
Interactions between S100A9 and Alpha-Synuclein: Insight from NMR Spectroscopy

 , , , ,
, , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
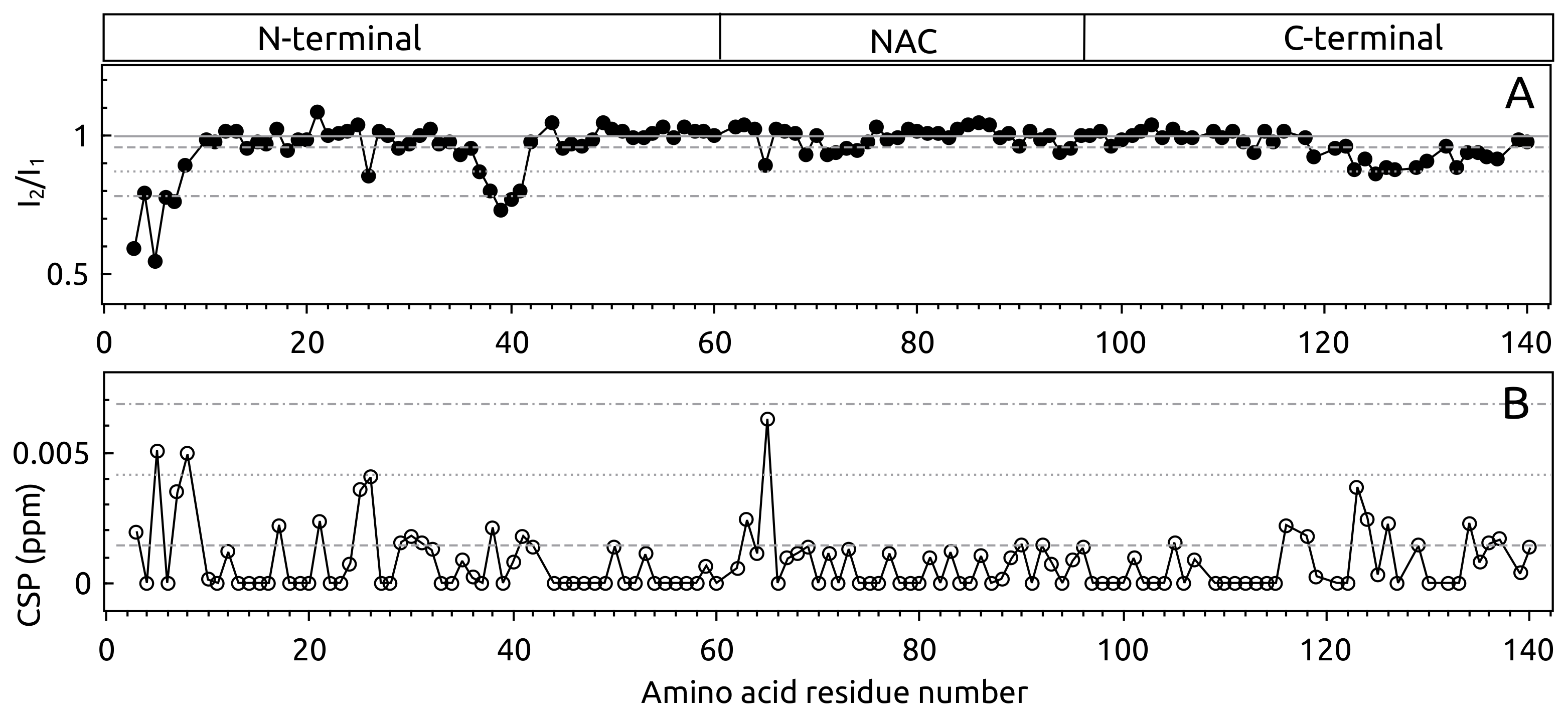
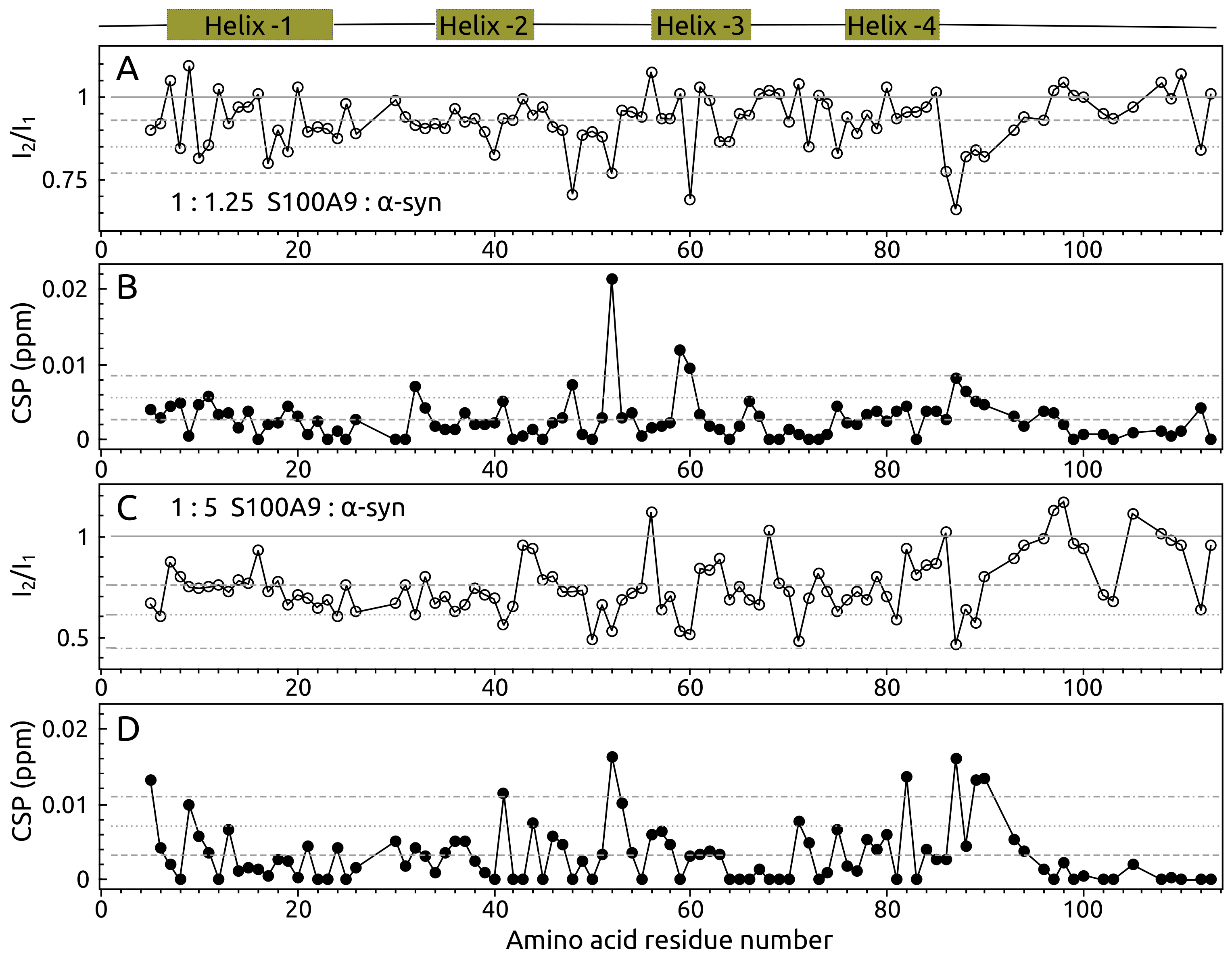
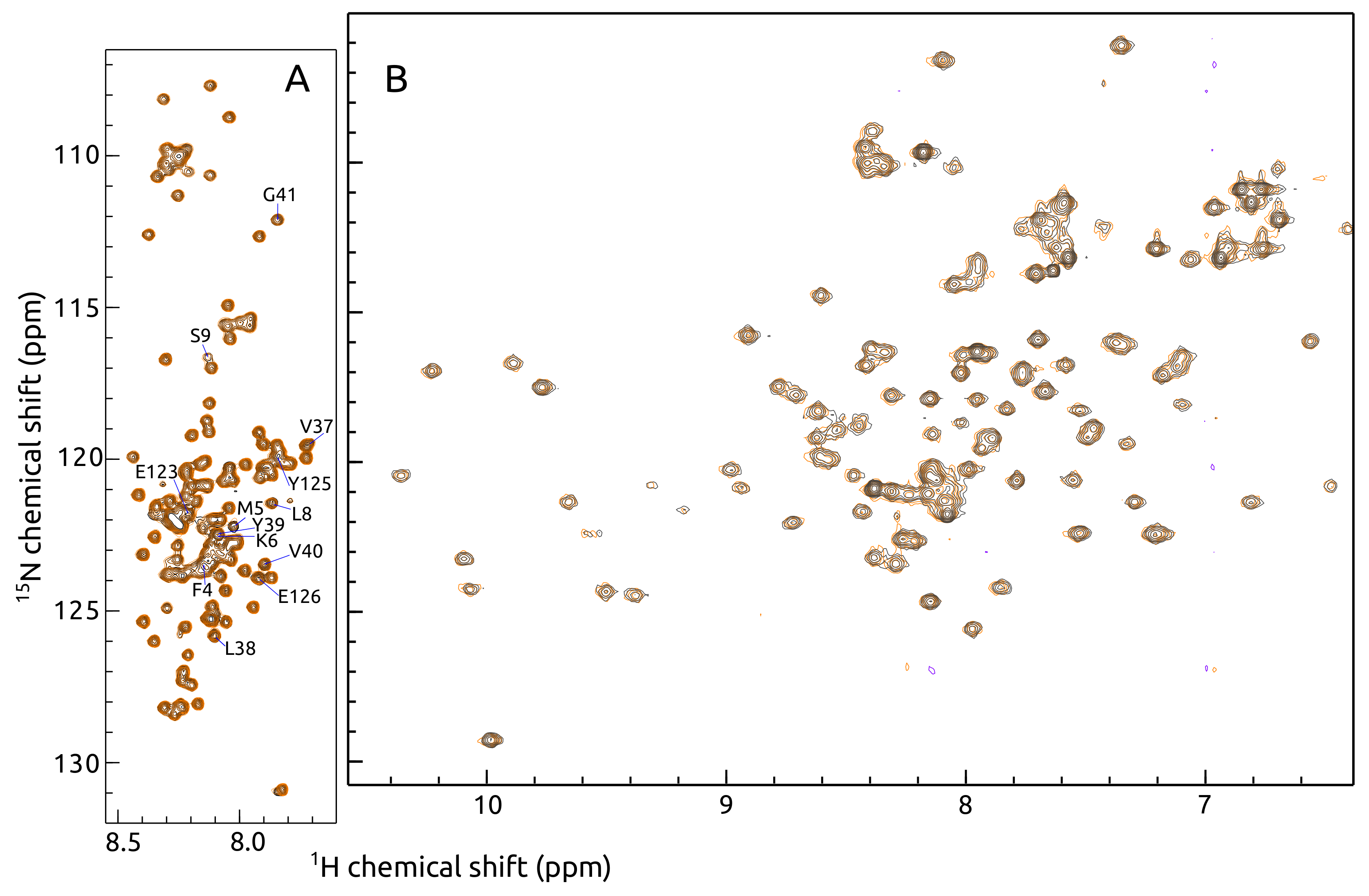
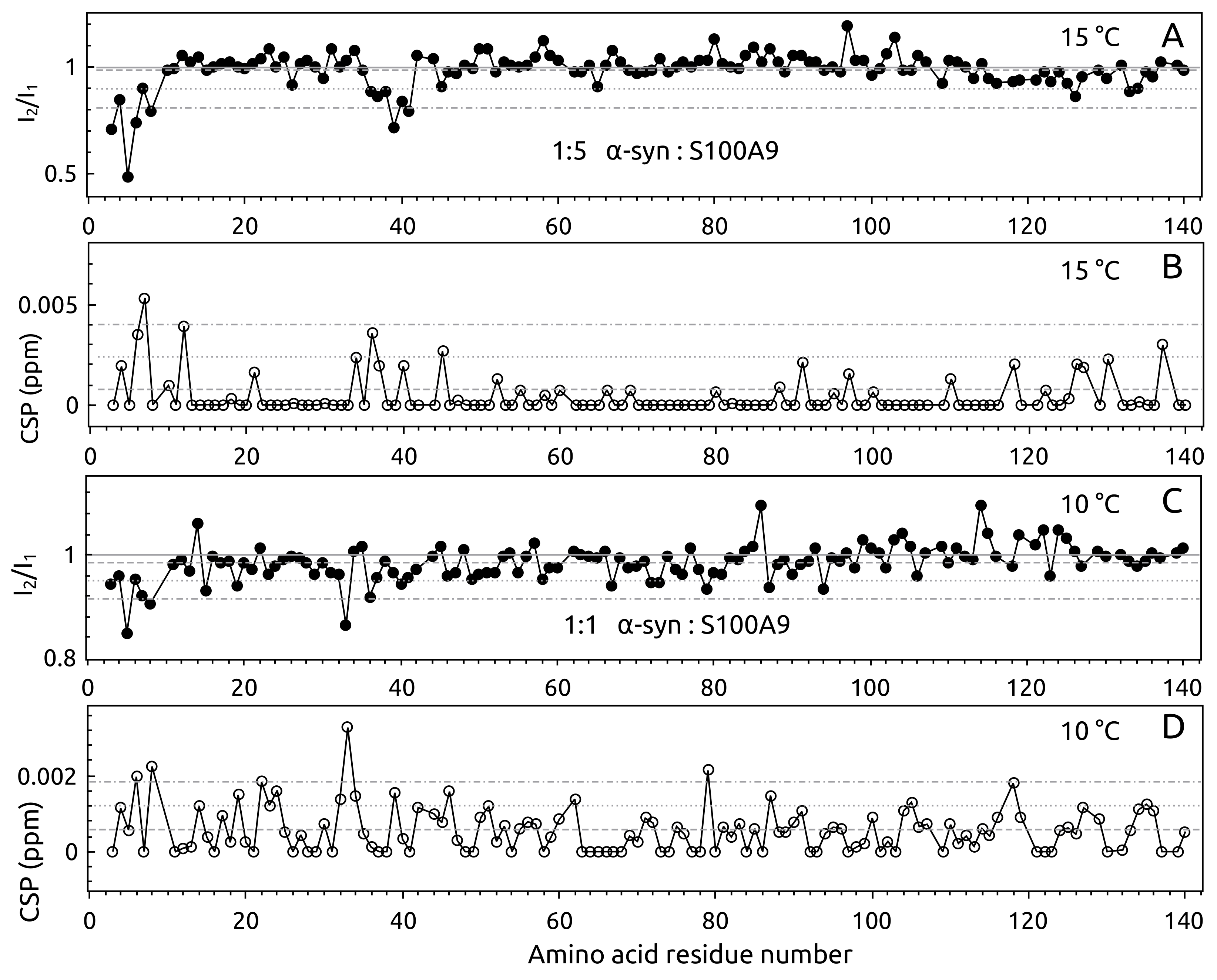
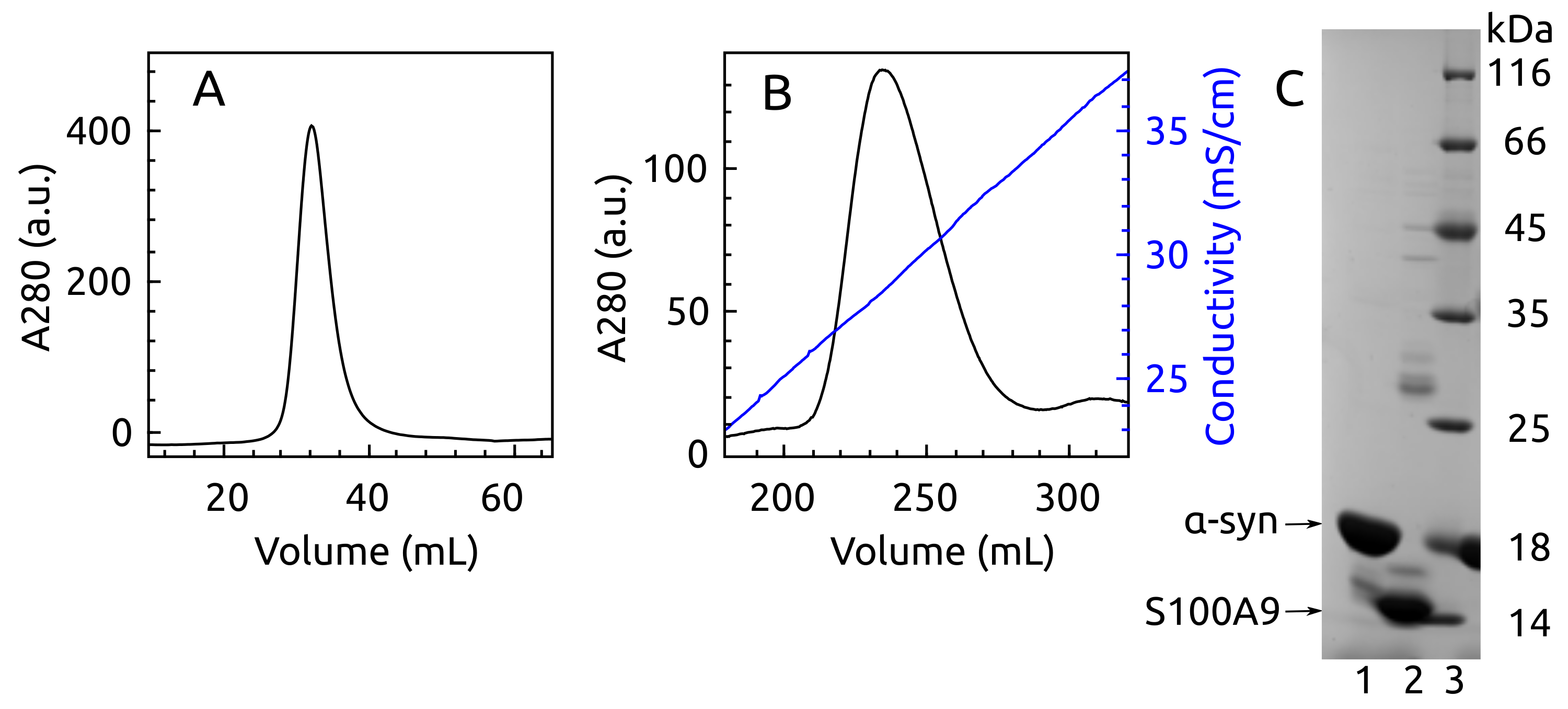
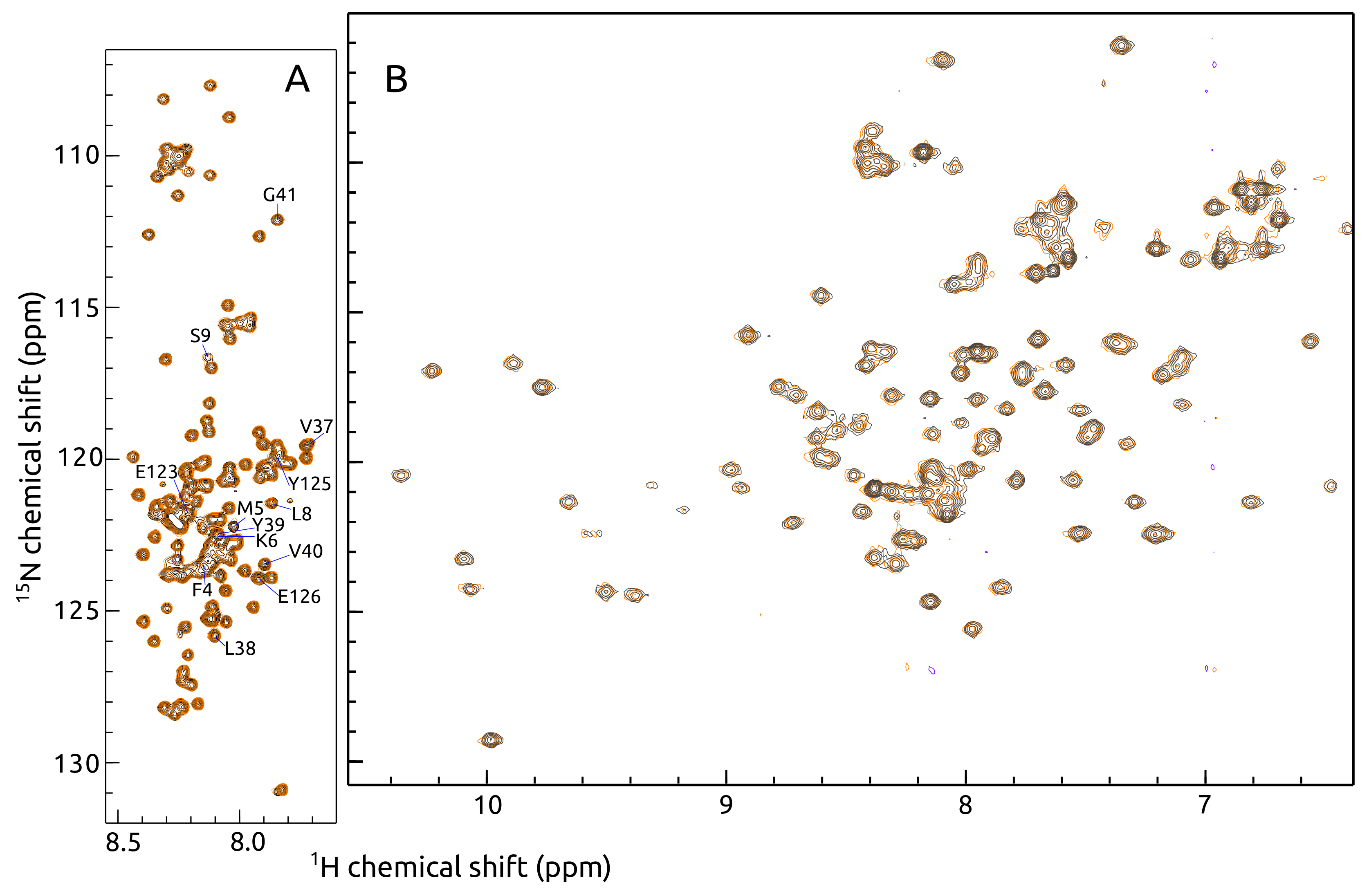
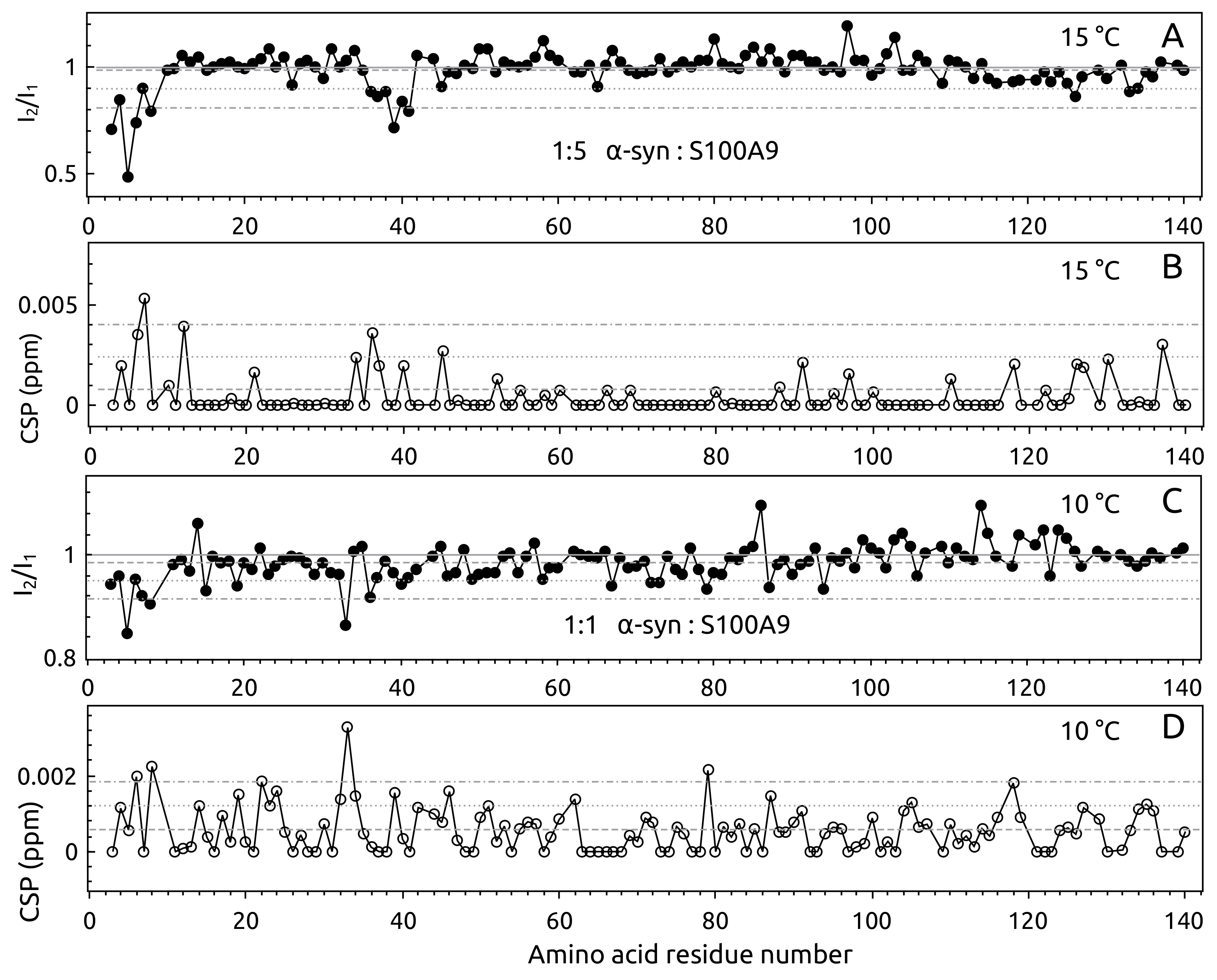
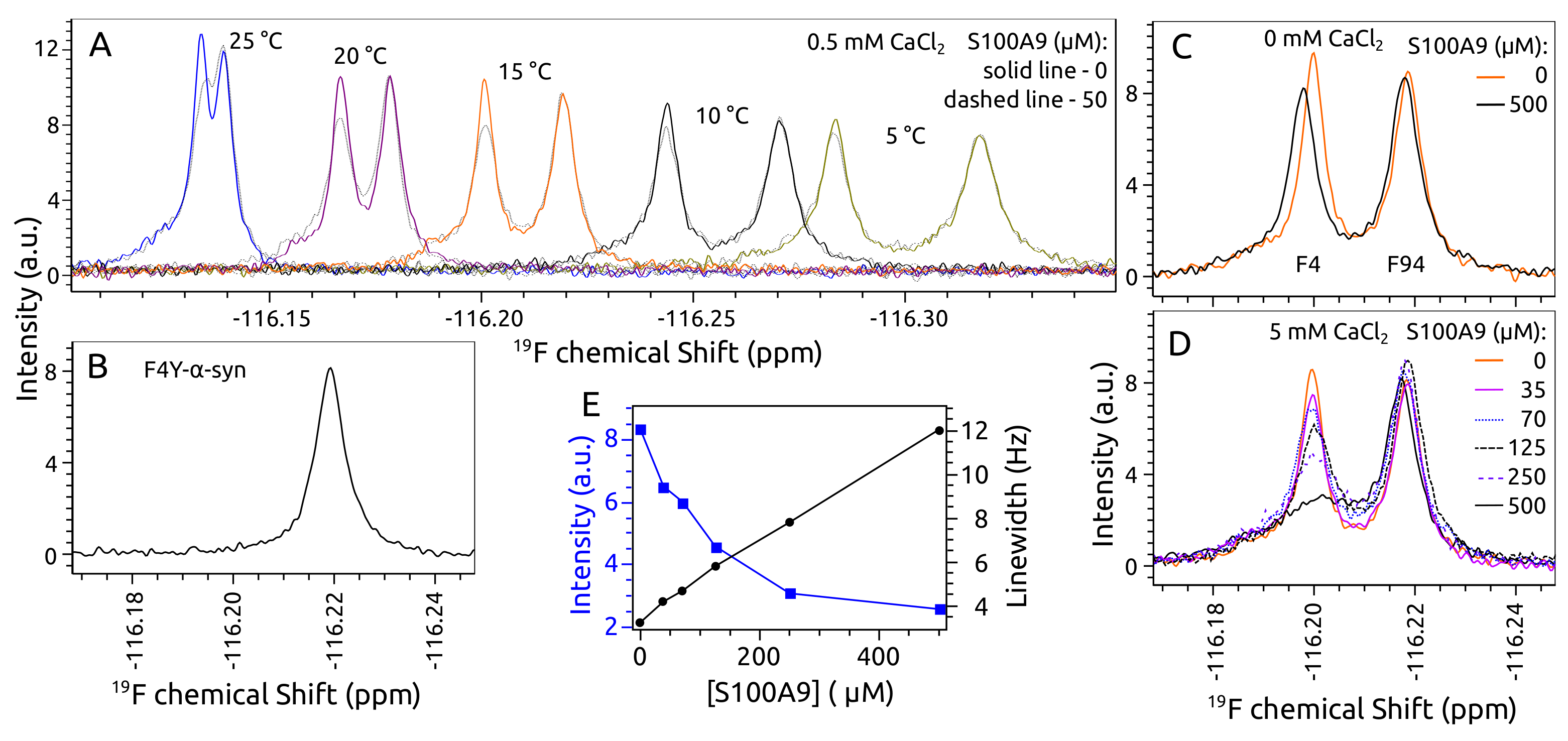
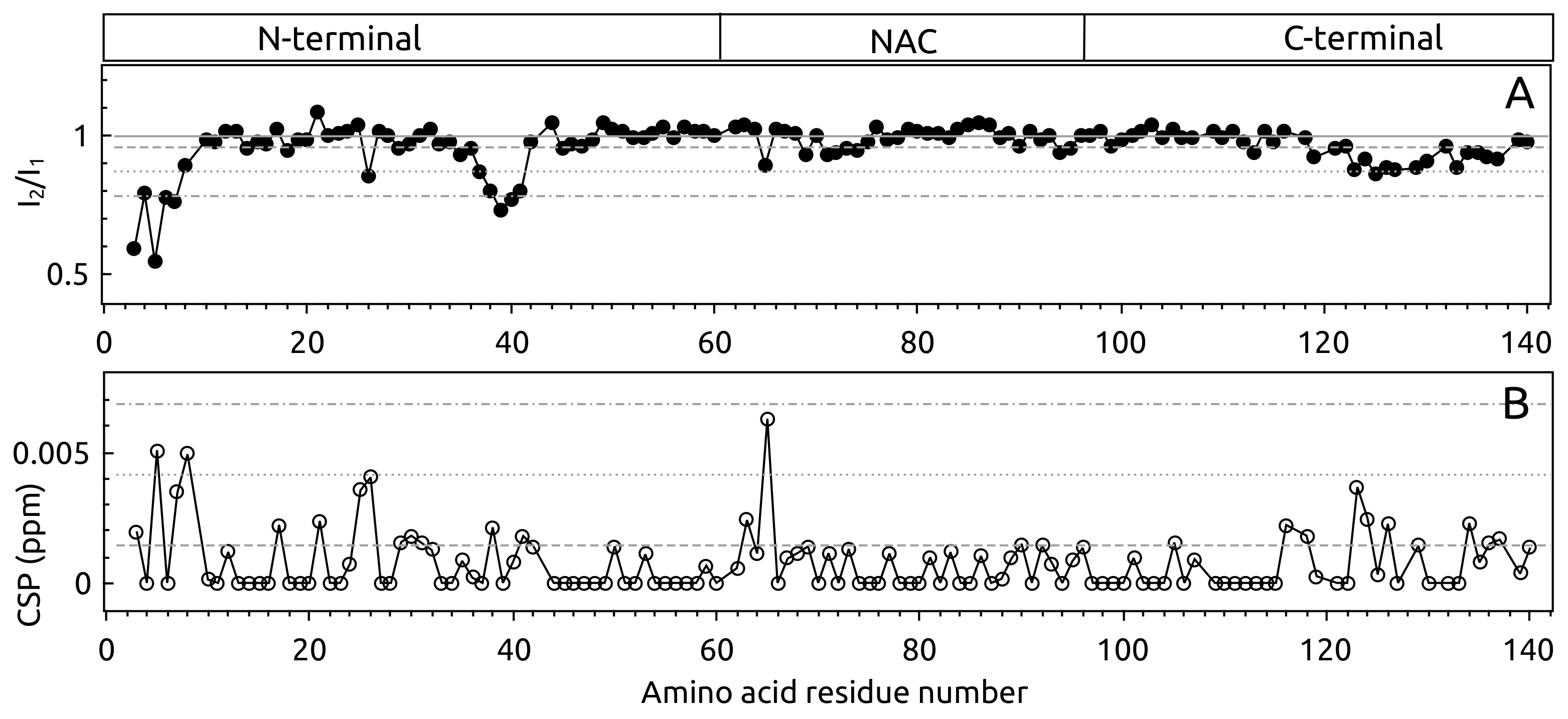
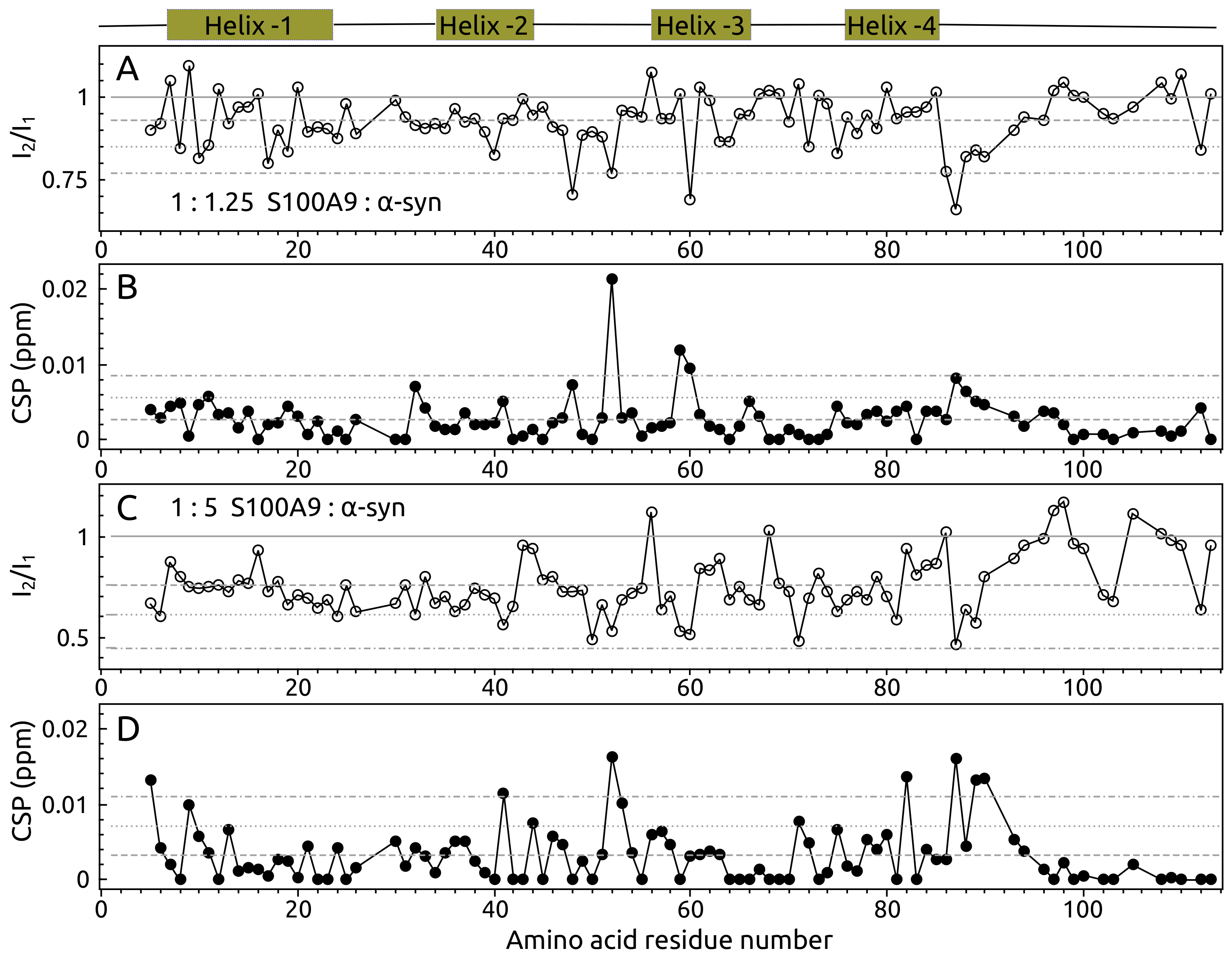
2.1. -syn Binding with S100A9
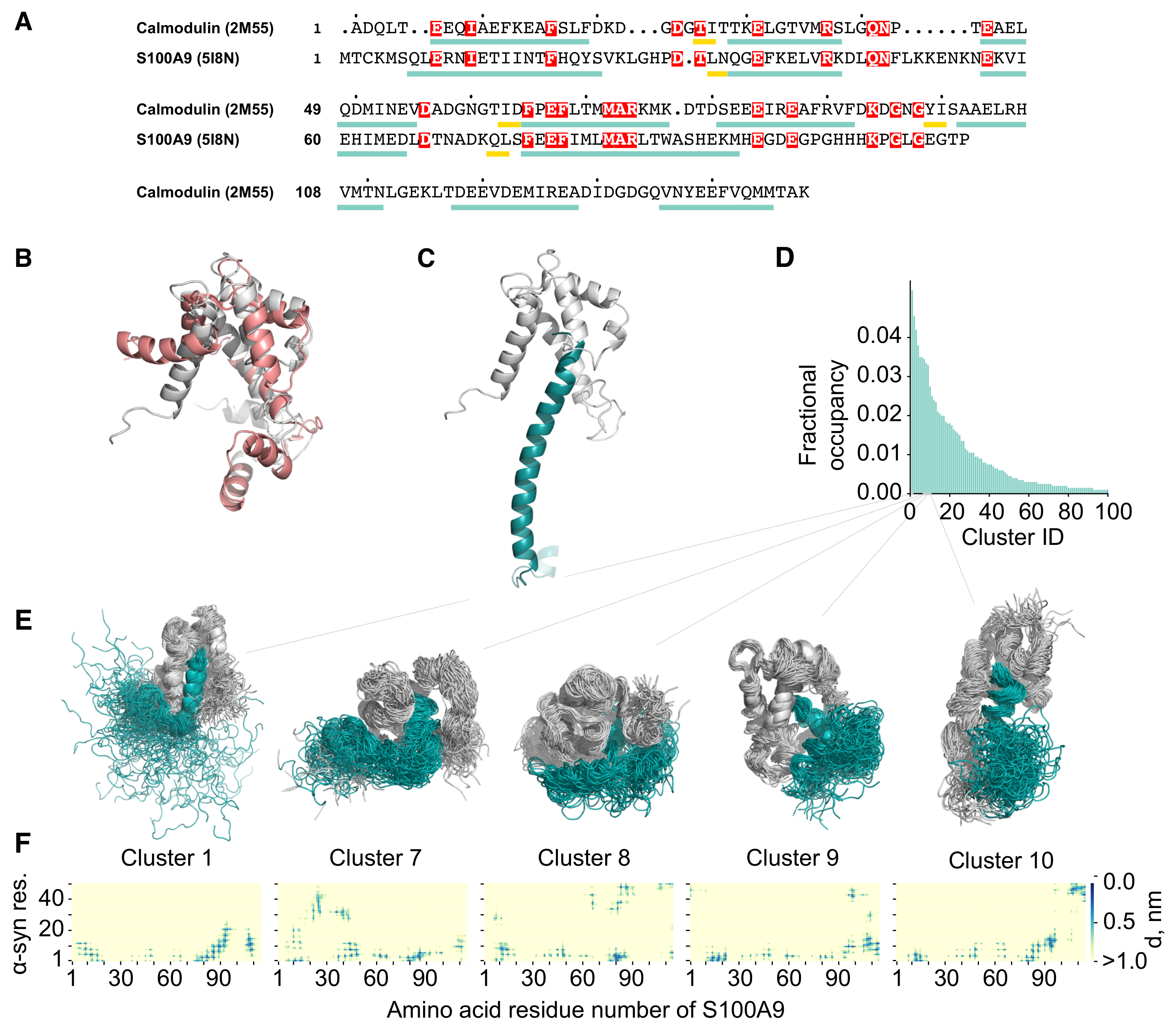
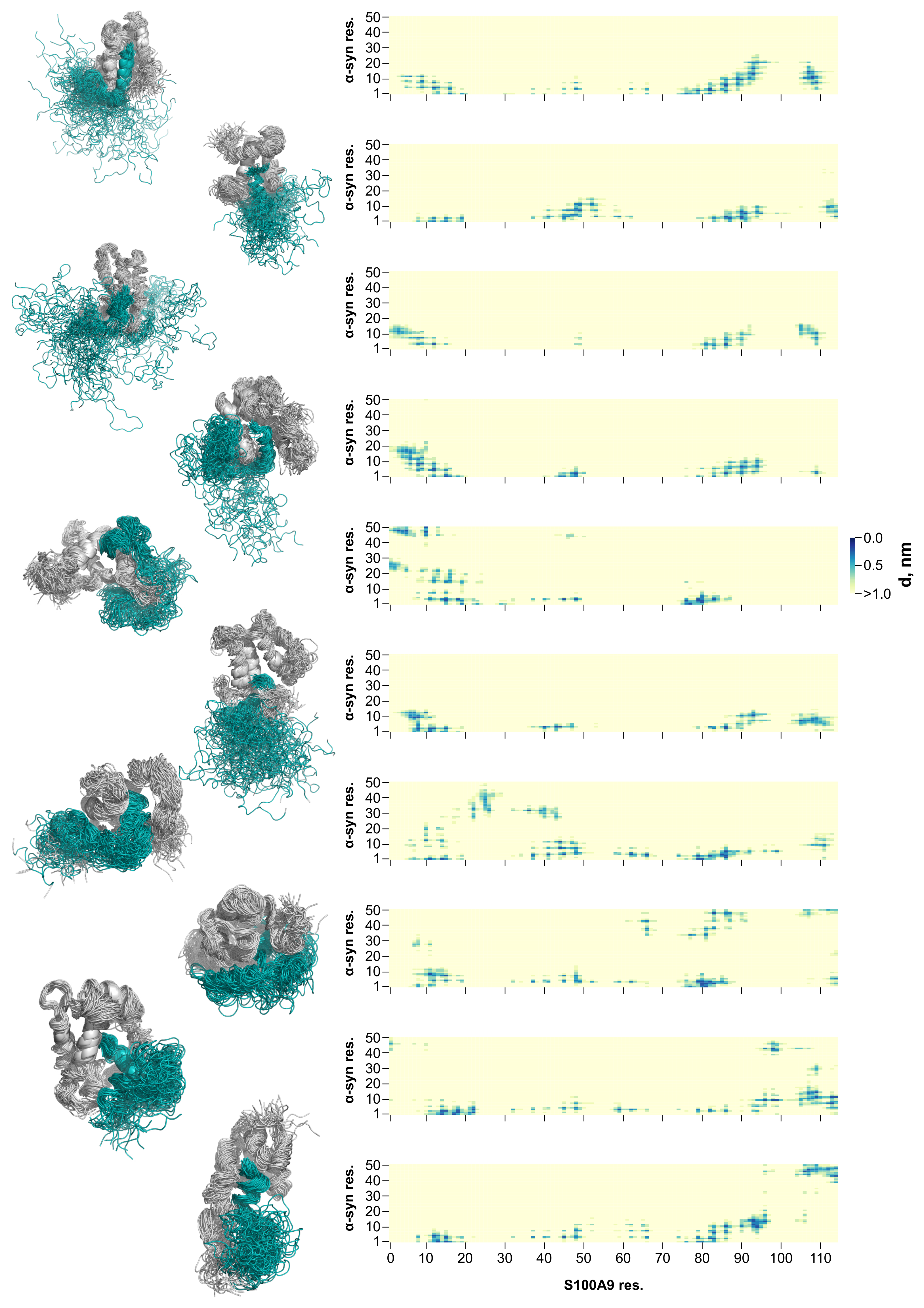
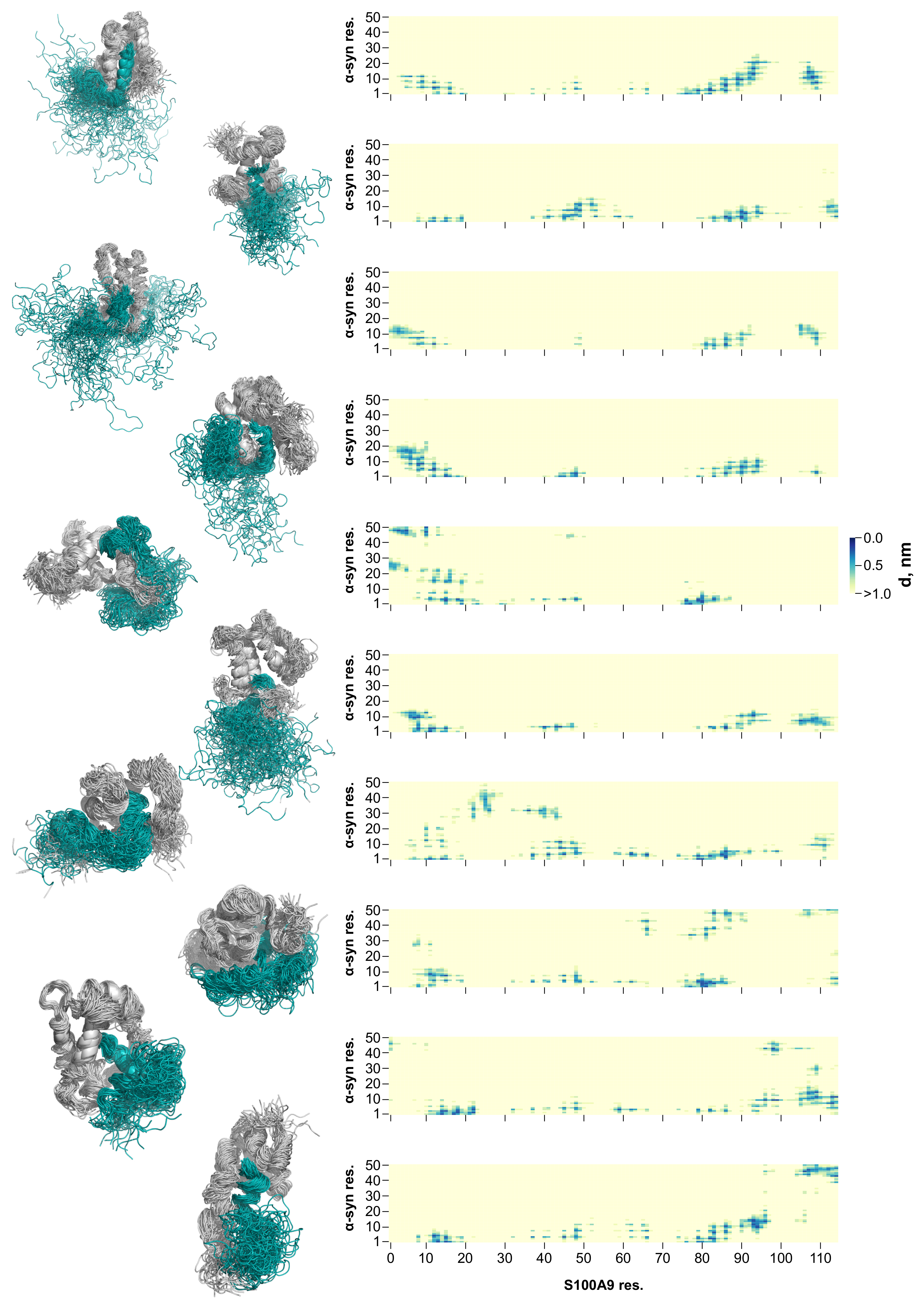
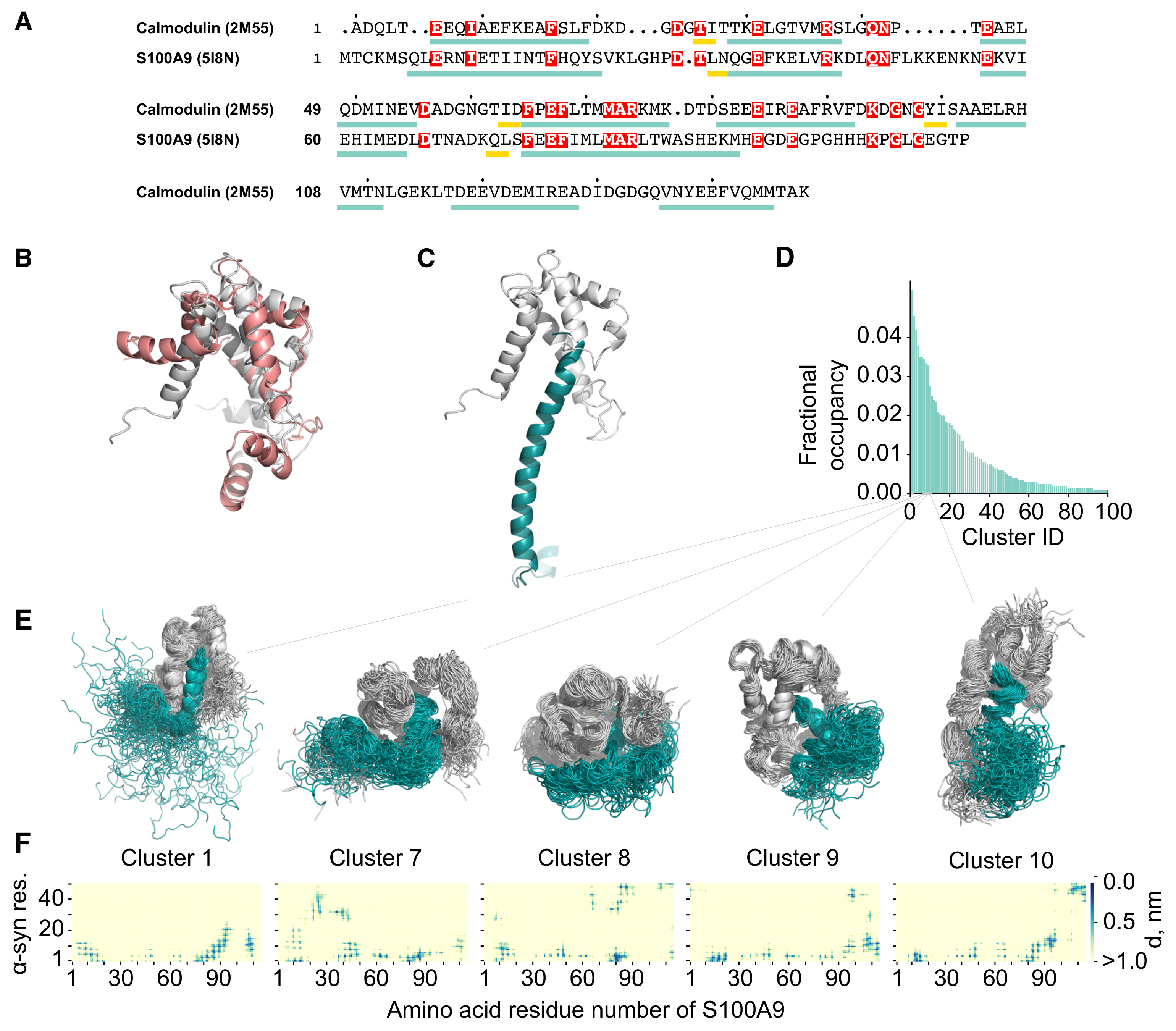
2.2. Molecular Dynamics Simulation of the -syn-S100A9 Complex
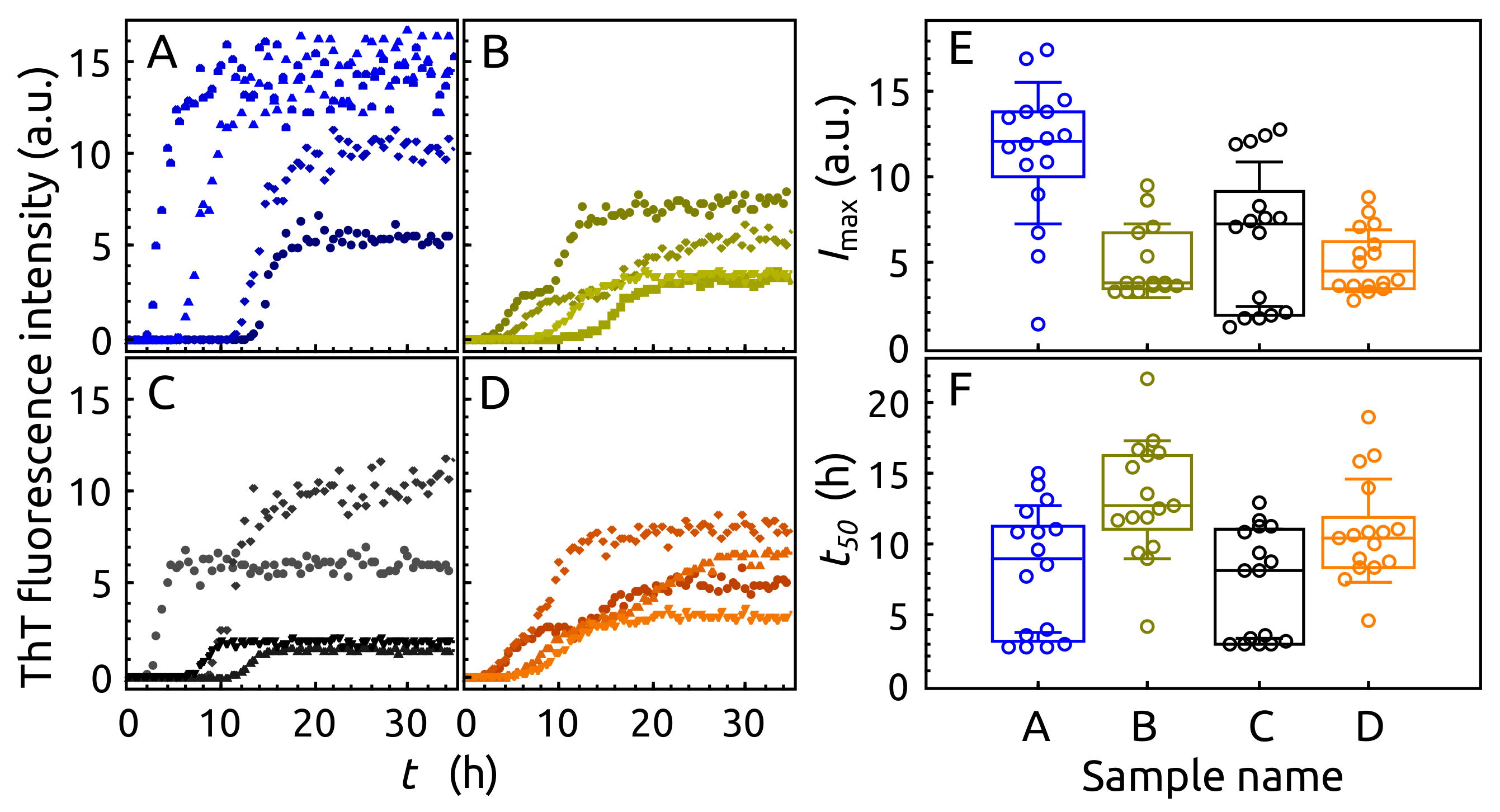
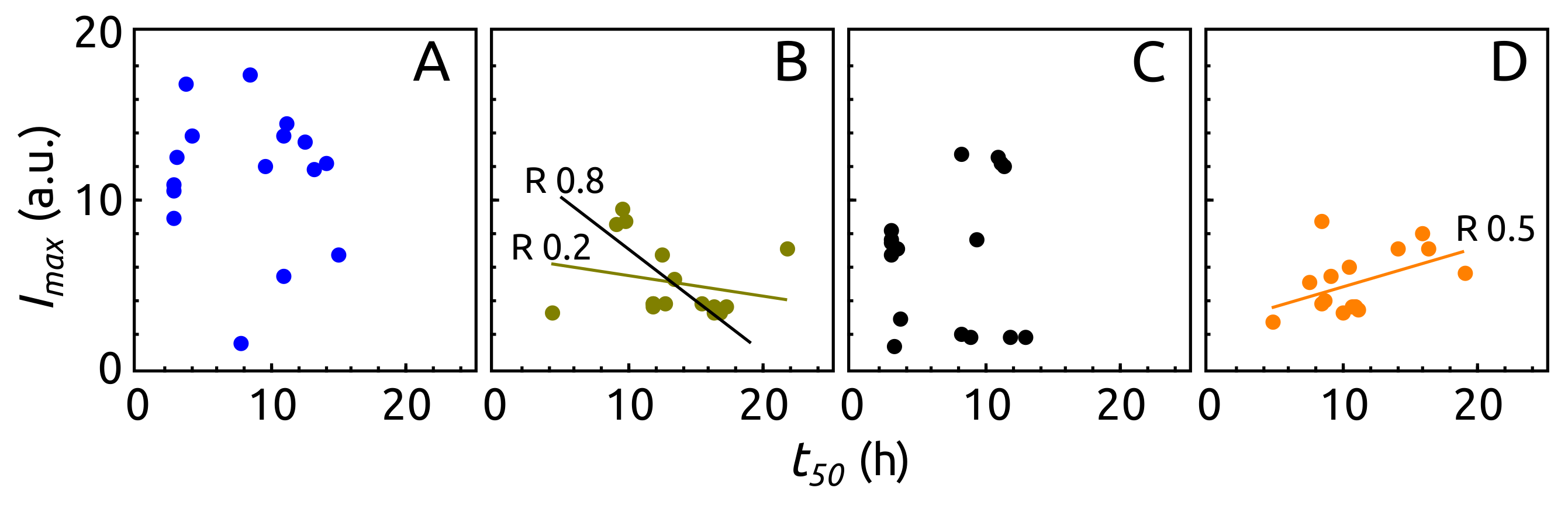
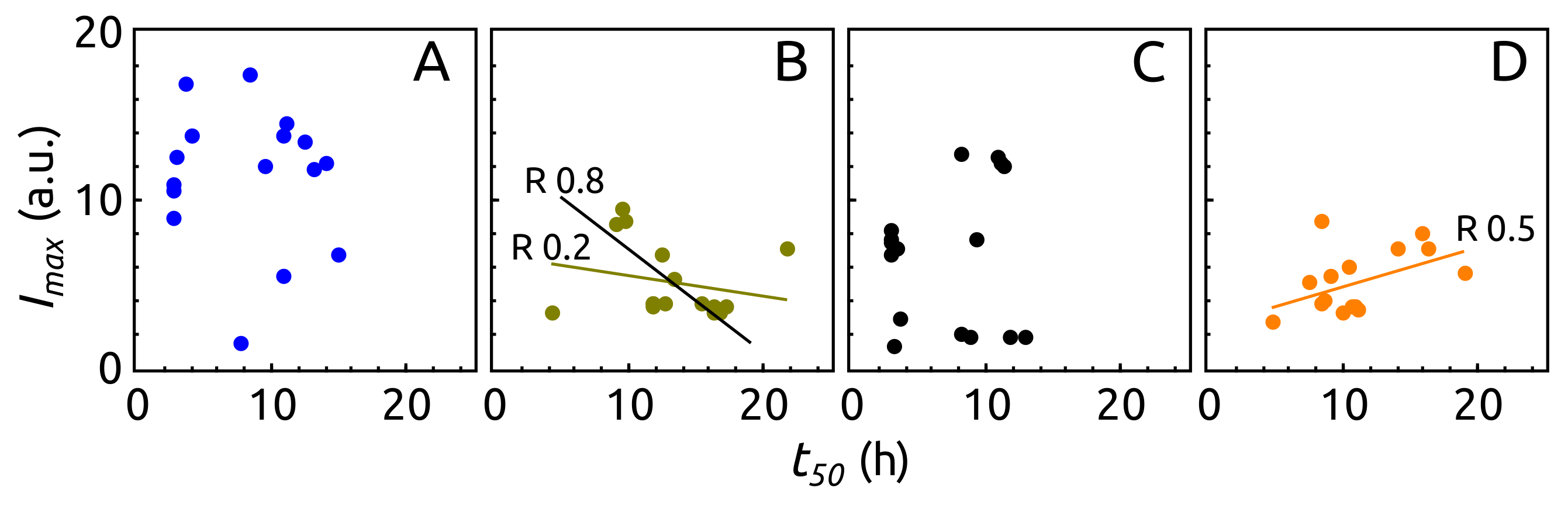
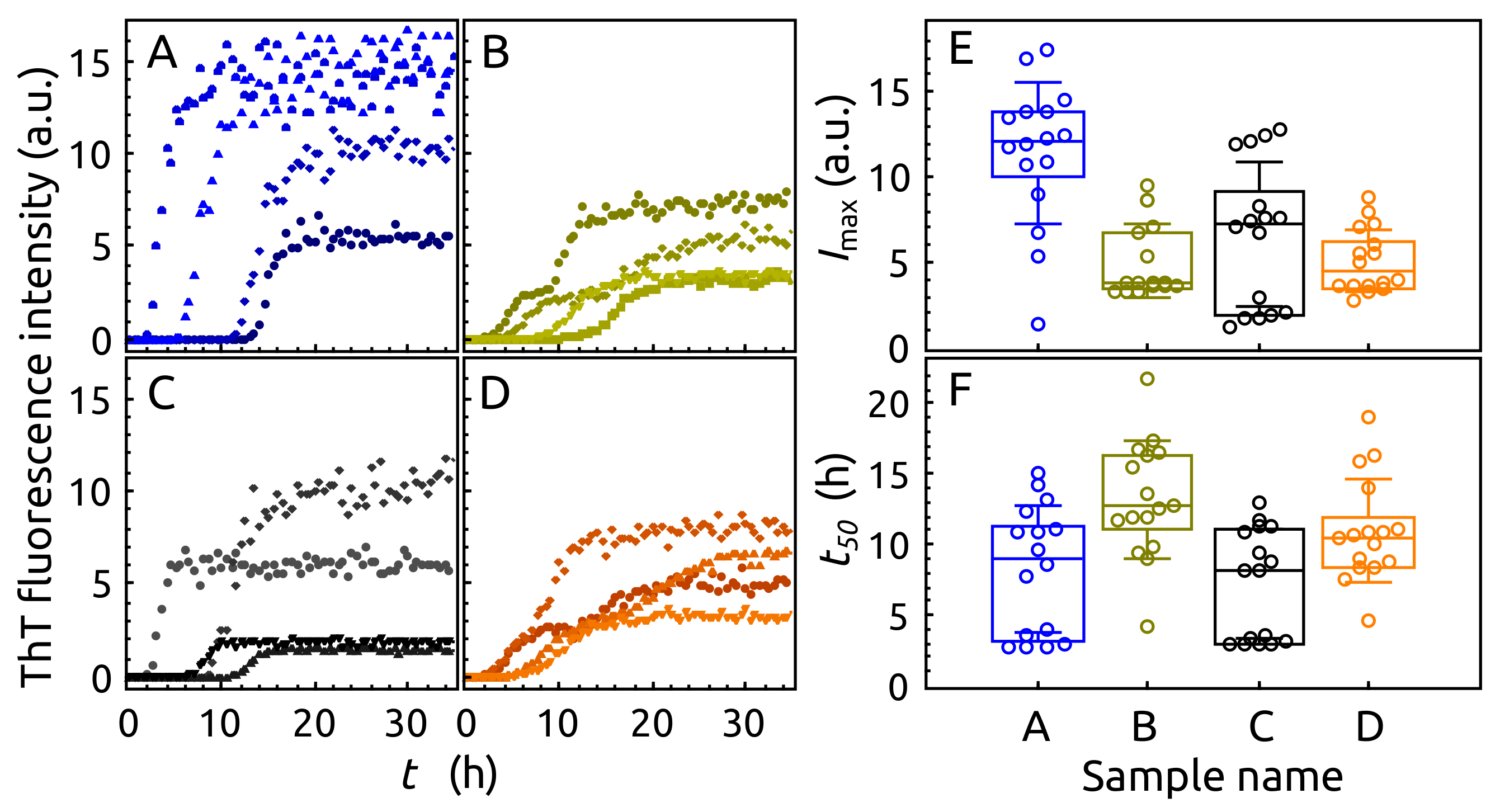
2.3. Aggregation of -syn by ThT Assay
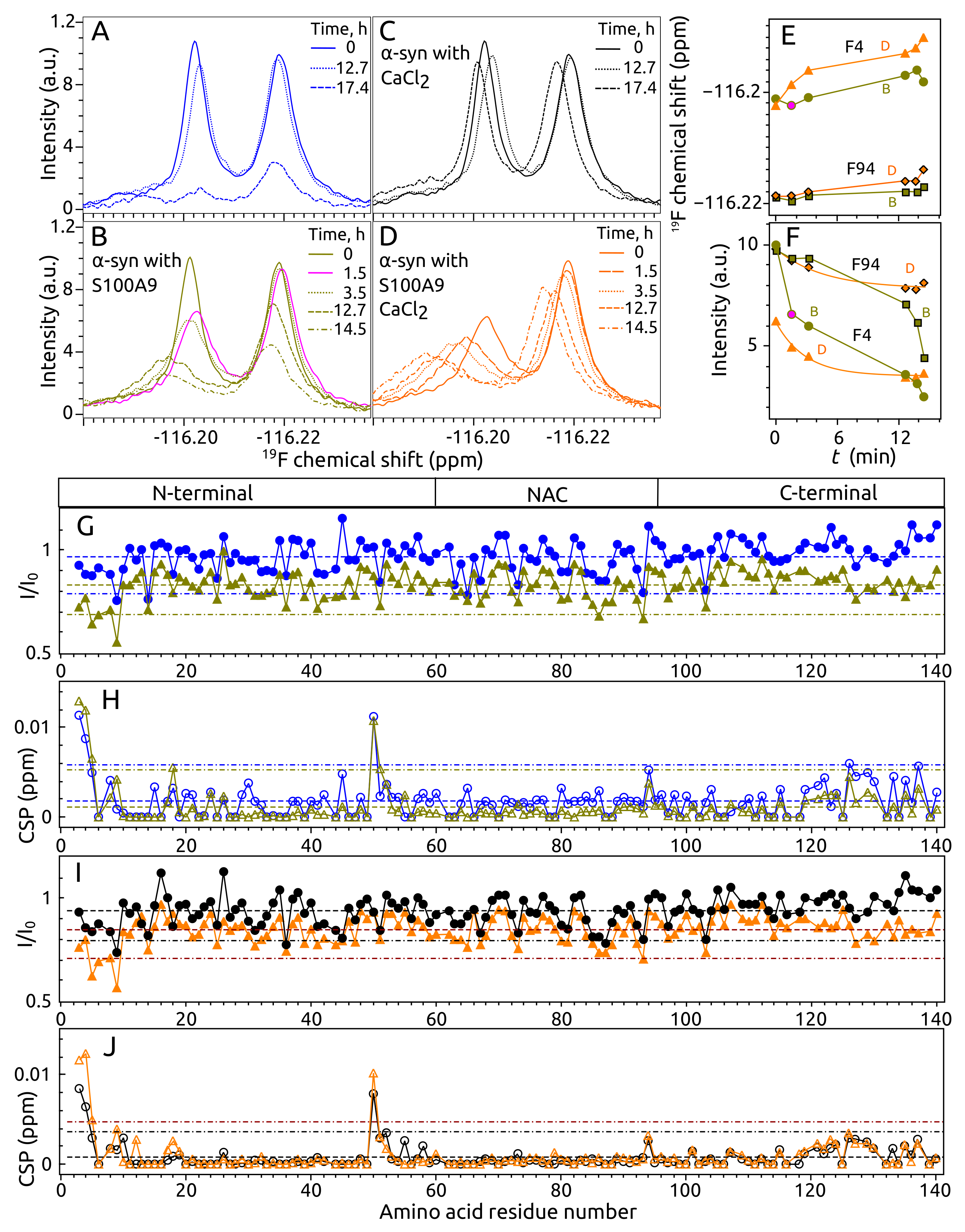
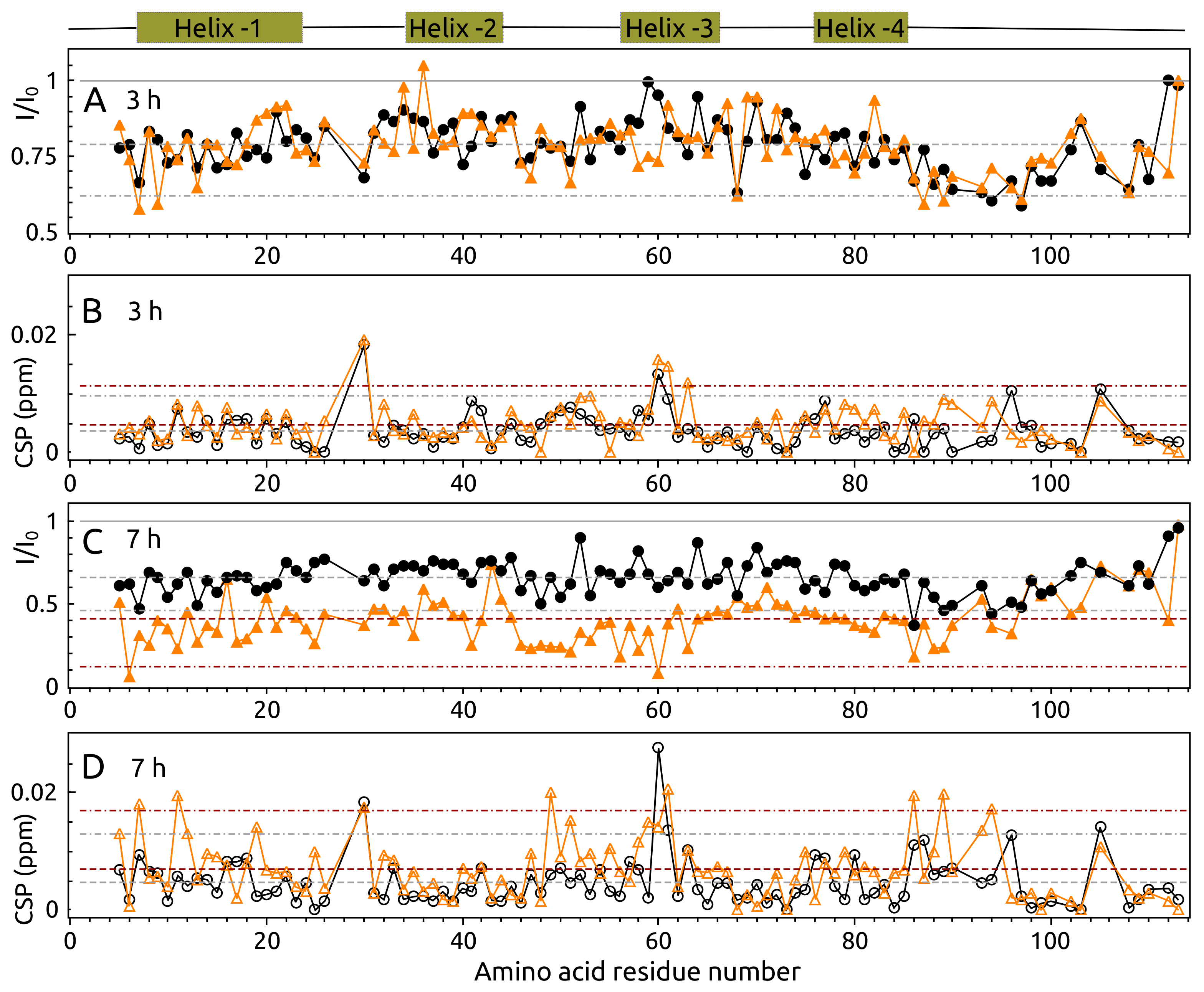
2.4. Aggregation of -syn by NMR Spectroscopy
2.5. Aggregation of S100A9 by NMR Spectroscopy
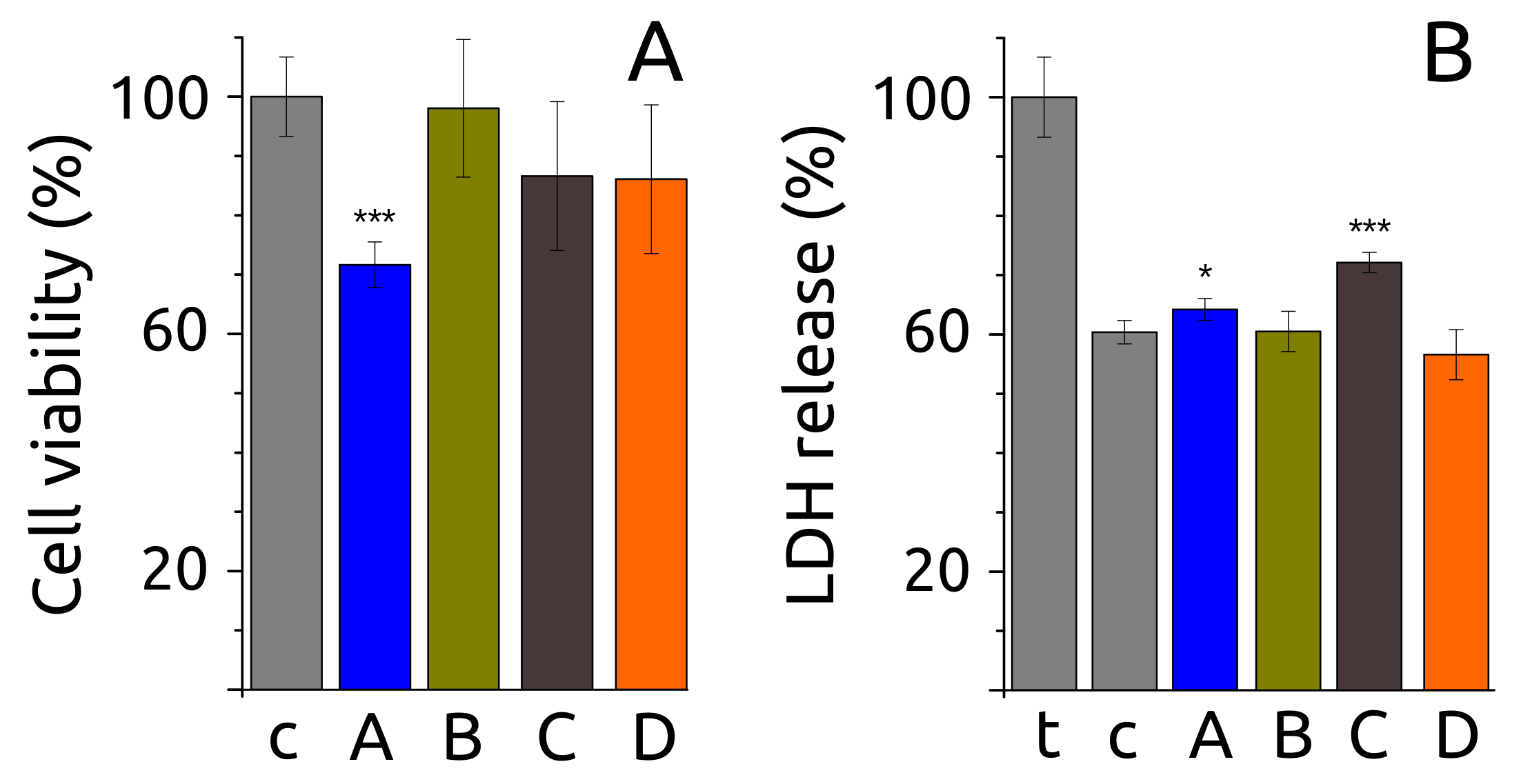
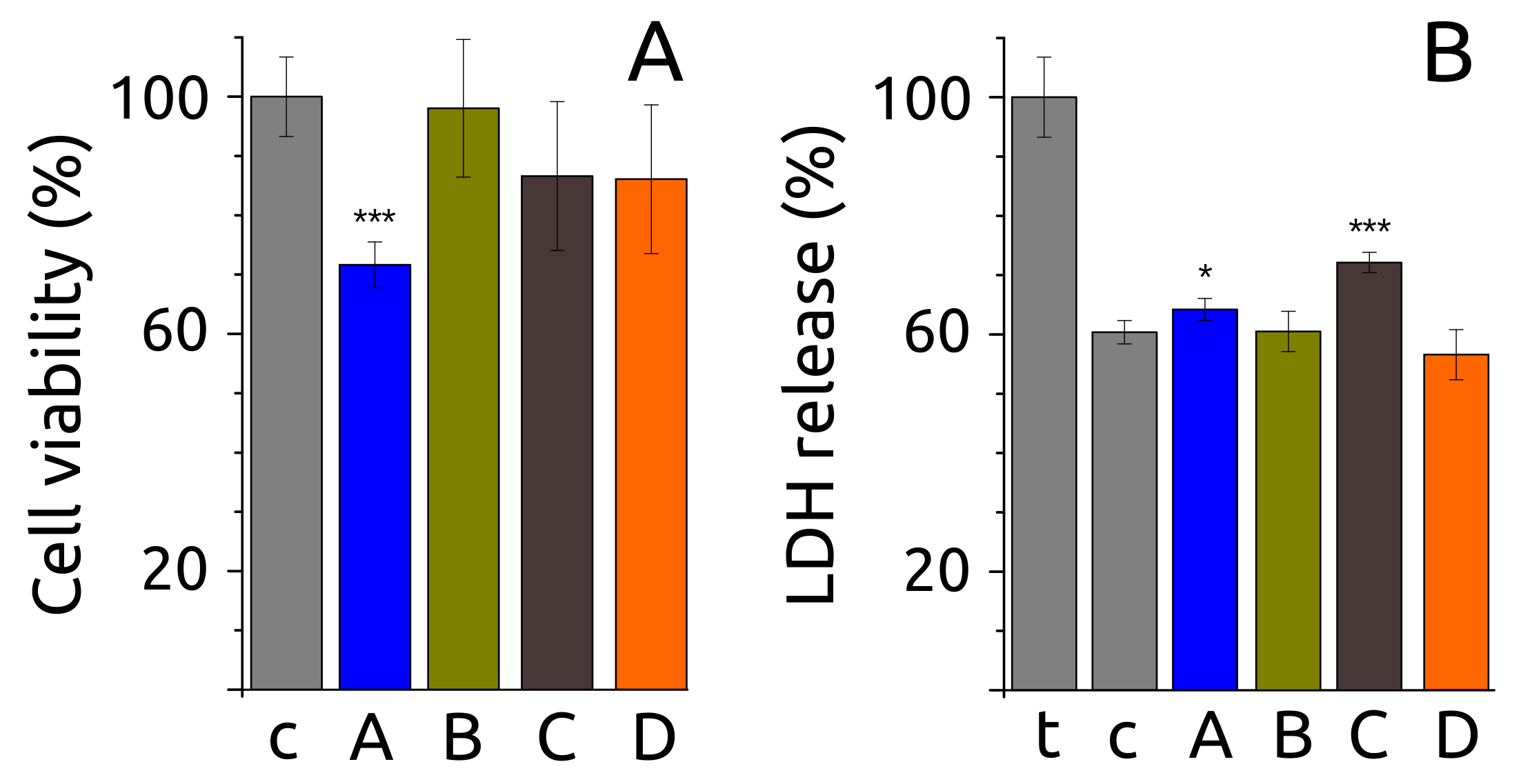
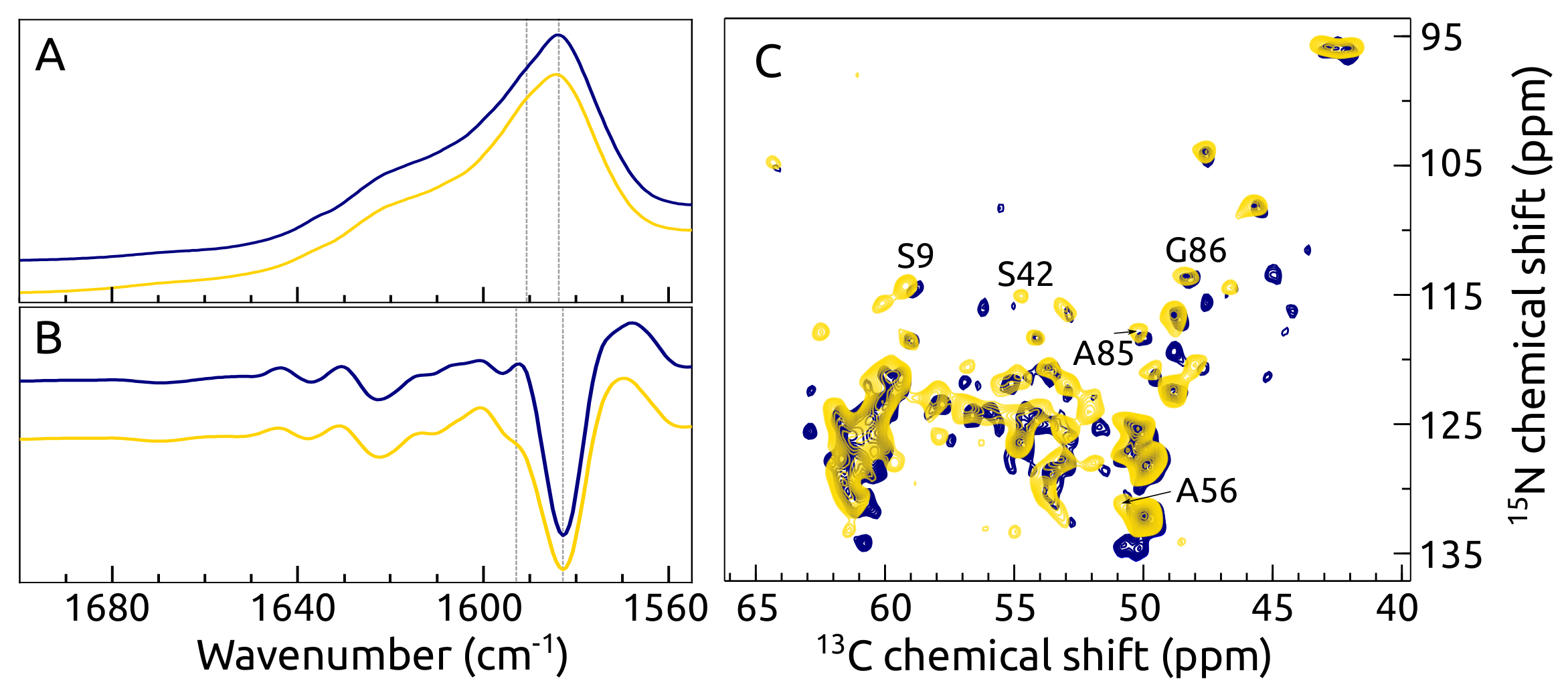
2.6. Fibril Morphology, Secondary Structure and Toxicity
2.7. C, N -syn Fibrils by FTIR and ssNMR
3. Discussion
4. Materials and Methods
4.1. Protein Production
4.2. Mutagenesis to Obtain F4Y -syn
4.3. -syn Binding with S100A9 by NMR Spectroscopy
4.4. MD Simulations
4.5. ThT Assay
4.6. -syn and S100A9 Aggregation by NMR Spectroscopy
4.7. Atomic Force Microscopy (AFM)
4.8. FTIR Spectroscopy
4.9. ssNMR Spectroscopy
4.10. Cell Culture, Cell Viability and Lactate Dehydrogenase (LDH) Release Tests
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

References
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The Amyloid State and Its Association with Protein Misfolding Diseases. Nat. Rev. Mol. Cell. Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.R.; Rice, L. The Amyloidoses: Clinical Features, Diagnosis and Treatment. Methodist Debakey Cardiovasc. J. 2012, 8, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer Disease in the United States (2010–2050) Estimated Using the 2010 Census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Egolum, U.; Parker, S.; Andrews, E.; Ombengi, D.; Ling, H. Tafamidis: A First-in-Class Transthyretin Stabilizer for Transthyretin Amyloid Cardiomyopathy. Ann. Pharmacother. 2020, 54, 470–477. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s Disease Drug Development Pipeline: 2020. Alzheimer’s Dementia Transl. Res. Clin. Interv. 2020, 6, e12050. [Google Scholar] [CrossRef]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why Do Trials for Alzheimer’s Disease Drugs Keep Failing? A Discontinued Drug Perspective for 2010–2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef]
- Alderson, T.R.; Markley, J.L. Biophysical Characterization of α-Synuclein and Its Controversial Structure. Intrinsically Disord. Proteins 2013, 1, e26255. [Google Scholar] [CrossRef]
- Lautenschläger, J.; Stephens, A.D.; Fusco, G.; Ströhl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-Terminal Calcium Binding of α-Synuclein Modulates Synaptic Vesicle Interaction. Nat. Commun. 2018, 9, 712. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Dai, C.; Bai, J.; Xu, G.; Liu, M.; Li, C. Ca2+ Modulating α-Synuclein Membrane Transient Interactions Revealed by Solution NMR Spectroscopy. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 853–858. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Jiang, X.; Xu, D.; Zheng, W.; Liu, M.; Li, C. Calcium Accelerates SNARE-mediated Lipid Mixing through Modulating α-Synuclein Membrane Interaction. Biochim. Biophys. Acta (BBA) Biomembr. 2018, 1860, 1848–1853. [Google Scholar] [CrossRef]
- Wang, L.; Das, U.; Scott, D.A.; Tang, Y.; McLean, P.J.; Roy, S. α-Synuclein Multimers Cluster Synaptic Vesicles and Attenuate Recycling. Curr. Biol. 2014, 24, 2319–2326. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Hayashi, S.; Kakita, A.; Yamada, M.; Toyoshima, Y.; Yoshimoto, M.; Takahashi, H. Accumulation of α-Synuclein/NACP Is a Cytopathological Feature Common to Lewy Body Disease and Multiple System Atrophy. Acta Neuropathol. 1998, 96, 445–452. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Yoshimoto, M.; Tsuji, S.; Takahashi, H. α-Synuclein Immunoreactivity in Glial Cytoplasmic Inclusions in Multiple System Atrophy. Neurosci. Lett. 1998, 249, 180–182. [Google Scholar] [CrossRef]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Böckmann, A.; Meier, B.H.; et al. Structural and Functional Characterization of Two Alpha-Synuclein Strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef] [Green Version]
- Gath, J.; Bousset, L.; Habenstein, B.; Melki, R.; Böckmann, A.; Meier, B.H. Unlike Twins: An NMR Comparison of Two α-Synuclein Polymorphs Featuring Different Toxicity. PLoS ONE 2014, 9, e90659. [Google Scholar] [CrossRef]
- Strohäker, T.; Jung, B.C.; Liou, S.H.; Fernandez, C.O.; Riedel, D.; Becker, S.; Halliday, G.M.; Bennati, M.; Kim, W.S.; Lee, S.J.; et al. Structural Heterogeneity of α-Synuclein Fibrils Amplified from Patient Brain Extracts. Nat. Commun. 2019, 10, 5535. [Google Scholar] [CrossRef] [Green Version]
- Van der Perren, A.; Gelders, G.; Fenyi, A.; Bousset, L.; Brito, F.; Peelaerts, W.; Van den Haute, C.; Gentleman, S.; Melki, R.; Baekelandt, V. The Structural Differences between Patient-Derived α-Synuclein Strains Dictate Characteristics of Parkinson’s Disease, Multiple System Atrophy and Dementia with Lewy Bodies. Acta Neuropathol. 2020, 139, 977–1000. [Google Scholar] [CrossRef]
- Morris, A.M.; Finke, R.G. α-Synuclein Aggregation Variable Temperature and Variable pH Kinetic Data: A Re-Analysis Using the Finke–Watzky 2-Step Model of Nucleation and Autocatalytic Growth. Biophys. Chem. 2009, 140, 9–15. [Google Scholar] [CrossRef]
- Roeters, S.J.; Iyer, A.; Pletikapić, G.; Kogan, V.; Subramaniam, V.; Woutersen, S. Evidence for Intramolecular Antiparallel Beta-Sheet Structure in Alpha-Synuclein Fibrils from a Combination of Two-Dimensional Infrared Spectroscopy and Atomic Force Microscopy. Sci. Rep. 2017, 7, 41051. [Google Scholar] [CrossRef]
- Ziaunys, M.; Sakalauskas, A.; Mikalauskaite, K.; Smirnovas, V. Polymorphism of Alpha-Synuclein Amyloid Fibrils Depends on Ionic Strength and Protein Concentration. Int. J. Mol. Sci. 2021, 22, 12382. [Google Scholar] [CrossRef]
- Afitska, K.; Fucikova, A.; Shvadchak, V.V.; Yushchenko, D.A. α-Synuclein Aggregation at Low Concentrations. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2019, 1867, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhang, S.; Ma, X.; Jia, C.; Liu, Z.; Huang, C.; Liu, C.; Li, D. Structural Basis of the Interplay between α-Synuclein and Tau in Regulating Pathological Amyloid Aggregation. J. Biol. Chem. 2020, 295, 7470–7480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köppen, J.; Schulze, A.; Machner, L.; Wermann, M.; Eichentopf, R.; Guthardt, M.; Hähnel, A.; Klehm, J.; Kriegeskorte, M.C.; Hartlage-Rübsamen, M.; et al. Amyloid-Beta Peptides Trigger Aggregation of Alpha-Synuclein In Vitro. Molecules 2020, 25, 580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rösener, N.S.; Gremer, L.; Wördehoff, M.M.; Kupreichyk, T.; Etzkorn, M.; Neudecker, P.; Hoyer, W. Clustering of Human Prion Protein and α-Synuclein Oligomers Requires the Prion Protein N-terminus. Commun. Biol. 2020, 3, 365. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.G.; Temido-Ferreira, M.; Vicente Miranda, H.; Batalha, V.L.; Coelho, J.E.; Szegö, É.M.; Marques-Morgado, I.; Vaz, S.H.; Rhee, J.S.; Schmitz, M.; et al. α-Synuclein Interacts with PrP C to Induce Cognitive Impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 2017, 20, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Rockenstein, E.; Inglis, C.; Adame, A.; Bett, C.; Lucero, M.; Sigurdson, C.J. Prion Infection Promotes Extensive Accumulation of α-Synuclein in Aged Human α-Synuclein Transgenic Mice. Prion 2012, 6, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Horvath, I.; Iashchishyn, I.A.; Moskalenko, R.A.; Wang, C.; Wärmländer, S.K.T.S.; Wallin, C.; Gräslund, A.; Kovacs, G.G.; Morozova-Roche, L.A. Co-Aggregation of pro-Inflammatory S100A9 with α-Synuclein in Parkinson’s Disease: Ex Vivo and in Vitro Studies. J. Neuroinflammation 2018, 15, 172. [Google Scholar] [CrossRef]
- Bellomo, G.; Bologna, S.; Cerofolini, L.; Paciotti, S.; Gatticchi, L.; Ravera, E.; Parnetti, L.; Fragai, M.; Luchinat, C. Dissecting the Interactions between Human Serum Albumin and α-Synuclein: New Insights on the Factors Influencing α-Synuclein Aggregation in Biological Fluids. J. Phys. Chem. B 2019, 123, 4380–4386. [Google Scholar] [CrossRef]
- Chaari, A.; Eliezer, D.; Ladjimi, M. The C-terminal α-Helices of Mammalian Hsc70 Play a Critical Role in the Stabilization of α-Synuclein Binding and Inhibition of Aggregation. Int. J. Biol. Macromol. 2016, 83, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.; Selig, E.; Griffin, M.D.W.; Carver, J.A.; Ecroyd, H. Small Heat-shock Proteins Prevent α-Synuclein Aggregation via Transient Interactions and Their Efficacy Is Affected by the Rate of Aggregation*. J. Biol. Chem. 2016, 291, 22618–22629. [Google Scholar] [CrossRef] [Green Version]
- Vogl, T.; Gharibyan, A.L.; Morozova-Roche, L.A. Pro-Inflammatory S100A8 and S100A9 Proteins: Self-Assembly into Multifunctional Native and Amyloid Complexes. Int. J. Mol. Sci. 2012, 13, 2893–2917. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, J.; Carson, W.E. Review of S100A9 Biology and Its Role in Cancer. Biochim. Biophys. Acta 2013, 1835, 100–109. [Google Scholar] [CrossRef] [Green Version]
- Hessian, P.A.; Edgeworth, J.; Hogg, N. MRP-8 and MRP-14, Two Abundant Ca2+-Binding Proteins of Neutrophils and Monocytes. J. Leukoc. Biol. 1993, 53, 197–204. [Google Scholar] [CrossRef]
- Itou, H.; Yao, M.; Fujita, I.; Watanabe, N.; Suzuki, M.; Nishihira, J.; Tanaka, I. The Crystal Structure of Human MRP14 (S100A9), a Ca2+-Dependent Regulator Protein in Inflammatory Process. J. Mol. Biol. 2002, 316, 265–276. [Google Scholar] [CrossRef]
- Damo, S.M.; Kehl-Fie, T.E.; Sugitani, N.; Holt, M.E.; Rathi, S.; Murphy, W.J.; Zhang, Y.; Betz, C.; Hench, L.; Fritz, G.; et al. Molecular Basis for Manganese Sequestration by Calprotectin and Roles in the Innate Immune Response to Invading Bacterial Pathogens. Proc. Natl. Acad. Sci. USA 2013, 110, 3841–3846. [Google Scholar] [CrossRef] [Green Version]
- Nakashige, T.G.; Zygiel, E.M.; Drennan, C.L.; Nolan, E.M. Nickel Sequestration by the Host-Defense Protein Human Calprotectin. J. Am. Chem. Soc. 2017, 139, 8828–8836. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Khan, I.; Tsai, K.L.; Li, H.; Yang, L.W.; Chou, R.H.; Yu, C. Blocking the Interaction between S100A9 and RAGE V Domain Using CHAPS Molecule: A Novel Route to Drug Development against Cell Proliferation. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2016, 1864, 1558–1569. [Google Scholar] [CrossRef]
- Iashchishyn, I.A.; Sulskis, D.; Nguyen Ngoc, M.; Smirnovas, V.; Morozova-Roche, L.A. Finke–Watzky Two-Step Nucleation–Autocatalysis Model of S100A9 Amyloid Formation: Protein Misfolding as “Nucleation” Event. ACS Chem. Neurosci. 2017, 8, 2152–2158. [Google Scholar] [CrossRef]
- Wang, C.; Klechikov, A.G.; Gharibyan, A.L.; Wärmländer, S.K.T.S.; Jarvet, J.; Zhao, L.; Jia, X.; Shankar, S.K.; Olofsson, A.; Brännström, T.; et al. The Role of Pro-Inflammatory S100A9 in Alzheimer’s Disease Amyloid-Neuroinflammatory Cascade. Acta Neuropathol. 2014, 127, 507–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, I.; Jia, X.; Johansson, P.; Wang, C.; Moskalenko, R.; Steinau, A.; Forsgren, L.; Wågberg, T.; Svensson, J.; Zetterberg, H.; et al. Pro-Inflammatory S100A9 Protein as a Robust Biomarker Differentiating Early Stages of Cognitive Impairment in Alzheimer’s Disease. ACS Chem. Neurosci. 2016, 7, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Iashchishyn, I.A.; Pansieri, J.; Nyström, S.; Klementieva, O.; Kara, J.; Horvath, I.; Moskalenko, R.; Rofougaran, R.; Gouras, G.; et al. S100A9-Driven Amyloid-Neuroinflammatory Cascade in Traumatic Brain Injury as a Precursor State for Alzheimer’s Disease. Sci. Rep. 2018, 8, 12836. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Iashchishyn, I.A.; Kara, J.; Foderà, V.; Vetri, V.; Sancataldo, G.; Marklund, N.; Morozova-Roche, L.A. Proinflammatory and Amyloidogenic S100A9 Induced by Traumatic Brain Injury in Mouse Model. Neurosci. Lett. 2019, 699, 199–205. [Google Scholar] [CrossRef]
- Toleikis, Z.; Ziaunys, M.; Baranauskiene, L.; Petrauskas, V.; Jaudzems, K.; Smirnovas, V. S100A9 Alters the Pathway of Alpha-Synuclein Amyloid Aggregation. Int. J. Mol. Sci. 2021, 22, 7972. [Google Scholar] [CrossRef]
- Pansieri, J.; Iashchishyn, I.A.; Fakhouri, H.; Ostojić, L.; Malisauskas, M.; Musteikyte, G.; Smirnovas, V.; Schneider, M.M.; Scheidt, T.; Xu, C.K.; et al. Templating S100A9 Amyloids on Aβ Fibrillar Surfaces Revealed by Charge Detection Mass Spectrometry, Microscopy, Kinetic and Microfluidic Analyses. Chem. Sci. 2020, 11, 7031–7039. [Google Scholar] [CrossRef]
- Boeszoermenyi, A.; Ogórek, B.; Jain, A.; Arthanari, H.; Wagner, G. The Precious Fluorine on the Ring: Fluorine NMR for Biological Systems. J. Biomol. NMR 2020, 74, 365–379. [Google Scholar] [CrossRef]
- Welte, H.; Zhou, T.; Mihajlenko, X.; Mayans, O.; Kovermann, M. What Does Fluorine Do to a Protein? Thermodynamic, and Highly-Resolved Structural Insights into Fluorine-Labelled Variants of the Cold Shock Protein. Sci. Rep. 2020, 10, 2640. [Google Scholar] [CrossRef]
- Kitevski-LeBlanc, J.L.; Evanics, F.; Scott Prosser, R. Optimizing 19F NMR Protein Spectroscopy by Fractional Biosynthetic Labeling. J. Biomol. NMR 2010, 48, 113–121. [Google Scholar] [CrossRef]
- Heise, H.; Hoyer, W.; Becker, S.; Andronesi, O.C.; Riedel, D.; Baldus, M. Molecular-Level Secondary Structure, Polymorphism, and Dynamics of Full-Length α-Synuclein Fibrils Studied by Solid-State NMR. Proc. Natl. Acad. Sci. USA 2005, 102, 15871–15876. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-State NMR Structure of a Pathogenic Fibril of Full-Length Human α-Synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef]
- De Giorgi, F.; Laferrière, F.; Zinghirino, F.; Faggiani, E.; Lends, A.; Bertoni, M.; Yu, X.; Grélard, A.; Morvan, E.; Habenstein, B.; et al. Novel Self-Replicating α-Synuclein Polymorphs That Escape ThT Monitoring Can Spontaneously Emerge and Acutely Spread in Neurons. Sci. Adv. 2020, 6, eabc4364. [Google Scholar] [CrossRef]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide Folding: When Simulation Meets Experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Ruggeri, F.S.; Šneideris, T.; Vendruscolo, M.; Knowles, T.P. Atomic Force Microscopy for Single Molecule Characterisation of Protein Aggregation. Arch. Biochem. Biophys. 2019, 664, 134–148. [Google Scholar] [CrossRef]
- Barth, A. Infrared Spectroscopy of Proteins. Biochim. Biophys. Acta (BBA) Bioenerg. 2007, 1767, 1073–1101. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the Lactate Dehydrogenase Assay. Cold Spring Harb Protoc. 2018, 2018, pdb.prot095497. [Google Scholar] [CrossRef]
- Tadesse, L.; Nazarbaghi, R.; Walters, L. Isotopically Enhanced Infrared Spectroscopy: A Novel Method for Examining Secondary Structure at Specific Sites in Conformationally Heterogeneous Peptides. J. Am. Chem. Soc. 1991, 113, 7036–7037. [Google Scholar] [CrossRef]
- Halverson, K.J.; Sucholeiki, I.; Ashburn, T.T.; Lansbury, P.T. Location of Beta-Sheet-Forming Sequences in Amyloid Proteins by FTIR. J. Am. Chem. Soc. 1991, 113, 6701–6703. [Google Scholar] [CrossRef]
- Katte, R.; Yu, C. Blocking the Interaction between S100A9 Protein and RAGE V Domain Using S100A12 Protein. PLoS ONE 2018, 13, e0198767. [Google Scholar] [CrossRef]
- Wang, G.F.; Li, C.; Pielak, G.J. 19F NMR Studies of α-Synuclein-Membrane Interactions. Protein Sci. 2010, 19, 1686–1691. [Google Scholar] [CrossRef] [Green Version]
- Zigoneanu, I.G.; Pielak, G.J. Interaction of α-Synuclein and a Cell Penetrating Fusion Peptide with Higher Eukaryotic Cell Membranes Assessed by 19F NMR. Mol. Pharm. 2012, 9, 1024–1029. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Lutz, E.A.; Slade, K.M.; Ruf, R.A.; Wang, G.F.; Pielak, G.J. 19F-NMR Studies of α-Synuclein Conformation and Fibrillation. Biochemistry 2009, 48, 8578–8584. [Google Scholar] [CrossRef] [Green Version]
- Gee, C.T.; Arntson, K.E.; Urick, A.K.; Mishra, N.K.; Hawk, L.M.L.; Wisniewski, A.J.; Pomerantz, W.C.K. Protein-Observed 19F-NMR for Fragment Screening, Affinity Quantification and Druggability Assessment. Nat. Protoc. 2016, 11, 1414–1427. [Google Scholar] [CrossRef]
- Hunter, M.J.; Chazin, W.J. High Level Expression and Dimer Characterization of the S100 EF-hand Proteins, Migration Inhibitory Factor-related Proteins 8 and 14. J. Biol. Chem. 1998, 273, 12427–12435. [Google Scholar] [CrossRef] [Green Version]
- Weiner, M.P.; Costa, G.L. Rapid PCR Site-Directed Mutagenesis. Genome Res. 1994, 4, S131–S136. [Google Scholar] [CrossRef]
- Rabhi, I.; Guedel, N.; Chouk, I.; Zerria, K.; Barbouche, M.R.; Dellagi, K.; Fathallah, D.M. A Novel Simple and Rapid PCR-based Site-Directed Mutagenesis Method. Mol. Biotechnol. 2004, 26, 27–34. [Google Scholar] [CrossRef]
- Schrödinger Inc. Maestro, Schrödinger Release 2020-4; Schrödinger Inc.: New York, NY, USA, 2020. [Google Scholar]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Inc. Prime, Schrödinger Release 2020-4; Schrödinger Inc.: New York, NY, USA, 2020. [Google Scholar]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-Chain Torsion Potentials for the Amber ff99SB Protein Force Field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Madura, J.D. Quantum and Statistical Mechanical Studies of Liquids. 25. Solvation and Conformation of Methanol in Water. J. Am. Chem. Soc. 1983, 105, 1407–1413. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Gunsteren, W.F.V.; Dinola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Gade Malmos, K.; Blancas-Mejia, L.M.; Weber, B.; Buchner, J.; Ramirez-Alvarado, M.; Naiki, H.; Otzen, D. ThT 101: A Primer on the Use of Thioflavin T to Investigate Amyloid Formation. Amyloid 2017, 24, 1–16. [Google Scholar] [CrossRef]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN Data Model for NMR Spectroscopy: Development of a Software Pipeline. Proteins 2005, 59, 687–696. [Google Scholar] [CrossRef]
- Teilum, K.; Kunze, M.B.A.; Erlendsson, S.; Kragelund, B.B. (S)Pinning down Protein Interactions by NMR. Protein Sci. 2017, 26, 436–451. [Google Scholar] [CrossRef] [Green Version]
- Nečas, D.; Klapetek, P. Gwyddion: An Open-Source Software for SPM Data Analysis. Open Phys. 2012, 10, 181–188. [Google Scholar] [CrossRef]
- Mikalauskaite, K.; Ziaunys, M.; Sneideris, T.; Smirnovas, V. Effect of Ionic Strength on Thioflavin-T Affinity to Amyloid Fibrils and Its Fluorescence Intensity. Int. J. Mol. Sci. 2020, 21, 8916. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toleikis, Z.; Bobrovs, R.; Janoniene, A.; Lends, A.; Ziaunys, M.; Baronaite, I.; Petrauskas, V.; Kitoka, K.; Smirnovas, V.; Jaudzems, K. Interactions between S100A9 and Alpha-Synuclein: Insight from NMR Spectroscopy. Int. J. Mol. Sci. 2022, 23, 6781. https://doi.org/10.3390/ijms23126781
Toleikis Z, Bobrovs R, Janoniene A, Lends A, Ziaunys M, Baronaite I, Petrauskas V, Kitoka K, Smirnovas V, Jaudzems K. Interactions between S100A9 and Alpha-Synuclein: Insight from NMR Spectroscopy. International Journal of Molecular Sciences. 2022; 23(12):6781. https://doi.org/10.3390/ijms23126781
Chicago/Turabian StyleToleikis, Zigmantas, Raitis Bobrovs, Agne Janoniene, Alons Lends, Mantas Ziaunys, Ieva Baronaite, Vytautas Petrauskas, Kristine Kitoka, Vytautas Smirnovas, and Kristaps Jaudzems. 2022. "Interactions between S100A9 and Alpha-Synuclein: Insight from NMR Spectroscopy" International Journal of Molecular Sciences 23, no. 12: 6781. https://doi.org/10.3390/ijms23126781
APA StyleToleikis, Z., Bobrovs, R., Janoniene, A., Lends, A., Ziaunys, M., Baronaite, I., Petrauskas, V., Kitoka, K., Smirnovas, V., & Jaudzems, K. (2022). Interactions between S100A9 and Alpha-Synuclein: Insight from NMR Spectroscopy. International Journal of Molecular Sciences, 23(12), 6781. https://doi.org/10.3390/ijms23126781