
Molecular Dynamics of DHHC20 Acyltransferase Suggests Principles of Lipid and Protein Substrate Selectivity

 , ,
, ,  , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Design of the Study
- Acyl-CoA (C12–C20) in the model membrane is the first DHHC substrate required for the autoacylation step.
- Free DHHC20 enzyme and two of its mutants (S29F and Y181A) describe the protein in the unbound state, prior to the auto- and transacylation reactions. These calculations provide information on the dynamics of the protein and its binding cavity when no substrate disturbs its interior.
- 3.
- Within its inner cavity DHHC20 (and the mutants) accommodates acyl-β-mercapto-ethylamine (MEA), an acyl-CoA analogue lacking a large 3′-phosphorylated ADP, without the formation of a chemical bond. This state corresponds to the pre-acylation step, when the substrate analogue has already entered DHHC’s cavity, but has not yet reacted and is thus free to move.
- 4.
- Acylated DHHC20 (and the mutants) models the autoacylation product, which is already activated for the transacylation of the substrate protein. In this case, DHHC20 topology was modified to form a chemical bond between the cysteine’s sulfur atom and the fatty acid’s Cα-atom, which restricts the movement of the fatty acid residue.
- 5.
- We performed about 15 in silico mutations to discover novel amino acid substitutions that shift hDHHC20 selectivity towards C18 (Table S1).
2.2. DHHC Avoids a Vacuum: The Central Cavity Is Mostly Occupied
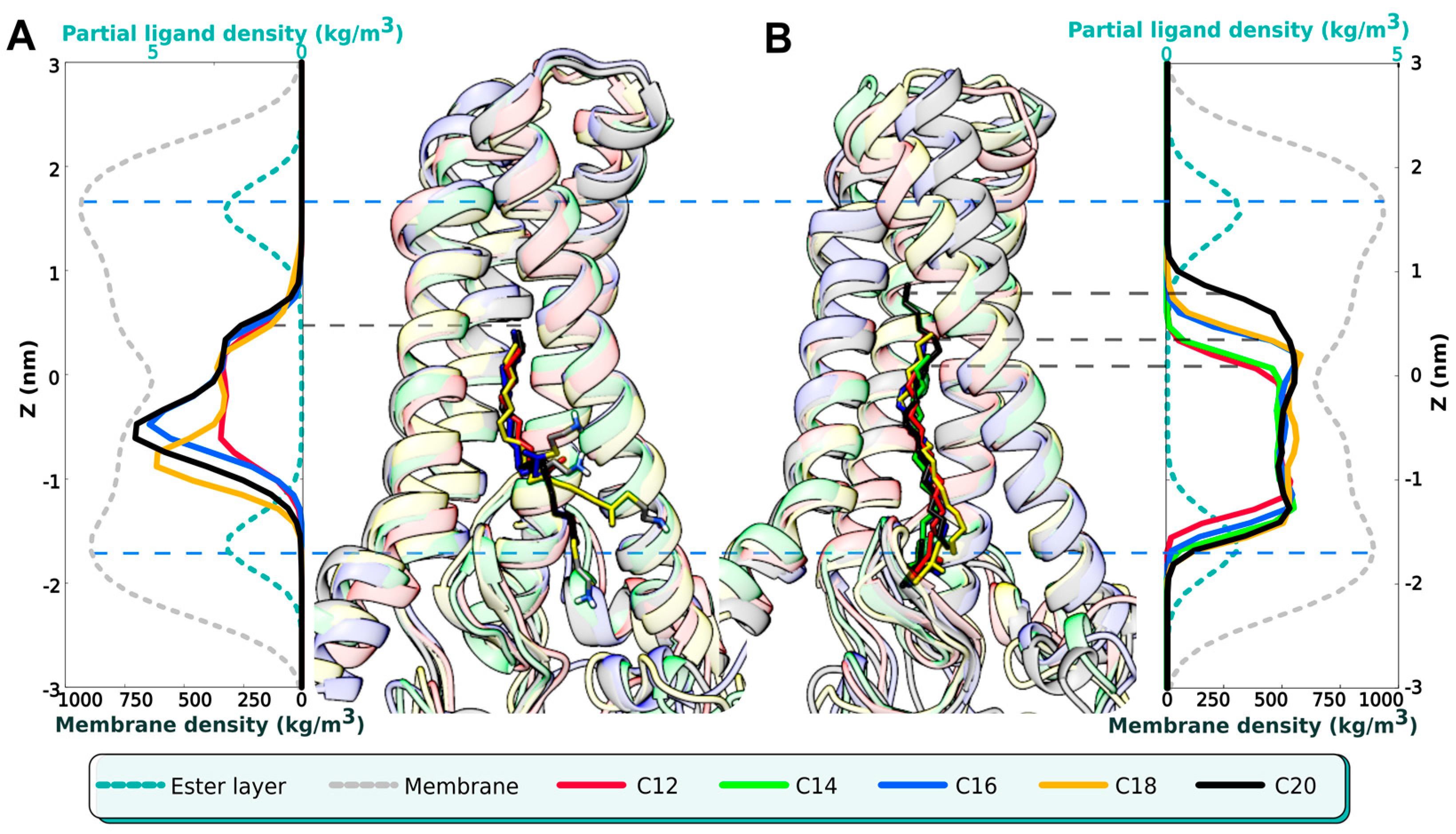
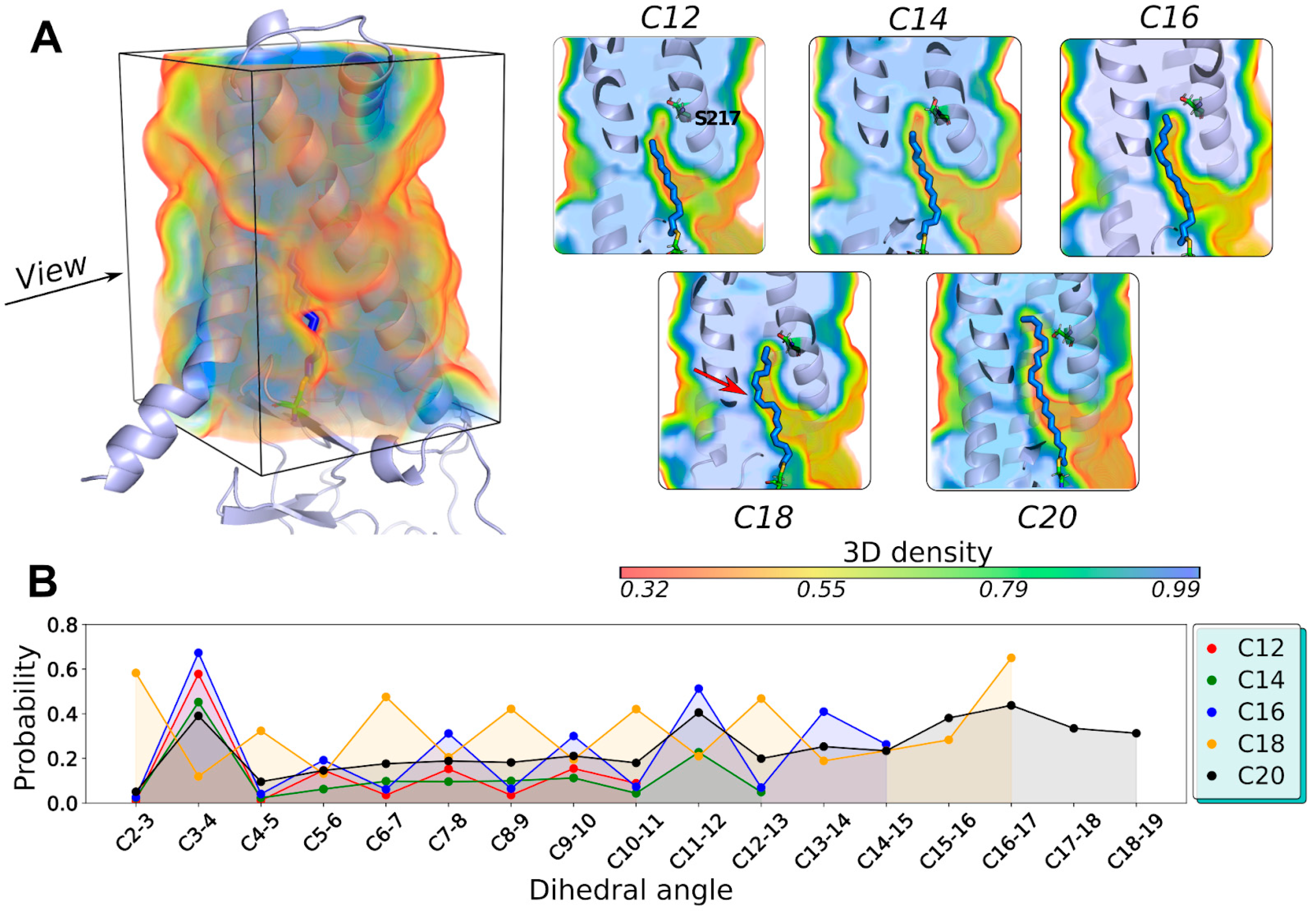
2.3. The DHHC Cavity Determines the Acyl Chain Length Preference
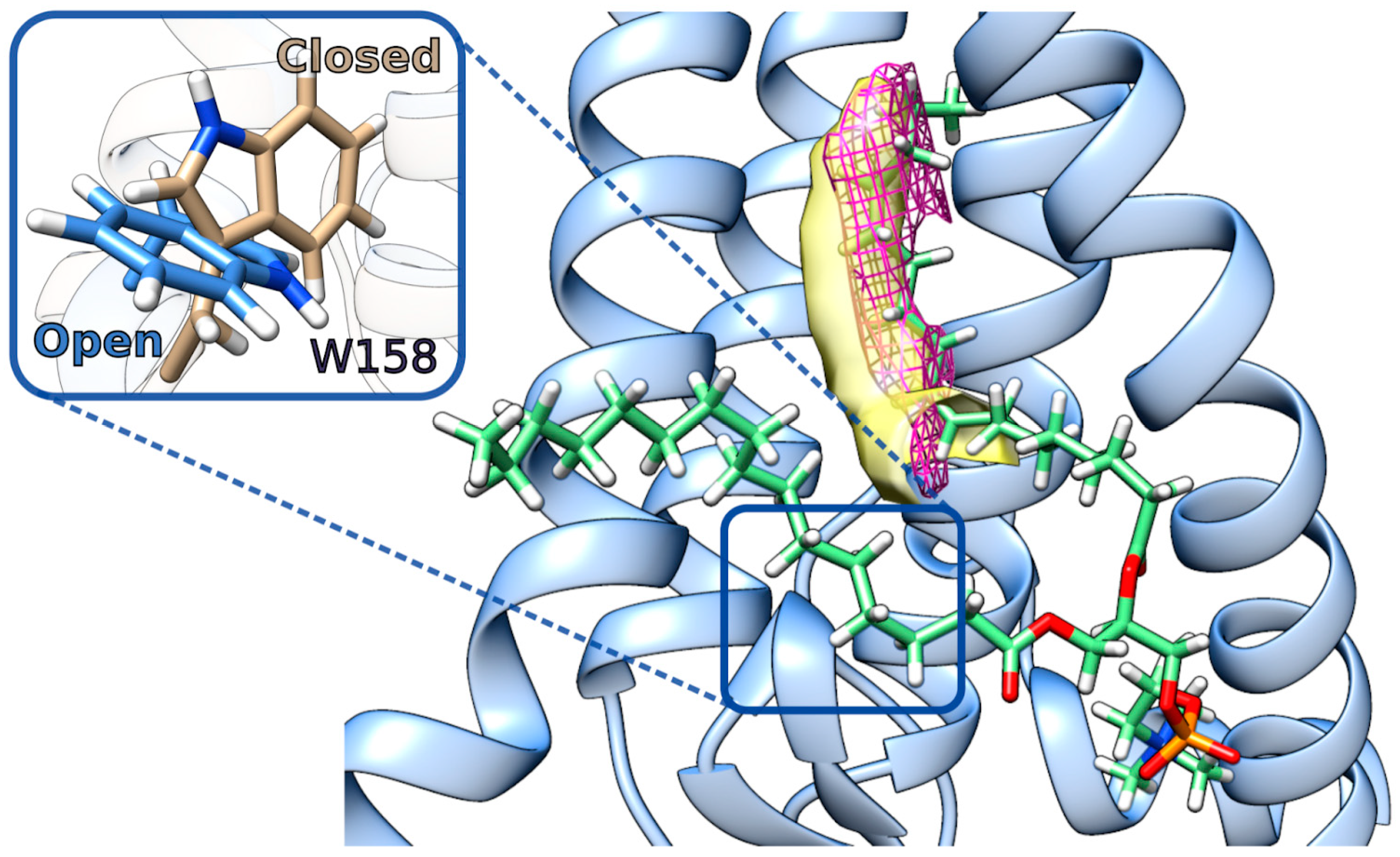
2.4. hDHHC20 Mutants S29F and Y181A Modify the Free Space of the Cavity
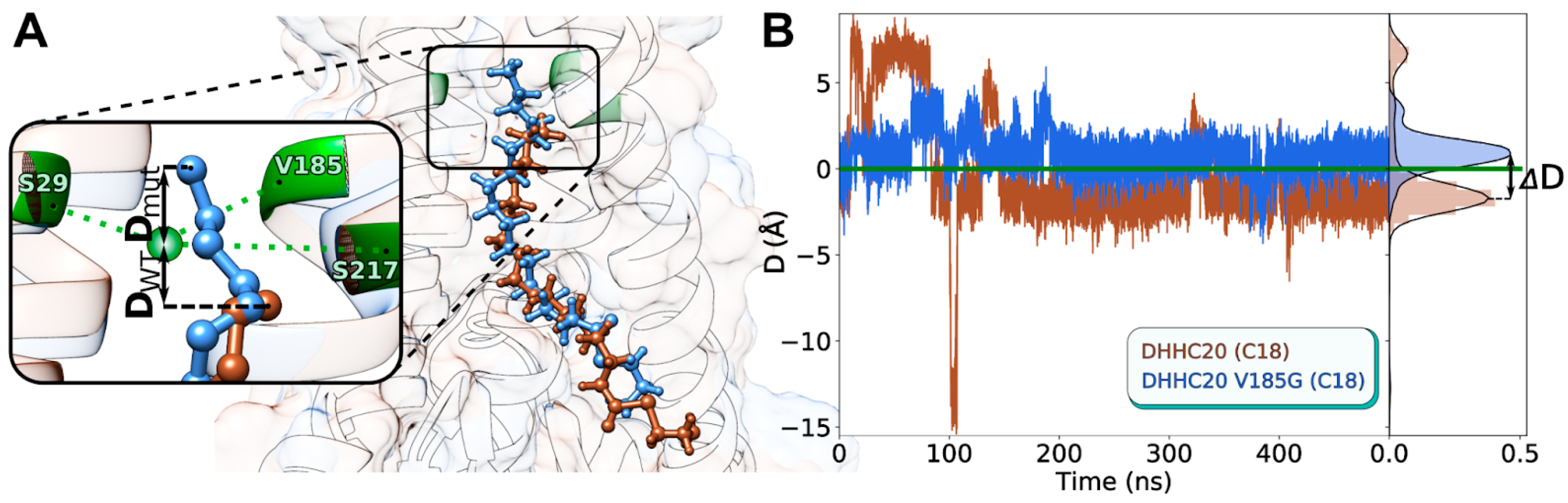
2.5. In Silico hDHHC20 V185G Mutant Exhibits C18-Selectivity
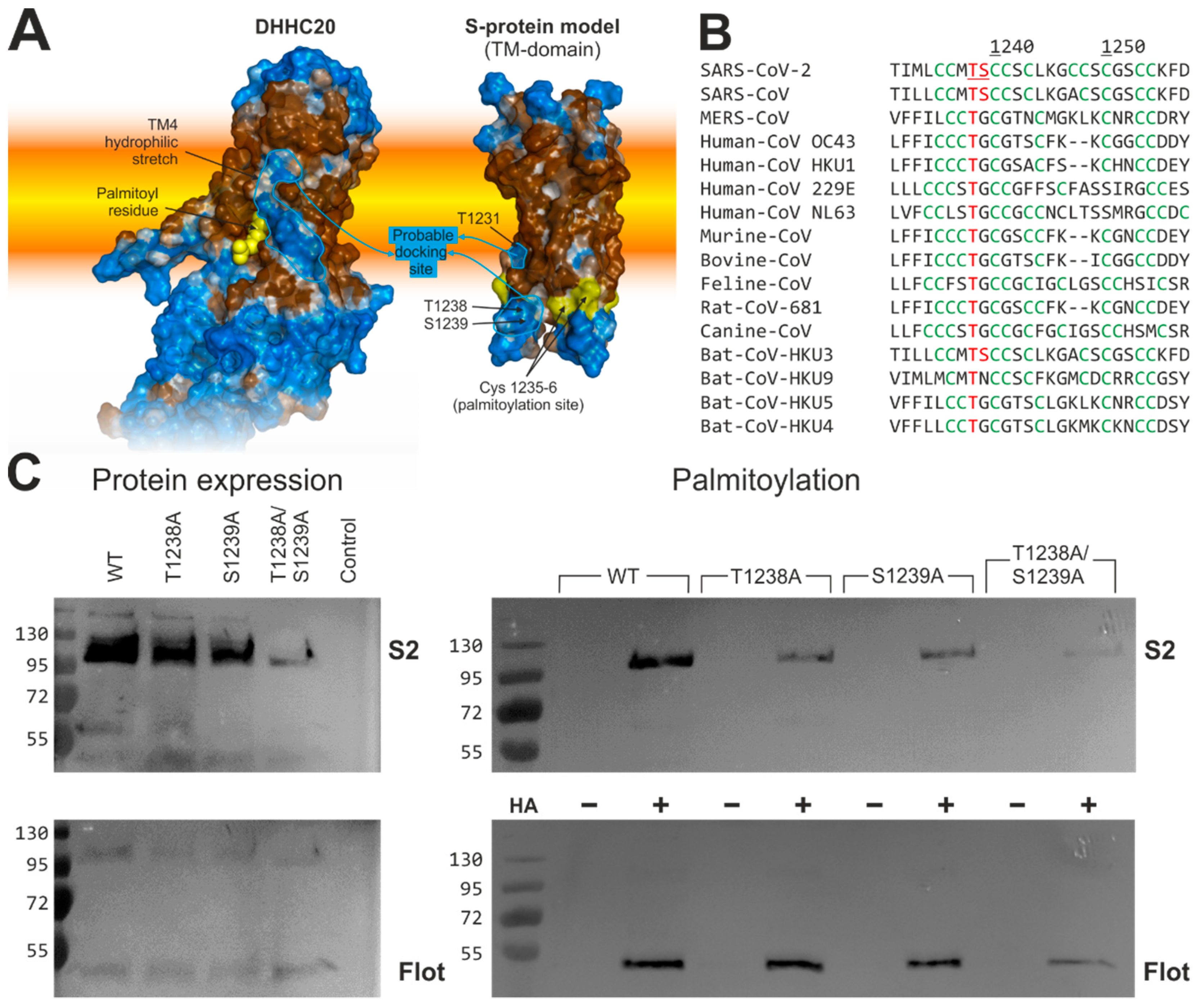
2.6. Human DHHC20 Might Recognize the Spike Protein of SARS-CoV-2via Hydrophilic Interactions between Their Transmembrane Regions
3. Discussion
4. Materials and Methods
4.1. Molecular Dynamics Simulations
4.1.1. Systems Setup
4.1.2. MD Simulation Parameters
4.1.3. MD Data Analysis
4.2. Spike Protein Transmembrane Domain Modeling
4.3. Mutagenesis, Expression, and Acylation Analysis of the S Protein of SARS-CoV-2
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blanc, M.; David, F.P.A.; van der Goot, F.G. SwissPalm 2: Protein S-Palmitoylation Database. Methods Mol. Biol. 2019, 2009, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, C.D.; Linder, M.E. Structure and Function of DHHC Protein S-Acyltransferases. Biochem. Soc. Trans. 2017, 45, 923–928. [Google Scholar] [CrossRef] [PubMed]
- González Montoro, A.; Quiroga, R.; Valdez Taubas, J. Zinc Co-Ordination by the DHHC Cysteine-Rich Domain of the Palmitoyltransferase Swf1. Biochem. J. 2013, 454, 427–435. [Google Scholar] [CrossRef] [Green Version]
- Zaballa, M.-E.; van der Goot, F.G. The Molecular Era of Protein S-Acylation: Spotlight on Structure, Mechanisms, and Dynamics. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 420–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, Y.; Kihara, A.; Sano, T.; Igarashi, Y. Intracellular Localization and Tissue-Specific Distribution of Human and Yeast DHHC Cysteine-Rich Domain-Containing Proteins. Biochim. Biophys. Acta 2006, 1761, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.M.; Syed, S.A.; Zaki, O.; Bottanelli, F.; Zheng, H.; Hacke, M.; Xi, Z.; Rivera-Molina, F.; Graham, M.; Rebane, A.A.; et al. S-Palmitoylation Sorts Membrane Cargo for Anterograde Transport in the Golgi. Dev. Cell 2018, 47, 479–493.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamberlain, L.H.; Shipston, M.J. The Physiology of Protein S-Acylation. Physiol. Rev. 2015, 95, 341–376. [Google Scholar] [CrossRef] [Green Version]
- Nůsková, H.; Serebryakova, M.V.; Ferrer-Caelles, A.; Sachsenheimer, T.; Lüchtenborg, C.; Miller, A.K.; Brügger, B.; Kordyukova, L.V.; Teleman, A.A. Stearic Acid Blunts Growth-Factor Signaling via Oleoylation of GNAI Proteins. Nat. Commun. 2021, 12, 4590. [Google Scholar] [CrossRef]
- Won, S.J.; Martin, B.R. Temporal Profiling Establishes a Dynamic S-Palmitoylation Cycle. ACS Chem. Biol. 2018, 13, 1560–1568. [Google Scholar] [CrossRef]
- Jennings, B.C.; Linder, M.E. DHHC Protein S-Acyltransferases Use Similar Ping-Pong Kinetic Mechanisms but Display Different Acyl-CoA Specificities. J. Biol. Chem. 2012, 287, 7236–7245. [Google Scholar] [CrossRef] [Green Version]
- Rana, M.S.; Kumar, P.; Lee, C.-J.; Verardi, R.; Rajashankar, K.R.; Banerjee, A. Fatty Acyl Recognition and Transfer by an Integral Membrane -Acyltransferase. Science 2018, 359, eaao6326. [Google Scholar] [CrossRef] [PubMed]
- Malgapo, M.I.P.; Linder, M.E. Substrate Recruitment by zDHHC Protein Acyltransferases. Open Biol. 2021, 11, 210026. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Lu, Y.; Neubert, T.A.; Resh, M.D. Mass Spectrometric Analysis of GAP-43/neuromodulin Reveals the Presence of a Variety of Fatty Acylated Species. J. Biol. Chem. 2002, 277, 33032–33040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordyukova, L.V.; Serebryakova, M.V.; Khrustalev, V.V.; Veit, M. Differential S-Acylation of Enveloped Viruses. Protein Pept. Lett. 2019, 26, 588–600. [Google Scholar] [CrossRef]
- Senyilmaz, D.; Virtue, S.; Xu, X.; Tan, C.Y.; Griffin, J.L.; Miller, A.K.; Vidal-Puig, A.; Teleman, A.A. Regulation of Mitochondrial Morphology and Function by Stearoylation of TFR1. Nature 2015, 525, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Jackson, C.L.; Walch, L.; Verbavatz, J.-M. Lipids and Their Trafficking: An Integral Part of Cellular Organization. Dev. Cell 2016, 39, 139–153. [Google Scholar] [CrossRef] [Green Version]
- Kordyukova, L.V.; Serebryakova, M.V.; Baratova, L.A.; Veit, M. Site-Specific Attachment of Palmitate or Stearate to Cytoplasmic versus Transmembrane Cysteines Is a Common Feature of Viral Spike Proteins. Virology 2010, 398, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Kordyukova, L.V.; Serebryakova, M.V.; Baratova, L.A.; Veit, M. S Acylation of the Hemagglutinin of Influenza Viruses: Mass Spectrometry Reveals Site-Specific Attachment of Stearic Acid to a Transmembrane Cysteine. J. Virol. 2008, 82, 9288–9292. [Google Scholar] [CrossRef] [Green Version]
- Brett, K.; Kordyukova, L.V.; Serebryakova, M.V.; Mintaev, R.R.; Alexeevski, A.V.; Veit, M. Site-Specific S-Acylation of Influenza Virus Hemagglutinin: The Location of the Acylation Site Relative to the Membrane Border Is the Decisive Factor for Attachment of Stearate. J. Biol. Chem. 2014, 289, 34978–34989. [Google Scholar] [CrossRef] [Green Version]
- Greaves, J.; Munro, K.R.; Davidson, S.C.; Riviere, M.; Wojno, J.; Smith, T.K.; Tomkinson, N.C.O.; Chamberlain, L.H. Molecular Basis of Fatty Acid Selectivity in the zDHHC Family of S-Acyltransferases Revealed by Click Chemistry. Proc. Natl. Acad. Sci. USA 2017, 114, E1365–E1374. [Google Scholar] [CrossRef] [Green Version]
- Siche, S.; Brett, K.; Möller, L.; Kordyukova, L.; Mintaev, R.; Alexeevski, A.; Veit, M. Two Cytoplasmic Acylation Sites and an Adjacent Hydrophobic Residue, but No Other Conserved Amino Acids in the Cytoplasmic Tail of HA from Influenza A Virus Are Crucial for Virus Replication. Viruses 2015, 7, 6458–6475. [Google Scholar] [CrossRef] [PubMed]
- Draper, J.M.; Smith, C.D. DHHC20: A Human Palmitoyl Acyltransferase That Causes Cellular Transformation. Mol. Membr. Biol. 2010, 27, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Kadry, Y.A.; Lee, J.-Y.; Witze, E.S. Regulation of EGFR Signalling by Palmitoylation and Its Role in Tumorigenesis. Open Biol. 2021, 11, 210033. [Google Scholar] [CrossRef] [PubMed]
- Abdulrahman, D.A.; Meng, X.; Veit, M. S-Acylation of Proteins of Coronavirus and Influenza Virus: Conservation of Acylation Sites in Animal Viruses and DHHC Acyltransferases in Their Animal Reservoirs. Pathogens 2021, 10, 669. [Google Scholar] [CrossRef] [PubMed]
- Gadalla, M.R.; Veit, M. Toward the Identification of ZDHHC Enzymes Required for Palmitoylation of Viral Protein as Potential Drug Targets. Expert Opin. Drug Discov. 2020, 15, 159–177. [Google Scholar] [CrossRef]
- Mesquita, F.S.; Abrami, L.; Sergeeva, O.; Turelli, P.; Qing, E.; Kunz, B.; Raclot, C.; Paz Montoya, J.; Abriata, L.A.; Gallagher, T.; et al. S-Acylation Controls SARS-CoV-2 Membrane Lipid Organization and Enhances Infectivity. Dev. Cell 2021, 56, 2790–2807.e8. [Google Scholar] [CrossRef]
- Gadalla, M.R.; Abrami, L.; van der Goot, F.G.; Veit, M. Hemagglutinin of Influenza A, but Not of Influenza B and C Viruses Is Acylated by ZDHHC2, 8, 15 and 20. Biochem. J. 2020, 477, 285–303. [Google Scholar] [CrossRef]
- Puthenveetil, R.; Lun, C.M.; Murphy, R.E.; Healy, L.B.; Vilmen, G.; Christenson, E.T.; Freed, E.O.; Banerjee, A. S-Acylation of SARS-CoV-2 Spike Protein: Mechanistic Dissection, in Vitro Reconstitution and Role in Viral Infectivity. J. Biol. Chem. 2021, 297, 101112. [Google Scholar] [CrossRef]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [Green Version]
- Torrens-Fontanals, M.; Stepniewski, T.M.; Aranda-García, D.; Morales-Pastor, A.; Medel-Lacruz, B.; Selent, J. How Do Molecular Dynamics Data Complement Static Structural Data of GPCRs. Int. J. Mol. Sci. 2020, 21, 5933. [Google Scholar] [CrossRef]
- Stix, R.; Song, J.; Banerjee, A.; Faraldo-Gómez, J.D. DHHC20 Palmitoyl-Transferase Reshapes the Membrane to Foster Catalysis. Biophys. J. 2020, 118, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Stix, R.; Lee, C.-J.; Faraldo-Gómez, J.D.; Banerjee, A. Structure and Mechanism of DHHC Protein Acyltransferases. J. Mol. Biol. 2020, 432, 4983–4998. [Google Scholar] [CrossRef] [PubMed]
- Chugunov, A.O.; Novoseletsky, V.N.; Nolde, D.E.; Arseniev, A.S.; Efremov, R.G. Method to Assess Packing Quality of Transmembrane α-Helices in Proteins. Part 1. Parametrization Using Structural Data. ChemInform 2007, 47, 1150–1162. [Google Scholar] [CrossRef]
- Chugunov, A.O.; Novoseletsky, V.N.; Nolde, D.E.; Arseniev, A.S.; Efremov, R.G. Method to Assess Packing Quality of Transmembrane α-Helices in Proteins. Part 2. Validation by “Correct vs. Misleading” Test. ChemInform 2007, 47, 1163–1170. [Google Scholar] [CrossRef]
- Efremov, R.G.; Chugunov, A.O.; Pyrkov, T.V.; Priestle, J.P.; Arseniev, A.S.; Jacoby, E. Molecular Lipophilicity in Protein Modeling and Drug Design. Curr. Med. Chem. 2007, 14, 393–415. [Google Scholar] [CrossRef] [PubMed]
- Pyrkov, T.V.; Chugunov, A.O.; Krylov, N.A.; Nolde, D.E.; Efremov, R.G. PLATINUM: A Web Tool for Analysis of Hydrophobic/hydrophilic Organization of Biomolecular Complexes. Bioinformatics 2009, 25, 1201–1202. [Google Scholar] [CrossRef]
- Wagner, R.; Herwig, A.; Azzouz, N.; Klenk, H.D. Acylation-Mediated Membrane Anchoring of Avian Influenza Virus Hemagglutinin Is Essential for Fusion Pore Formation and Virus Infectivity. J. Virol. 2005, 79, 6449–6458. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.J.; Takeda, M.; Lamb, R.A. Influenza Virus Hemagglutinin (H3 Subtype) Requires Palmitoylation of Its Cytoplasmic Tail for Assembly: M1 Proteins of Two Subtypes Differ in Their Ability to Support Assembly. J. Virol. 2005, 79, 13673–13684. [Google Scholar] [CrossRef] [Green Version]
- The PyMOL Molecular Graphics System; Schrödinger, LLC: New York, NY, USA, 2020.
- Ray, A.; Gräter, F.; Thukral, L. Probing Molecular Forces in Multi-Component Physiological Membranes. Phys. Chem. Chem. Phys. 2018, 20, 2155–2161. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone φ, ψ and Side-Chain χ(1) and χ(2) Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klauda, J.B.; Monje, V.; Kim, T.; Im, W. Improving the CHARMM Force Field for Polyunsaturated Fatty Acid Chains. J. Phys. Chem. B 2012, 116, 9424–9431. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1998, 79, 926. [Google Scholar] [CrossRef]
- Beglov, D.; Roux, B. Finite Representation of an Infinite Bulk System: Solvent Boundary Potential for Computer Simulations. J. Chem. Phys. 1998, 100, 9050. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiser, A.; Sali, A. Modeller: Generation and Refinement of Homology-Based Protein Structure Models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Fu, Q.; Pan, L.; Piai, A.; Chou, J.J. The Diversity and Similarity of Transmembrane Trimerization of TNF Receptors. Front. Cell Dev. Biol. 2020, 8, 569684. [Google Scholar] [CrossRef] [PubMed]
- Werno, M.W.; Chamberlain, L.H. S-Acylation of the Insulin-Responsive Aminopeptidase (IRAP): Quantitative Analysis and Identification of Modified Cysteines. Sci. Rep. 2015, 5, 12413. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System Composition | MD Length (ns) | Number of Replicas |
|---|---|---|
| Acyl-CoA in Lipid Bilayer | ||
| Acyl: C12/C14/C16/C18/C20 | ||
| Acyl-CoA2/POPC176/POPE84/POPI28/Water25481/Na+88/Cl−50 | 300 | 5 |
| C16-CoA2/POPC288/Water25481/Na+88/Cl−50 | 300 | 1 |
| DHHC Enzyme in Lipid Bilayer | ||
| Protein: hDHHC20/hDHHC20S29F/hDHHC20Y181A (starting structure PDB ID: 6BMN) | ||
| Protein/POPC144/POPE72/POPI25/Water24565/Na+72/Zn2+2/Cl−59 | 1000 | 3 |
| Protein/POPC240/Water25004/Na+60/Zn2+2/Cl−72 | 700–900 | 3 |
| DHHC Enzyme with Acyl-MEA in Lipid Bilayer | ||
| Protein: hDHHC20/hDHHC20S29F/hDHHC20Y181A (starting structure PDB ID: 6BML) | ||
| Acyl: C12/C14/C16/C18/C20 | ||
| Protein/Acyl-MEA/POPC145/POPE69/POPI26/Water24558/Na+76/Cl−58 | 500 | 15 |
| Acylated DHHC Enzyme in Lipid Bilayer | ||
| Protein: hDHHC20/hDHHC20S29F/hDHHC20Y181A (starting structure PDB ID: 6BML) | ||
| Acyl: C12/C14/C16/C18/C20 | ||
| Acyl-Protein/POPC146/POPE71/POPI26/Water24555/Na+72/Zn2+2/Cl−58 | 500 | 15 |
| DHHC Mutants (Predicted in Silico) with C18(16)-MEA in Lipid Bilayer | ||
| Mutant: V185G/S29A/V185A/S217A/L213A/S29A+V185A/V185G+S217A/A32L/Y33W/F58W/F62W/F184W/V185I/A186L/L213F (starting structure PDB ID: 6BML) | ||
| Mutant/POPC145/POPE69/POPI26/Water24570/Na+80/Cl−62 | 100–500 * | 15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panina, I.; Krylov, N.; Gadalla, M.R.; Aliper, E.; Kordyukova, L.; Veit, M.; Chugunov, A.; Efremov, R. Molecular Dynamics of DHHC20 Acyltransferase Suggests Principles of Lipid and Protein Substrate Selectivity. Int. J. Mol. Sci. 2022, 23, 5091. https://doi.org/10.3390/ijms23095091
Panina I, Krylov N, Gadalla MR, Aliper E, Kordyukova L, Veit M, Chugunov A, Efremov R. Molecular Dynamics of DHHC20 Acyltransferase Suggests Principles of Lipid and Protein Substrate Selectivity. International Journal of Molecular Sciences. 2022; 23(9):5091. https://doi.org/10.3390/ijms23095091
Chicago/Turabian StylePanina, Irina, Nikolay Krylov, Mohamed Rasheed Gadalla, Elena Aliper, Larisa Kordyukova, Michael Veit, Anton Chugunov, and Roman Efremov. 2022. "Molecular Dynamics of DHHC20 Acyltransferase Suggests Principles of Lipid and Protein Substrate Selectivity" International Journal of Molecular Sciences 23, no. 9: 5091. https://doi.org/10.3390/ijms23095091
APA StylePanina, I., Krylov, N., Gadalla, M. R., Aliper, E., Kordyukova, L., Veit, M., Chugunov, A., & Efremov, R. (2022). Molecular Dynamics of DHHC20 Acyltransferase Suggests Principles of Lipid and Protein Substrate Selectivity. International Journal of Molecular Sciences, 23(9), 5091. https://doi.org/10.3390/ijms23095091