
Development and Biochemical Characterization of Self-Immolative Linker Containing GnRH-III-Drug Conjugates

 ,
, 
 , and
, and 
Abstract
:1. Introduction
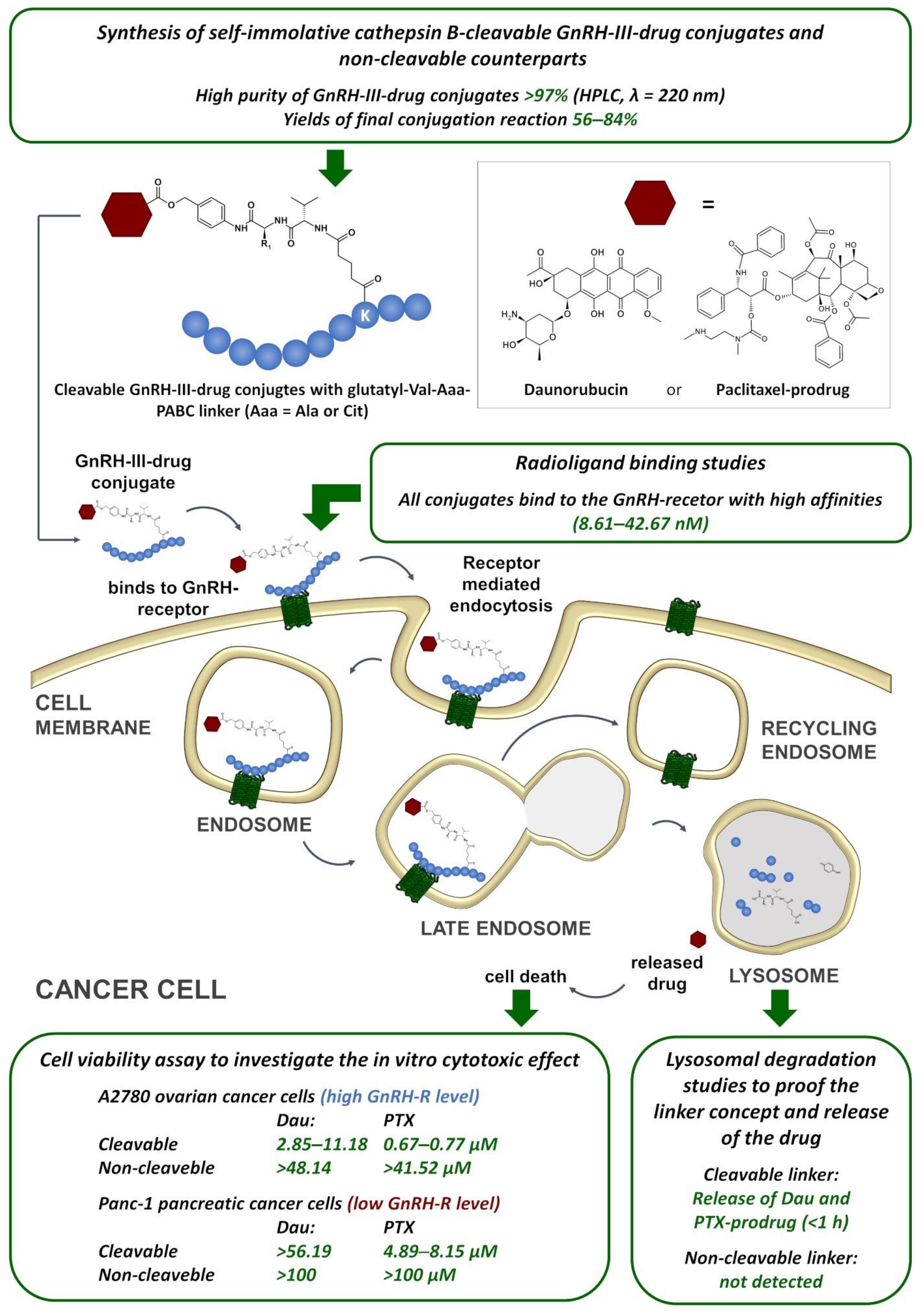
2. Results
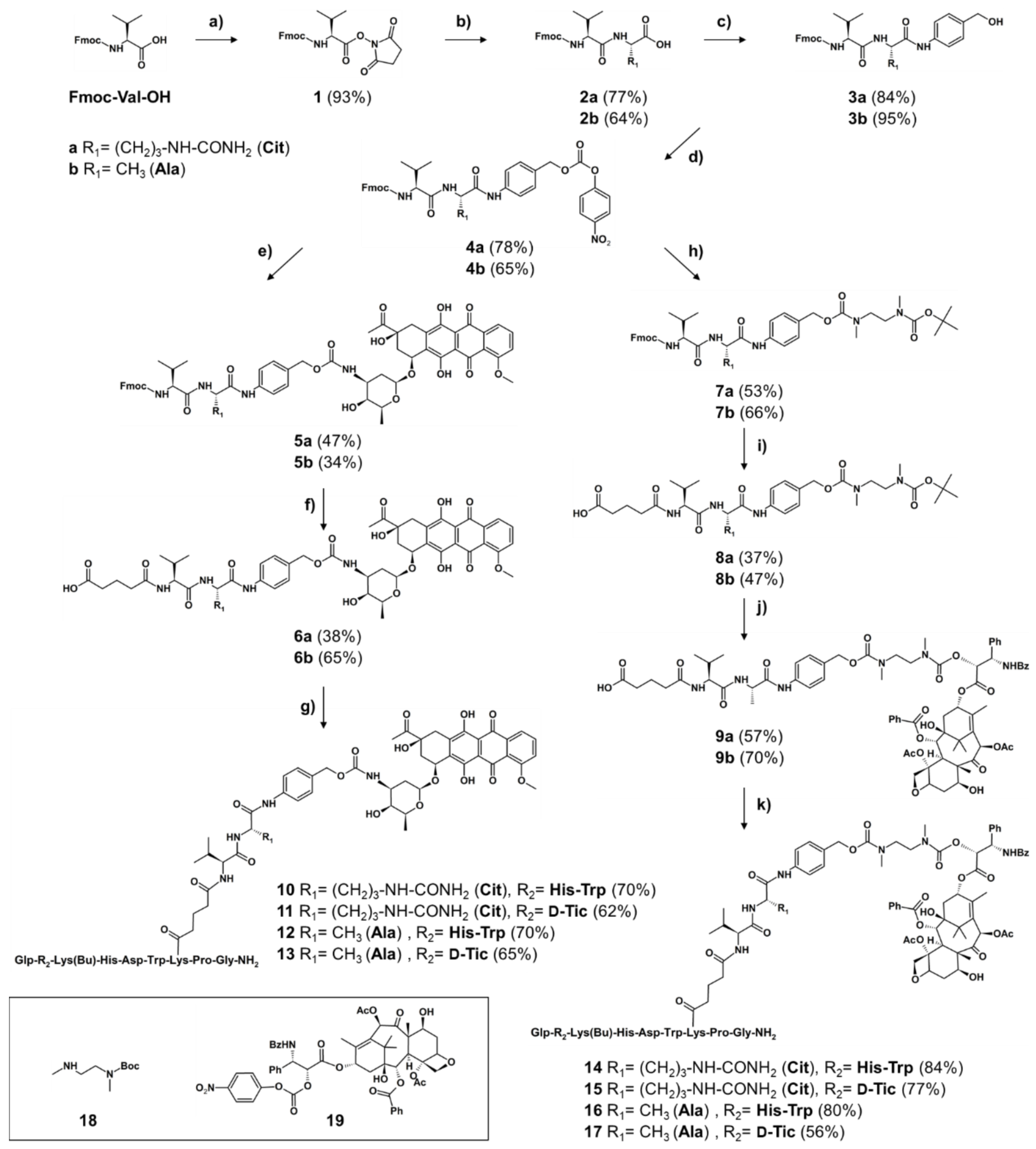
2.1. Synthesis
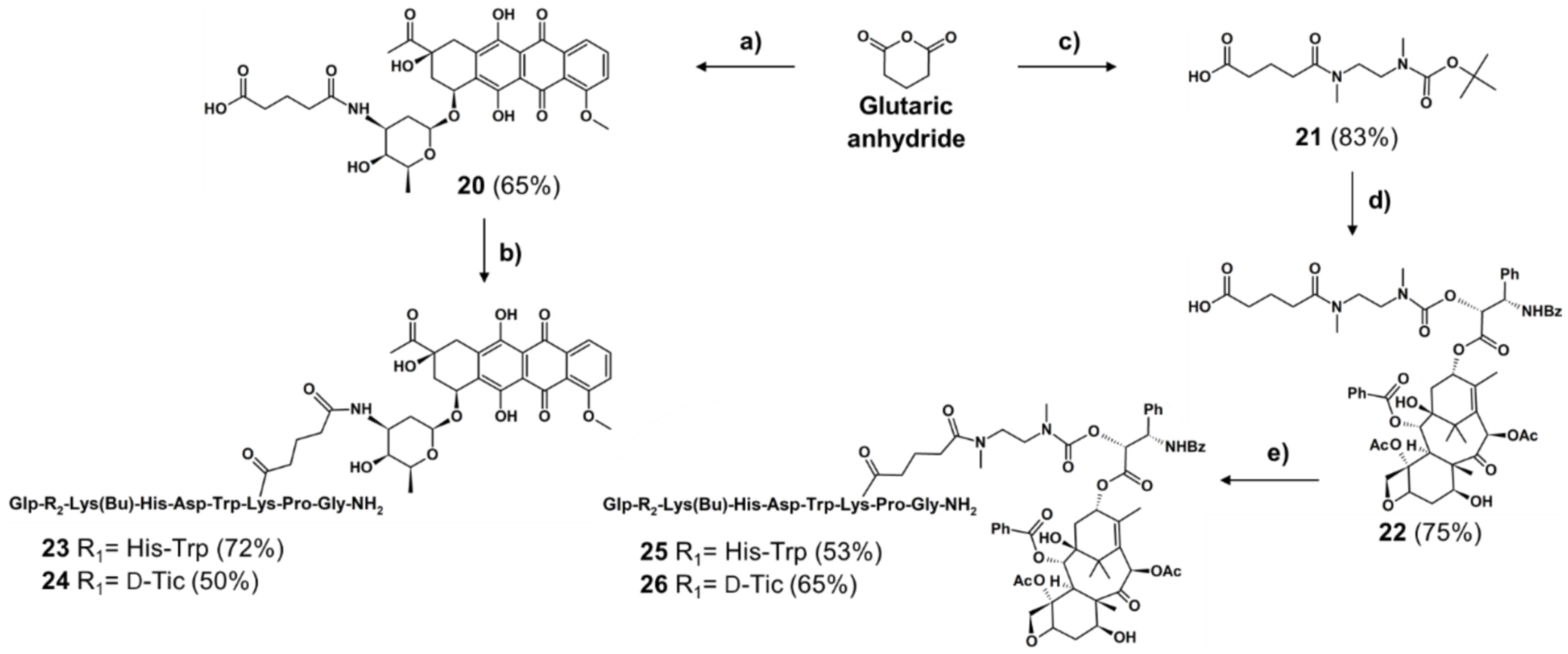
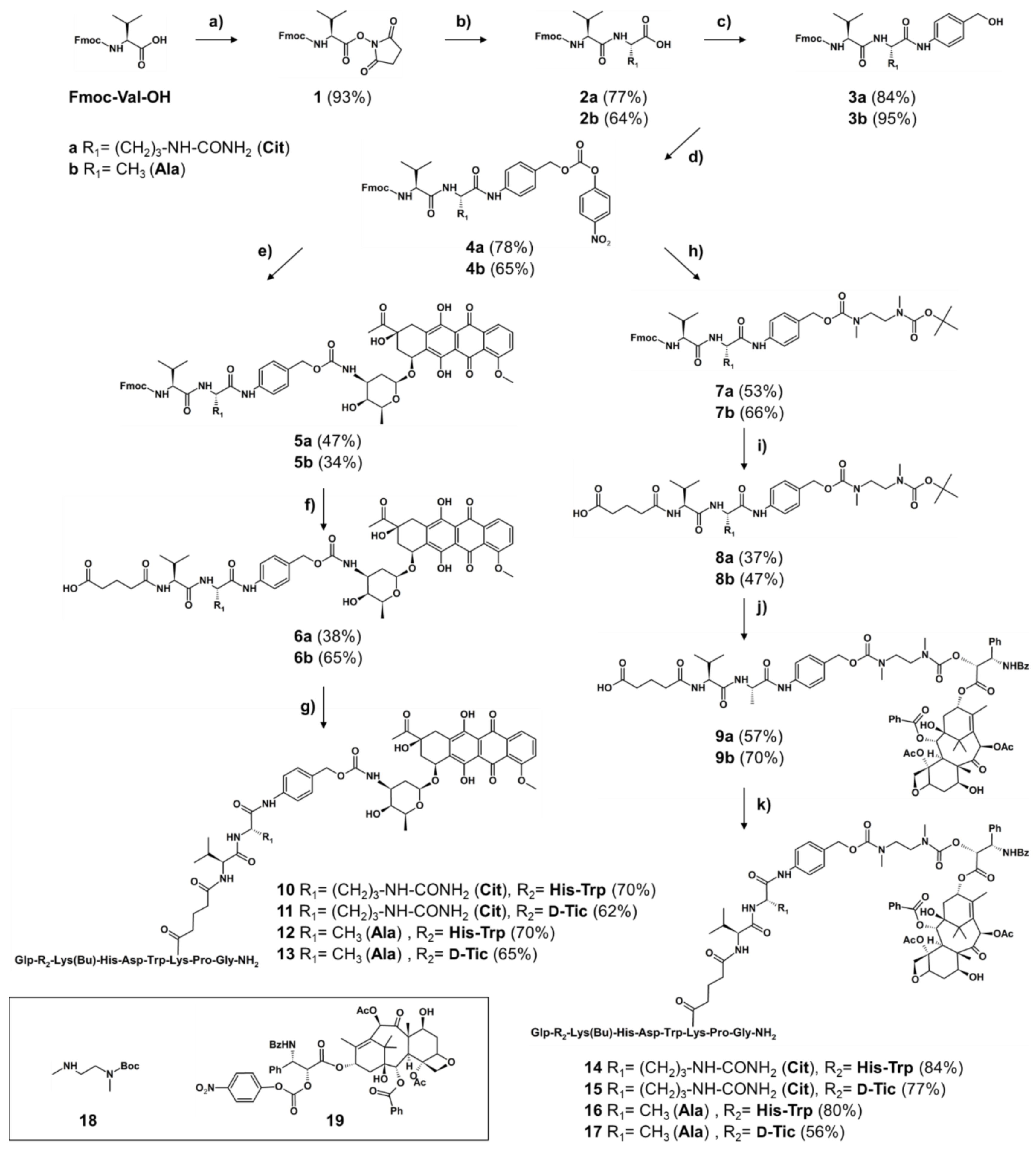
2.1.1. Synthesis of Cleavable Linker Containing GnRH-Drug Conjugates
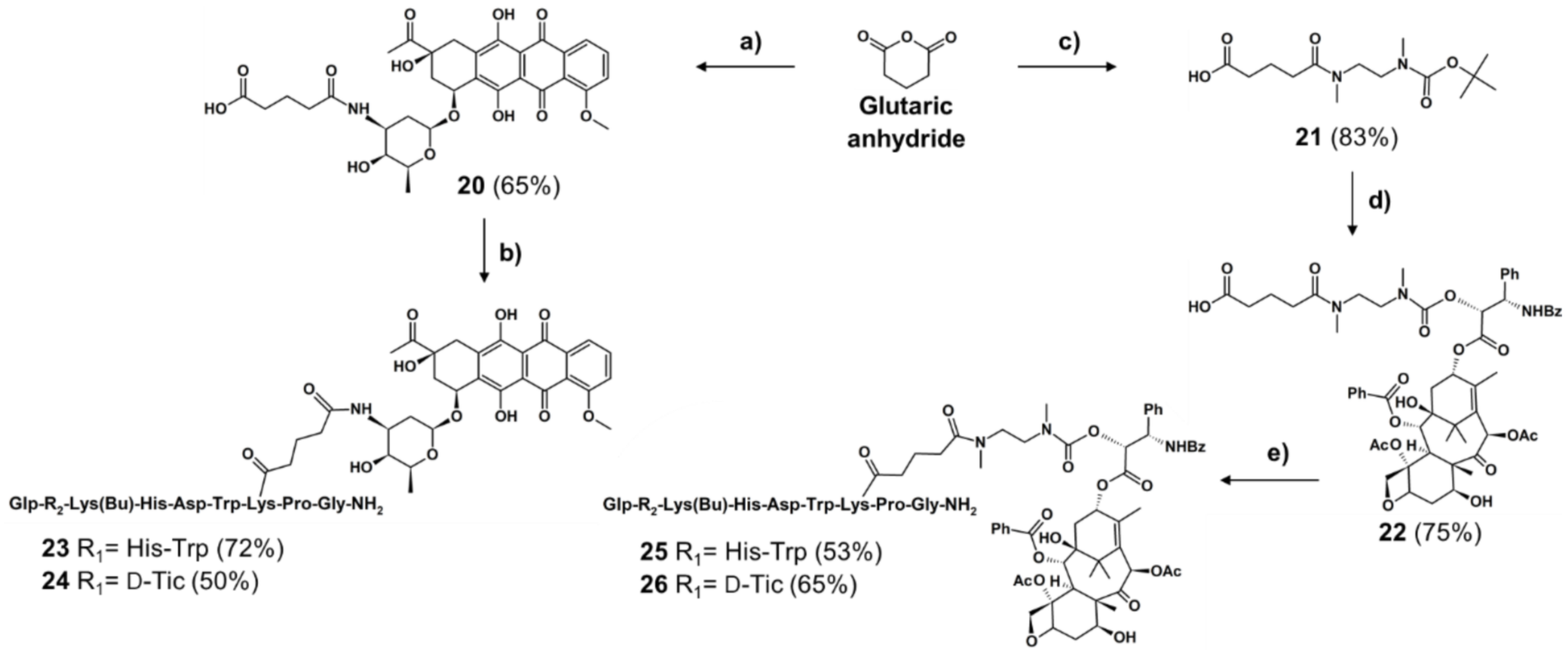
2.1.2. Synthesis of Non-Cleavable Linker-Containing GnRH-III-Drug Conjugates
2.2. In Vitro Cytotoxic Effect
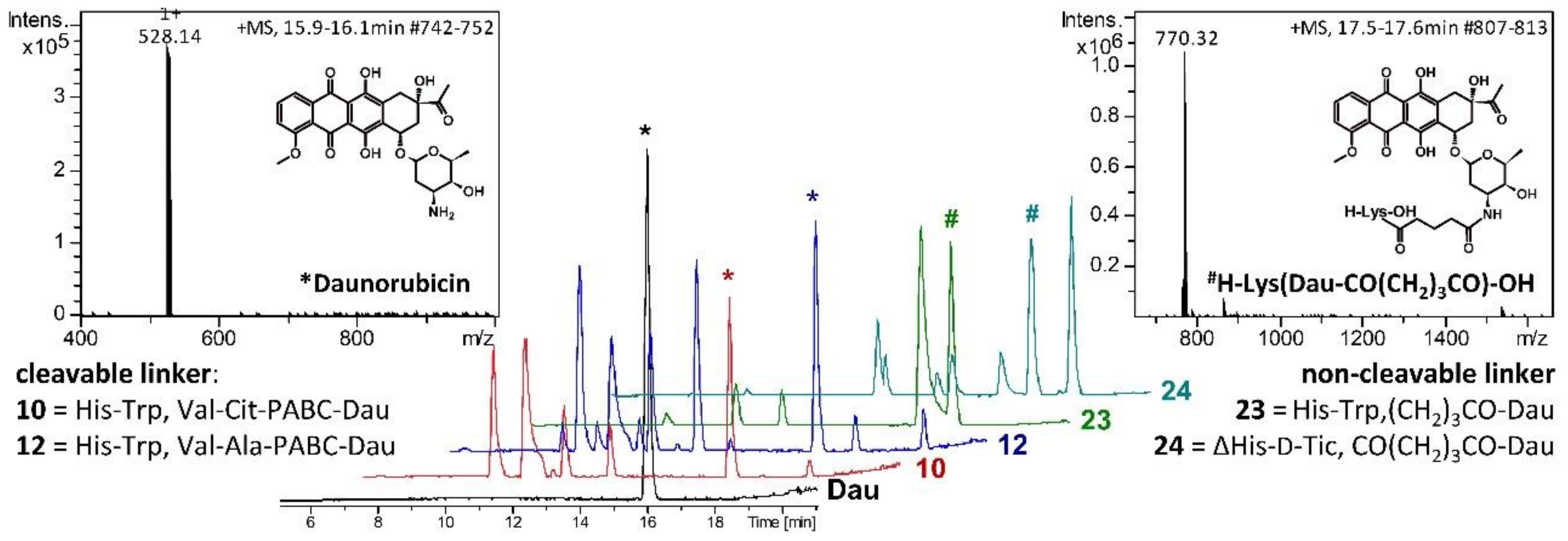
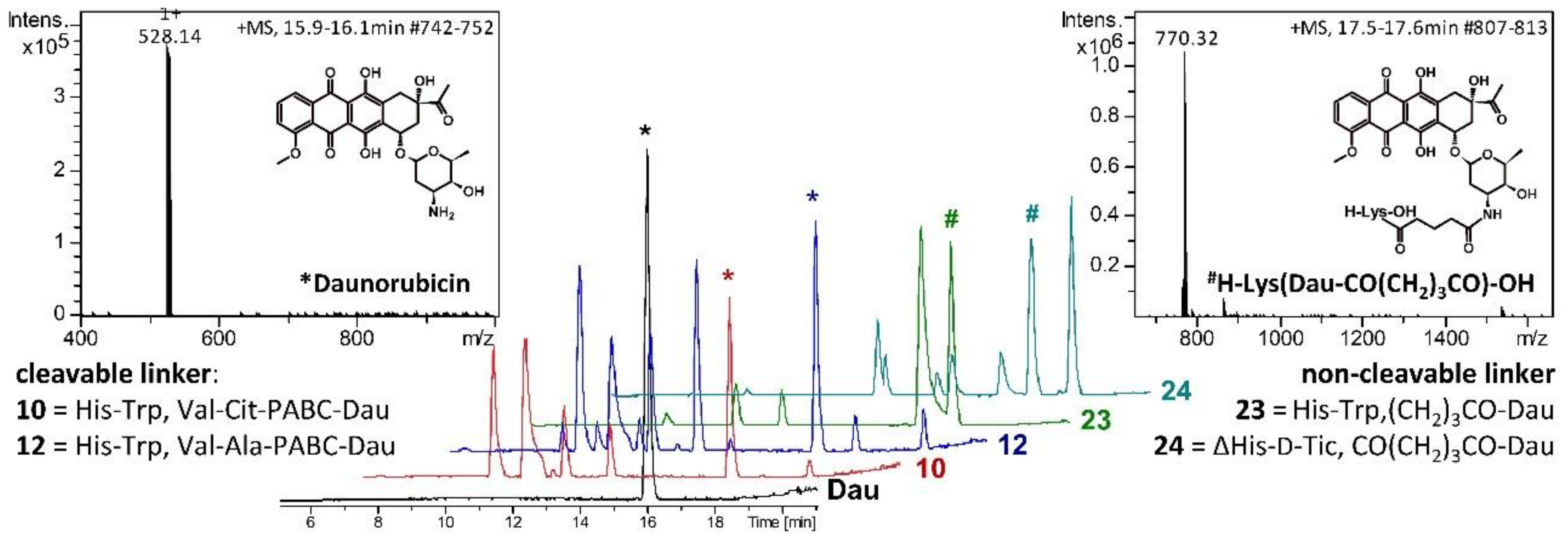
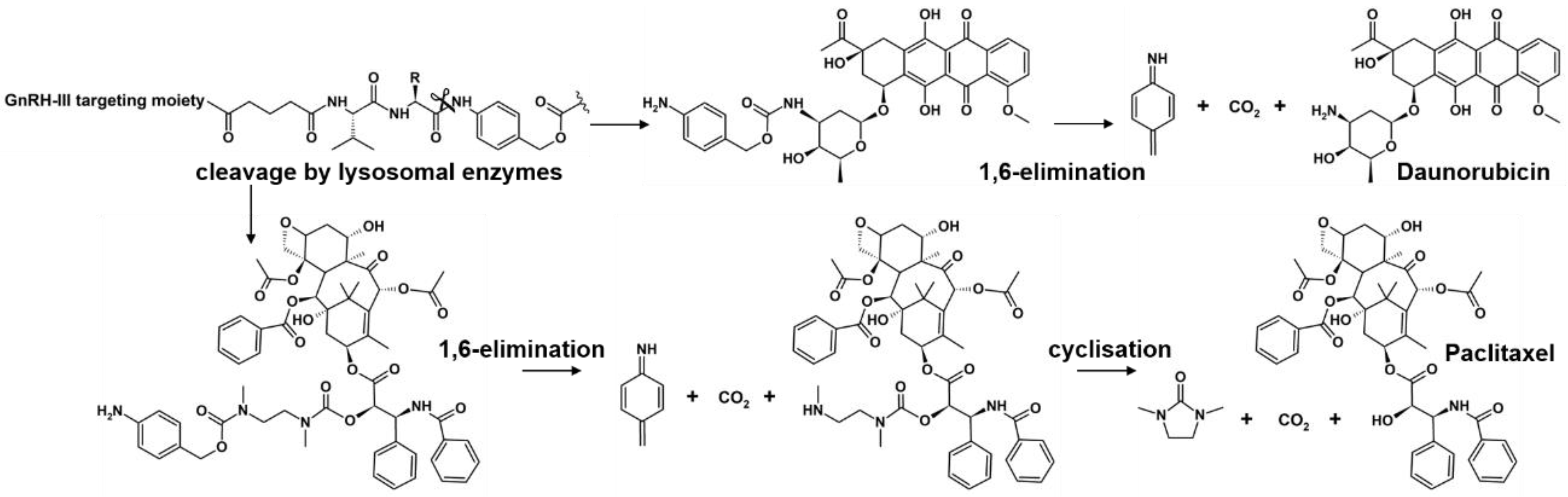
2.3. Lysosomal Degradation in Presence of Rat Liver Lysosomal Homogenate
2.4. Radioligand Binding Studies
3. Discussion
4. Materials and Methods
4.1. Synthesis
4.1.1. Synthesis of Peptide Carriers
4.1.2. Synthesis of Self-Immolative and Non-Cleavable Drug Linker
4.1.3. Conjugation Reaction of Drug-Linker and GnRH-III Peptide
4.2. In Vitro Antiproliferative Activity
4.3. Degradation of Drug Conjugates in Presence of Rat Liver Lysosomal Homogenate
4.4. Western Blotting
4.5. Radioligand Binding Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fosgerau, K.; Hoffmann, T. Peptide Therapeutics: Current Status and Future Directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagimori, M.; Fuchigami, Y.; Kawakami, S. Peptide-Based Cancer-Targeted DDS and Molecular Imaging. Chem. Pharm. Bull. 2017, 65, 618–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, A.; Schally, A.V. Targeting of Cytotoxic Luteinizing Hormone-Releasing Hormone Analogs to Breast, Ovarian, Endometrial, and Prostate Cancers. Biol. Reprod. 2005, 73, 851–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sower, S.A.; Chiang, Y.C.; Lovas, S.; Conlon, J.M. Primary Structure and Biological Activity of a Third Gonadotropin-Releasing Hormone from Lamprey Brain. Endocrinology 1993, 132, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Lovas, S.; Pályi, I.; Vincze, B.; Horváth, J.; Kovács, M.; Mezö, I.; Tóth, G.; Teplán, I.; Murphy, R.F. Direct Anticancer Activity of Gonadotropin-Releasing Hormone-III. J. Pept. Res. 1998, 52, 384–389. [Google Scholar] [CrossRef]
- Kovács, M.; Vincze, B.; Horváth, J.E.; Seprődi, J. Structure–Activity Study on the LH- and FSH-Releasing and Anticancer Effects of Gonadotropin-Releasing Hormone (GnRH)-III Analogs. Peptides 2007, 28, 821–829. [Google Scholar] [CrossRef]
- Mezo, G.; Manea, M.; Szabí, I.; Vincze, B.; Kovács, M. New Derivatives of GnRH as Potential Anticancer Therapeutic Agents. Curr. Med. Chem. 2008, 15, 2366–2379. [Google Scholar] [CrossRef] [Green Version]
- Schlage, P.; Mezo, G.; Orbán, E.; Bosze, S.; Manea, M. Anthracycline-GnRH Derivative Bioconjugates with Different Linkages: Synthesis, in Vitro Drug Release and Cytostatic Effect. J. Control Release 2011, 156, 170–178. [Google Scholar] [CrossRef]
- Manea, M.; Leurs, U.; Orbán, E.; Baranyai, Z.; Öhlschläger, P.; Marquardt, A.; Schulcz, Á.; Tejeda, M.; Kapuvári, B.; Tóvári, J.; et al. Enhanced Enzymatic Stability and Antitumor Activity of Daunorubicin-GnRH-III Bioconjugates Modified in Position 4. Bioconjug. Chem. 2011, 22, 1320–1329. [Google Scholar] [CrossRef]
- Hegedüs, R.; Manea, M.; Orbán, E.; Szabó, I.; Kiss, É.; Sipos, É.; Halmos, G.; Mező, G. Enhanced Cellular Uptake and in Vitro Antitumor Activity of Short-Chain Fatty Acid Acylated Daunorubicin–GnRH-III Bioconjugates. Eur. J. Med. Chem. 2012, 56, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.; Biri-Kovács, B.; Szeder, B.; Farkas, V.; Buday, L.; Szabó, Z.; Halmos, G.; Mező, G. Synthesis and in Vitro Biochemical Evaluation of Oxime Bond-Linked Daunorubicin-GnRH-III Conjugates Developed for Targeted Drug Delivery. Beilstein J. Org. Chem. 2018, 14, 756–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, S.; Biri-Kovács, B.; Szeder, B.; Buday, L.; Gardi, J.; Szabó, Z.; Halmos, G.; Mező, G. Enhanced In Vitro Antitumor Activity of GnRH-III-Daunorubicin Bioconjugates Influenced by Sequence Modification. Pharmaceutics 2018, 10, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranđelović, I.; Schuster, S.; Kapuvári, B.; Fossati, G.; Steinkühler, C.; Mező, G.; Tóvári, J. Improved In Vivo Anti-Tumor and Anti-Metastatic Effect of GnRH-III-Daunorubicin Analogs on Colorectal and Breast Carcinoma Bearing Mice. Int. J. Mol. Sci. 2019, 20, 4763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.M.; Chari, R.V.J. Ado-Trastuzumab Emtansine (T-DM1): An Antibody-Drug Conjugate (ADC) for HER2-Positive Breast Cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef] [PubMed]
- Von Minckwitz, G.; Huang, C.-S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med. 2018, 380, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.-O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab Mafodotin for Relapsed or Refractory Multiple Myeloma (DREAMM-2): A Two-Arm, Randomised, Open-Label, Phase 2 Study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody–Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef]
- Tong, J.T.W.; Harris, P.W.R.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-Labile Dipeptide Linkers for Lysosomal Release of Doxorubicin from Internalizing Immunoconjugates: Model Studies of Enzymatic Drug Release and Antigen-Specific in Vitro Anticancer Activity. Bioconjug. Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef]
- Dal Corso, A.; Caruso, M.; Belvisi, L.; Arosio, D.; Piarulli, U.; Albanese, C.; Gasparri, F.; Marsiglio, A.; Sola, F.; Troiani, S.; et al. Synthesis and Biological Evaluation of RGD Peptidomimetic–Paclitaxel Conjugates Bearing Lysosomally Cleavable Linkers. Chem.—Eur. J. 2015, 21, 6921–6929. [Google Scholar] [CrossRef]
- Tsuchikama, K.; An, Z. Antibody-Drug Conjugates: Recent Advances in Conjugation and Linker Chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staudacher, A.H.; Brown, M.P. Antibody Drug Conjugates and Bystander Killing: Is Antigen-Dependent Internalisation Required? Br. J. Cancer 2017, 117, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Sharma, S.; Shah, D.K. Quantitative Characterization of In Vitro Bystander Effect of Antibody-Drug Conjugates. J. Pharmacokinet. Pharmacodyn. 2016, 43, 567–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Groot, F.M.H.; Loos, W.J.; Koekkoek, R.; van Berkom, L.W.A.; Busscher, G.F.; Seelen, A.E.; Albrecht, C.; de Bruijn, P.; Scheeren, H.W. Elongated Multiple Electronic Cascade and Cyclization Spacer Systems in Activatible Anticancer Prodrugs for Enhanced Drug Release. J. Org. Chem. 2001, 66, 8815–8830. [Google Scholar] [CrossRef] [PubMed]
- Hochdörffer, K.; Abu Ajaj, K.; Schäfer-Obodozie, C.; Kratz, F. Development of Novel Bisphosphonate Prodrugs of Doxorubicin for Targeting Bone Metastases That Are Cleaved PH Dependently or by Cathepsin B: Synthesis, Cleavage Properties, and Binding Properties to Hydroxyapatite As Well As Bone Matrix. J. Med. Chem. 2012, 55, 7502–7515. [Google Scholar] [CrossRef] [PubMed]
- Dal Corso, A.; Borlandelli, V.; Corno, C.; Perego, P.; Belvisi, L.; Pignataro, L.; Gennari, C. Fast Cyclization of a Proline-Derived Self-Immolative Spacer Improves the Efficacy of Carbamate Prodrugs. Angew. Chem. Int. Ed. 2020, 59, 4176–4181. [Google Scholar] [CrossRef]
- Kapuvári, B.; Hegedüs, R.; Schulcz, Á.; Manea, M.; Tóvári, J.; Gacs, A.; Vincze, B.; Mező, G. Improved in Vivo Antitumor Effect of a Daunorubicin—GnRH-III Bioconjugate Modified by Apoptosis Inducing Agent Butyric Acid on Colorectal Carcinoma Bearing Mice. Investig. New Drugs 2016, 34, 416–423. [Google Scholar] [CrossRef] [Green Version]
- Orbán, E.; Mezo, G.; Schlage, P.; Csík, G.; Kulić, Z.; Ansorge, P.; Fellinger, E.; Möller, H.M.; Manea, M. In Vitro Degradation and Antitumor Activity of Oxime Bond-Linked Daunorubicin-GnRH-III Bioconjugates and DNA-Binding Properties of Daunorubicin-Amino Acid Metabolites. Amino Acids 2011, 41, 469–483. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Radia, S. Monomethoxytrityl (MMT) as a Versatile Amino Protecting Group for Complex Prodrugs of Anticancer Compounds Sensitive to Strong Acids, Bases and Nucleophiles. Tetrahedron Lett. 1997, 38, 5257–5260. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Hayashi, Y.; Kiso, Y. Paclitaxel Prodrugs: Toward Smarter Delivery of Anticancer Agents. J. Med. Chem. 2006, 49, 7253–7269. [Google Scholar] [CrossRef]
- Colombo, R.; Mingozzi, M.; Belvisi, L.; Arosio, D.; Piarulli, U.; Carenini, N.; Perego, P.; Zaffaroni, N.; De Cesare, M.; Castiglioni, V.; et al. Synthesis and Biological Evaluation (in Vitro and in Vivo) of Cyclic Arginine–Glycine–Aspartate (RGD) Peptidomimetic–Paclitaxel Conjugates Targeting Integrin AVβ3. J. Med. Chem. 2012, 55, 10460–10474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.; Lv, Q.; Lu, J.; Yao, H.; Lv, X.; Jiang, F.; Lu, A.; Zhang, G. Prodrug Strategies for Paclitaxel. Int. J. Mol. Sci. 2016, 17, 796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo Moreira Dias, A.; Pina, A.; Dal Corso, A.; Arosio, D.; Belvisi, L.; Pignataro, L.; Caruso, M.; Gennari, C. Multivalency Increases the Binding Strength of RGD Peptidomimetic-Paclitaxel Conjugates to Integrin AVβ3. Chem.—Eur. J. 2017, 23, 14410–14415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.H.J.; Ughetto, G.; Quigley, G.J.; Rich, A. Interactions between an Anthracycline Antibiotic and DNA: Molecular Structure of Daunomycin Complexed to d(CpGpTpApCpG) at 1.2-.ANG. Resolution. Biochemistry 1987, 26, 1152–1163. [Google Scholar] [CrossRef]
- Gao, Y.G.; Liaw, Y.C.; Li, Y.K.; van der Marel, G.A.; van Boom, J.H.; Wang, A.H. Facile Formation of a Crosslinked Adduct between DNA and the Daunorubicin Derivative MAR70 Mediated by Formaldehyde: Molecular Structure of the MAR70-d(CGTnACG) Covalent Adduct. Proc. Natl. Acad. Sci. USA 1991, 88, 4845–4849. [Google Scholar] [CrossRef] [Green Version]
- Frederick, C.A.; Williams, L.D.; Ughetto, G.; van der Marel, G.A.; van Boom, J.H.; Rich, A.; Wang, A.H. Structural Comparison of Anticancer Drug-DNA Complexes: Adriamycin and Daunomycin. Biochemistry 1990, 29, 2538–2549. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, Y.G.; van der Marel, G.A.; van Boom, J.H.; Wang, A.H. Simultaneous Incorporations of Two Anticancer Drugs into DNA. The Structures of Formaldehyde-Cross-Linked Adducts of Daunorubicin-d(CG(AraC)GCG) and Doxorubicin-d(CA(AraC)GTG) Complexes at High Resolution. J. Biol. Chem. 1993, 268, 10095–10101. [Google Scholar] [CrossRef]
- Halmos, G.; Arencibia, J.M.; Schally, A.V.; Davis, R.; Bostwick, D.G. High Incidence of Receptors for Luteinizing Hormone-Releasing Hormone (Lhrh) and Lhrh Receptor Gene Expression in Human Prostate Cancers. J. Urol. 2000, 163, 623–629. [Google Scholar] [CrossRef]
- Rozsa, B.; Nadji, M.; Schally, A.V.; Dezso, B.; Flasko, T.; Toth, G.; Mile, M.; Block, N.L.; Halmos, G. Receptors for Luteinizing Hormone-Releasing Hormone (LHRH) in Benign Prostatic Hyperplasia (BPH) as Potential Molecular Targets for Therapy with LHRH Antagonist Cetrorelix. Prostate 2011, 71, 445–452. [Google Scholar] [CrossRef]
- Sharma, S.; Lagisetti, C.; Poliks, B.; Coates, R.M.; Kingston, D.G.I.; Bane, S. Dissecting Paclitaxel-Microtubule Association: Quantitative Assessment of the 2’-OH Group. Biochemistry 2013, 52, 2328–2336. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated PH: A Perfect Storm for Cancer Progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Chand, S.; O’Hayer, K.; Blanco, F.F.; Winter, J.M.; Brody, J.R. The Landscape of Pancreatic Cancer Therapeutic Resistance Mechanisms. Int. J. Biol. Sci. 2016, 12, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Xie, K.; Wolff, R.; Abbruzzese, J.L. Pancreatic Cancer. Lancet 2004, 363, 1049–1057. [Google Scholar] [CrossRef]
- Stern, L.; Giese, N.; Hackert, T.; Strobel, O.; Schirmacher, P.; Felix, K.; Gaida, M.M. Overcoming Chemoresistance in Pancreatic Cancer Cells: Role of the Bitter Taste Receptor T2R10. J. Cancer 2018, 9, 711–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, B.; Page, M.; Noel, C. A new fluorometric assay for cytotoxicity measurements in-vitro. Int. J. Oncol. 1993, 3, 473–476. [Google Scholar] [CrossRef]
- Halmos, G.; Wittliff, J.L.; Schally, A.V. Characterization of Bombesin/Gastrin-Releasing Peptide Receptors in Human Breast Cancer and Their Relationship to Steroid Receptor Expression. Cancer Res. 1995, 55, 280–287. [Google Scholar]
- Hunter, W.M.; Greenwood, F.C. Preparation of Iodine-131 Labelled Human Growth Hormone of High Specific Activity. Nature 1962, 194, 495–496. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | GnRH-III-[4Lys(Bu), 8Lys(linker-drug)] | Linker | A2780 IC50 [µM] | Panc-1 IC50 [µM] |
|---|---|---|---|---|
| Dau | 0.21 ± 0.01 | 2.43 ± 0.58 | ||
| 10 | [2His-3Trp] | Val-Cit | 11.18 ± 0.38 | 85.57 ± 24.33 |
| 11 | [2ΔHis-3D-Tic] | Val-Cit | 4.24 ± 1.09 | >100 |
| 12 | [2His-3Trp] | Val-Ala | 7.48 ± 0.66 | 56.19 ± 17.28 |
| 13 | [2ΔHis-3D-Tic] | Val-Ala | 2.85 ± 0.90 | >100 |
| 23 | [2His-3Trp] | non-cleavable | 67.88 ± 25.36 | >100 |
| 24 | [2ΔHis-3D-Tic] | non-cleavable | 48.14 ± 0.47 | >100 |
| PTX | 0.02 ± 0.001 | 0.17 ± 0.01 | ||
| 14 | [2His-3Trp] | Val-Cit | 0.67 ± 0.07 | 5.03 ± 1.91 |
| 15 | [2ΔHis-3D-Tic] | Val-Cit | 0.51 ±0.11 | 6.44 ± 1.22 |
| 16 | [2His-3Trp] | Val-Ala | 0.66 ± 0.18 | 4.89 ± 1.08 |
| 17 | [2ΔHis-3D-Tic] | Val-Ala | 0.77 ± 0.08 | 8.15 ± 3.22 |
| 25 | [2His-3Trp] | non-cleavable | 41.52 ± 9.83 | >100 |
| 26 | [2ΔHis-3D-Tic] | non-cleavable | >100 | >100 |
| Code | GnRH-III-[4Lys(Bu), 8Lys(linker-drug)] | Linker | Drug | IC50 [nM] | |
|---|---|---|---|---|---|
| Pituitary | Prostate Cancer | ||||
| I | [2His-3Trp] | none | none | 2.72 ± 0.95 | 3.11 ± 0.76 |
| 10 | [2His-3Trp] | Val-Cit | Dau | 16.12 ± 2.03 | 13.66 ± 1.87 |
| 11 | [2ΔHis-3D-Tic] | Val-Cit | 11.70 ± 0.42 | 10.97 ± 1.23 | |
| 12 | [2His-3Trp] | Val-Ala | 17.04 ± 1.04 | 15.46 ± 0.83 | |
| 13 | [2ΔHis-3D-Tic] | Val-Ala | 24.77 ± 1.73 | 20.54 ± 1.46 | |
| 23 | [2His-3Trp] | non-cleavable | 9.70 ± 1.07 | 8.61 ± 1.08 | |
| 24 | [2ΔHis-3D-Tic] | non-cleavable | 36.29 ± 3.17 | 42.67 ± 7.04 | |
| 14 | [2His-3Trp] | Val-Cit | PTX | 10.54 ± 2.01 | 10.65 ± 1.82 |
| 15 | [2ΔHis-3D-Tic] | Val-Cit | 18.49 ± 2.72 | 14.88 ± 0.33 | |
| 16 | [2His-3Trp] | Val-Ala | 9.86 ± 0.82 | 8.47 ± 1.06 | |
| 17 | [2ΔHis-3D-Tic] | Val-Ala | 10.82 ± 1.98 | 12.73 ± 2.23 | |
| 25 | [2His-3Trp] | non-cleavable | n.d. | n.d. | |
| 26 | [2ΔHis-3D-Tic] | non-cleavable | 23.43 ± 1.67 | 22.21 ± 0.96 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schuster, S.; Juhász, É.; Halmos, G.; Neundorf, I.; Gennari, C.; Mező, G. Development and Biochemical Characterization of Self-Immolative Linker Containing GnRH-III-Drug Conjugates. Int. J. Mol. Sci. 2022, 23, 5071. https://doi.org/10.3390/ijms23095071
Schuster S, Juhász É, Halmos G, Neundorf I, Gennari C, Mező G. Development and Biochemical Characterization of Self-Immolative Linker Containing GnRH-III-Drug Conjugates. International Journal of Molecular Sciences. 2022; 23(9):5071. https://doi.org/10.3390/ijms23095071
Chicago/Turabian StyleSchuster, Sabine, Éva Juhász, Gábor Halmos, Ines Neundorf, Cesare Gennari, and Gábor Mező. 2022. "Development and Biochemical Characterization of Self-Immolative Linker Containing GnRH-III-Drug Conjugates" International Journal of Molecular Sciences 23, no. 9: 5071. https://doi.org/10.3390/ijms23095071
APA StyleSchuster, S., Juhász, É., Halmos, G., Neundorf, I., Gennari, C., & Mező, G. (2022). Development and Biochemical Characterization of Self-Immolative Linker Containing GnRH-III-Drug Conjugates. International Journal of Molecular Sciences, 23(9), 5071. https://doi.org/10.3390/ijms23095071