. A Vesicular Stomatitis Virus Replicon-Based Bioassay for the Rapid and Sensitive Determination of Multi-Species Type I Interferon. PLoS ONE 2011, 6, e25858. [Google Scholar] [CrossRef] [Green Version]
- Baer, A.; Kehn-Hall, K. Viral Concentration Determination through Plaque Assays: Using Traditional and Novel Overlay Systems. J. Vis. Exp. 2014, e52065. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Meng, F.; Kondo, N.; Iwamoto, A.; Matsuda, Z. Generation of a Dual-Functional Split-Reporter Protein for Monitoring Membrane Fusion Using Self-Associating Split GFP. Protein Eng. Des. Sel. 2012, 25, 813–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottolini, D.; Calì, T.; Brini, M. Methods to Measure Intracellular Ca2+ Fluxes with Organelle-Targeted Aequorin-Based Probes. Methods Enzymol. 2014, 543, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Miyauchi, K.; Meng, F.; Iwamoto, A.; Matsuda, Z. Conformational Changes of the HIV-1 Envelope Protein during Membrane Fusion Are Inhibited by the Replacement of Its Membrane-Spanning Domain. J. Biol. Chem. 2010, 285, 14681–14688. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caputo, I.; Caroccia, B.; Frasson, I.; Poggio, E.; Zamberlan, S.; Morpurgo, M.; Seccia, T.M.; Calì, T.; Brini, M.; Richter, S.N.; et al. Angiotensin II Promotes SARS-CoV-2 Infection via Upregulation of ACE2 in Human Bronchial Cells. Int. J. Mol. Sci. 2022, 23, 5125. https://doi.org/10.3390/ijms23095125
Caputo I, Caroccia B, Frasson I, Poggio E, Zamberlan S, Morpurgo M, Seccia TM, Calì T, Brini M, Richter SN, et al. Angiotensin II Promotes SARS-CoV-2 Infection via Upregulation of ACE2 in Human Bronchial Cells. International Journal of Molecular Sciences. 2022; 23(9):5125. https://doi.org/10.3390/ijms23095125
Chicago/Turabian StyleCaputo, Ilaria, Brasilina Caroccia, Ilaria Frasson, Elena Poggio, Stefania Zamberlan, Margherita Morpurgo, Teresa M. Seccia, Tito Calì, Marisa Brini, Sara N. Richter, and et al. 2022. "Angiotensin II Promotes SARS-CoV-2 Infection via Upregulation of ACE2 in Human Bronchial Cells" International Journal of Molecular Sciences 23, no. 9: 5125. https://doi.org/10.3390/ijms23095125
APA StyleCaputo, I., Caroccia, B., Frasson, I., Poggio, E., Zamberlan, S., Morpurgo, M., Seccia, T. M., Calì, T., Brini, M., Richter, S. N., & Rossi, G. P. (2022). Angiotensin II Promotes SARS-CoV-2 Infection via Upregulation of ACE2 in Human Bronchial Cells. International Journal of Molecular Sciences, 23(9), 5125. https://doi.org/10.3390/ijms23095125