
The Use of Molecular Dynamics Simulation Method to Quantitatively Evaluate the Affinity between HBV Antigen T Cell Epitope Peptides and HLA-A Molecules

 ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Selection of HLA Structures for Affinity Analysis
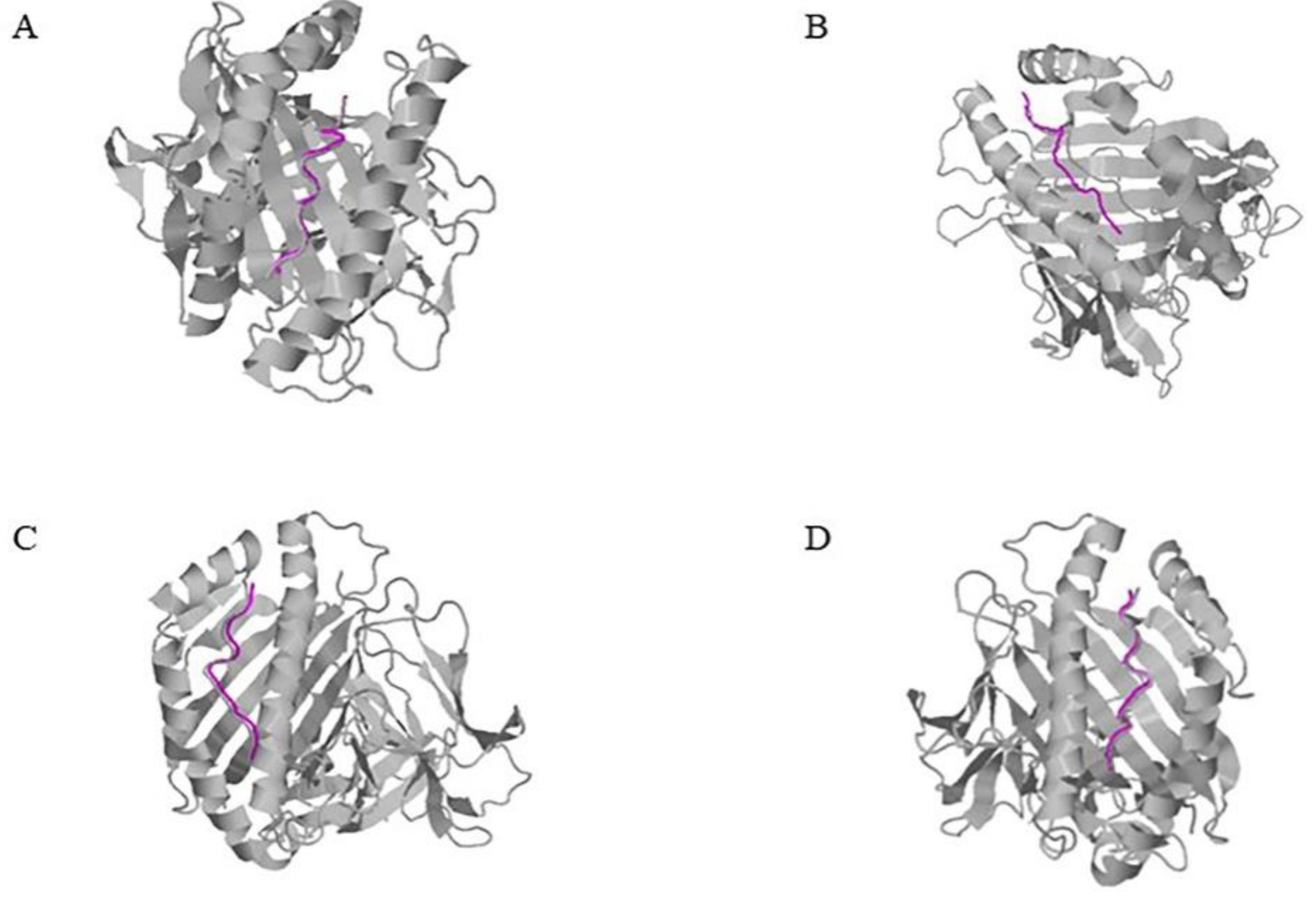
2.2. Acquisition of the Initial Conformation of HBV Peptide-HLA Docking
2.3. Analysis of Binding between HBV Peptides and HLA Molecules
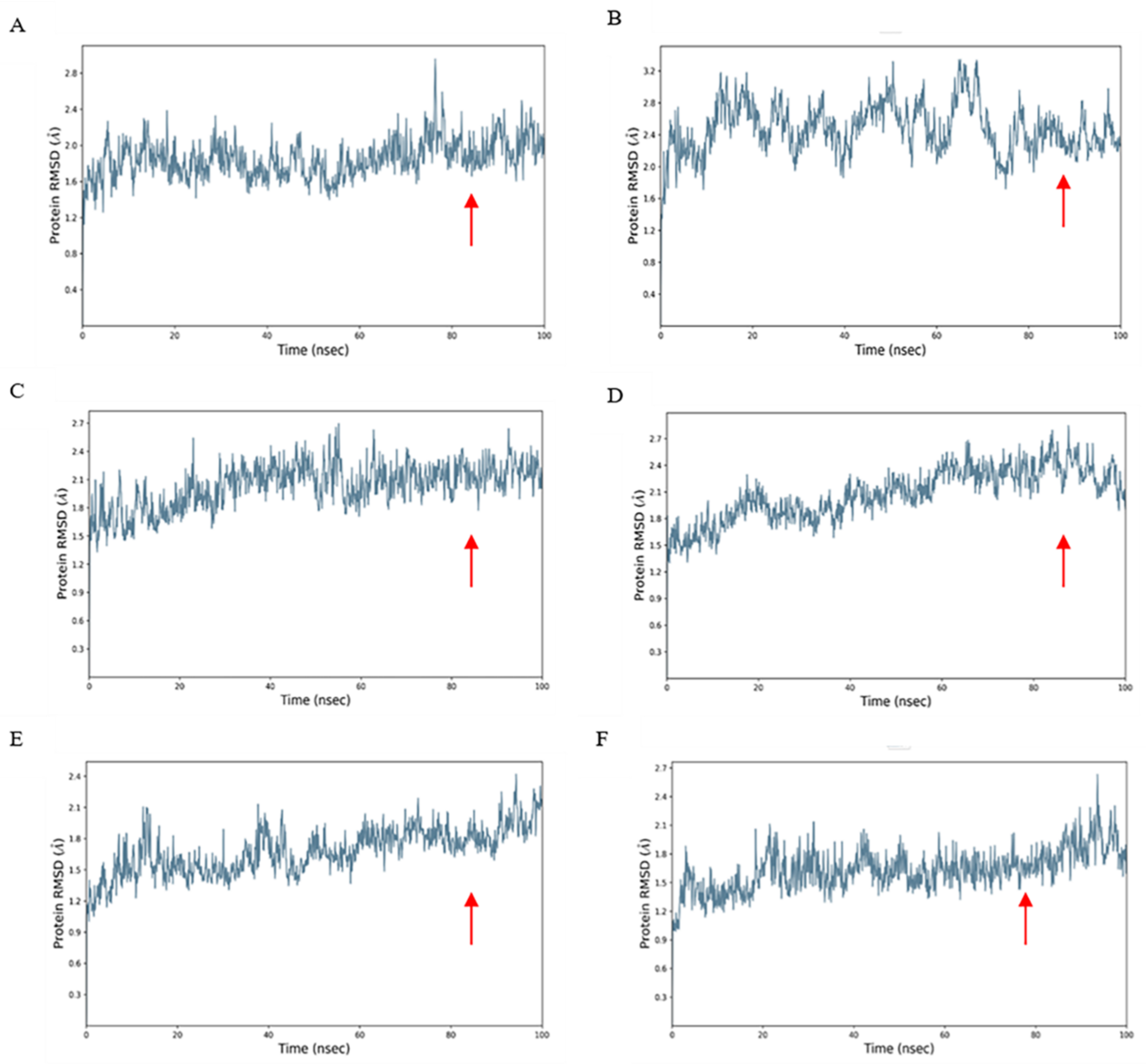
2.4. Dynamics Simulation
2.5. Binding Analysis of 45 HBV Epitopes and HLA-A Alleles
3. Discussion
4. Materials and Methods
4.1. HLA Structure Source
4.2. HBV Positive Peptide
4.3. Selection of HLA Structure
4.4. Scanning Amino Acid Mutation
4.5. Molecular Docking
4.6. Molecular Dynamics Simulation
4.7. Protein Ligand Interaction Analysis
4.8. Binding Free Energy Calculation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ortega-Prieto, A.M.; Cherry, C.; Gunn, H.; Dorner, M. In Vivo Model Systems for Hepatitis B Virus Research. ACS Infect. Dis. 2019, 5, 688–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.; Altstetter, S.M.; Protzer, U. Innate immune recognition and modulation in hepatitis D virus infection. World J. Gastroenterol. 2020, 26, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guojin, O.U.; Wang, J.; Liu, Z. Research progress on the correlation between HLA and HBV infection. Chin. J. Blood Transfus. 2018, 31, 1321–1326. [Google Scholar]
- Sun, S.F.; Li, Y.; Han, S.Y.; Jia, H.T.; Li, X.L.; Li, X.F. A comprehensive genome-wide profiling comparison between HBV and HCV infected hepatocellular carcinoma. BMC Med. Genom. 2019, 12, 147. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.D.; Ding, Y.; Shen, C.L. A Systematic Review of T Cell Epitopes Defined from the Proteome of Hepatitis B Virus. Vaccines 2022, 10, 257. [Google Scholar] [CrossRef]
- Wang, L.; Zou, Z.Q.; Wang, K. Clinical Relevance of HLA Gene Variants in HBV Infection. J. Immunol. Res. 2016, 2016, 9069375. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Tang, S.T.; Stryhn, A.; Justesen, S.; Larsen, M.V.; Dziegiel, M.H.; Lewinsohn, D.M.; Buus, S.; Lund, O.; Claesson, M.H. Identification of MHC class II restricted T-cell-mediated reactivity against MHC class I binding Mycobacterium tuberculosis peptides. Immunology 2011, 132, 482–491. [Google Scholar] [CrossRef]
- Tian, Z.J.; Shen, Y.; Li, X.R.; Wei, Y.N.; Fan, H.; Ren, Q.K. Increased interleukin-32, interleukin-1, and interferon-γ levels in serum from hepatitis B patients and in HBV-stimulated peripheral blood mononuclear cells from healthy volunteers. J. Infect. Public Health 2019, 12, 7–12. [Google Scholar] [CrossRef]
- Chen, Y.; Lan-Juan, L.I.; Lou, B.; Chen-Huai, X.U. Determination of circulating HBV specific CD8~+ cells by tetramer staining flow cytometry. Chin. J. Lab. Med. 2004, 4, 16–18. [Google Scholar]
- Boppana, S.; Sterrett, S.; Files, J.; Qin, K.; Fiore-Gartland, A.; Cohen, K.W.; De Rosa, S.C.; Bansal, A.; Goepfert, P.A. HLA-I Associated Adaptation Dampens CD8 T-Cell Responses in HIV Ad5-Vectored Vaccine Recipients. J. Infect. Dis. 2019, 220, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Antunes, D.A.; Abella, J.R.; Devaurs, D.; Rigo, M.M.; Kavraki, L.E. Structure-based Methods for Binding Mode and Binding Affinity Prediction for Peptide-MHC Complexes. Curr. Top. Med. Chem. 2018, 18, 2239–2255. [Google Scholar] [CrossRef] [PubMed]
- Rammensee, H.; Bachmann, J.; Emmerich, N.P.; Bachor, O.A.; Stevanović, S. SYFPEITHI: Database for MHC ligands and peptide motifs. Immunogenetics 1999, 50, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Lundegaard, C.; Lamberth, K.; Harndahl, M.; Buus, S.; Lund, O.; Nielsen, M. NetMHC-3.0: Accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 2008, 36, W509–W512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Dönnes, P.; Elofsson, A. Prediction of MHC class I binding peptides, using SVMHC. BMC Bioinform. 2002, 3, 25. [Google Scholar] [CrossRef]
- Parker, K.C.; Bednarek, M.A.; Coligan, J.E. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J. Immunol. 1994, 152, 163–175. [Google Scholar]
- Doytchinova, I.A.; Guan, P.; Flower, D.R. EpiJen: A server for multistep T cell epitope prediction. BMC Bioinform. 2006, 7, 131. [Google Scholar] [CrossRef] [Green Version]
- Van Rosmalen, M.; Krom, M.; Merkx, M. Tuning the Flexibility of Glycine-Serine Linkers To Allow Rational Design of Multidomain Proteins. Biochemistry 2017, 56, 6565–6574. [Google Scholar] [CrossRef]
- Huang, C.; Chen, J.; Ding, F.; Yang, L.; Zhang, S.; Wang, X.; Shi, Y.; Zhu, Y. Related parameters of affinity and stability prediction of HLA-A * 2402 restricted antigen peptides based on molecular docking. Ann. Transl. Med. 2021, 9, 673. [Google Scholar] [CrossRef]
- Kong, R.; Liu, R.R.; Xu, X.M.; Zhang, D.W.; Xu, X.S.; Shi, H.; Chang, S. Template-based modeling and ab-initio docking using CoDock in CAPRI. Proteins 2020, 88, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Wang, F.; Zhang, J.; Wang, F.; Chang, S. CoDockPP: A Multistage Approach for Global and Site-Specific Protein-Protein Docking. J. Chem. Inf. Model. 2019, 59, 3556–3564. [Google Scholar] [CrossRef] [PubMed]
- Lensink, M.F.; Brysbaert, G.; Nadzirin, N.; Velankar, S.; Chaleil, R.A.G.; Gerguri, T.; Bates, P.A.; Laine, E.; Carbone, A.; Grudinin, S.; et al. Blind prediction of homo- and hetero-protein complexes: The CASP13-CAPRI experiment. Proteins 2019, 87, 1200–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Zhou, Z.N.; Li, X.Y.; Zhao, C.; Jin, X.X.; Liu, X.T.; Wu, Y.D.; Mei, X.Y.; Li, J.; Qiu, J.; et al. Screening and identification of HBV epitopes restricted by multiple prevalent HLA-A allotypes. Front. Immunol. 2022, 13, 847105. [Google Scholar] [CrossRef]
- Mei, S.; Li, F.; Leier, A.; Marquez-Lago, T.T.; Giam, K.; Croft, N.P.; Akutsu, T.; Smith, A.L.; Li, J.; Rossjohn, J.; et al. A comprehensive review and performance evaluation of bioinformatics tools for HLA class I peptide-binding prediction. Brief. Bioinform. 2020, 21, 1119–1135. [Google Scholar] [CrossRef]
- Comber, J.D.; Karabudak, A.; Shetty, V.; Testa, J.S.; Huang, X.; Philip, R. MHC Class I Presented T Cell Epitopes as Potential Antigens for Therapeutic Vaccine against HBV Chronic Infection. Hepat. Res. Treat. 2014, 2014, 860562. [Google Scholar] [CrossRef]
- Lumley, S.; Noble, H.; Hadley, M.J.; Callow, L.; Malik, A.; Chua, Y.Y.; Duffey, O.J.; Grolmusova, N.; Kumar, A.; Ravenscroft, S.; et al. Hepitopes: A live interactive database of HLA class I epitopes in hepatitis B virus. Wellcome Open Res. 2016, 1, 9. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos Francisco, R.; Buhler, S.; Nunes, J.M.; Bitarello, B.D.; França, G.S.; Meyer, D.; Sanchez-Mazas, A. HLA supertype variation across populations: New insights into the role of natural selection in the evolution of HLA-A and HLA-B polymorphisms. Immunogenetics 2015, 67, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Duan, J.; Lupyan, D.; Wang, L. Improving the Accuracy of Protein Thermostability Predictions for Single Point Mutations. Biophys. J. 2020, 119, 115–127. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Cob-Calan, N.N.; Chi-Uluac, L.A.; Ortiz-Chi, F.; Cerqueda-García, D.; Navarrete-Vázquez, G.; Ruiz-Sánchez, E.; Hernández-Núñez, E. Molecular Docking and Dynamics Simulation of Protein β-Tubulin and Antifungal Cyclic Lipopeptides. Molecules 2019, 24, 3387. [Google Scholar] [CrossRef] [Green Version]
- Reif, M.M.; Oostenbrink, C. Net charge changes in the calculation of relative ligand-binding free energies via classical atomistic molecular dynamics simulation. J. Comput. Chem. 2014, 35, 227–243. [Google Scholar] [CrossRef]
- Wallraven, K.; Holmelin, F.L.; Glas, A.; Hennig, S.; Frolov, A.I.; Grossmann, T.N. Adapting free energy perturbation simulations for large macrocyclic ligands: How to dissect contributions from direct binding and free ligand flexibility. Chem. Sci. 2020, 11, 2269–2276. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Positive Peptide Sequence | HLA-A Genotype | PDB-ID | Peptide | Resolution (Å) |
|---|---|---|---|---|
| FLWEWASVR | A * 02:01 | 1AO7 | LLFGYPVYV | 2.6 |
| 1BD2 | LLFGYPVYV | 2.5 | ||
| 1DUZ | LLFGYPVYV | 1.8 | ||
| 1QEW | FLWGPRALV | 2.2 | ||
| 1JHT | ALGIGILTV | 2.15 | ||
| 3I6G | GLMWLSYFV | 2.2 | ||
| 5ENW | GLKEGIPAL | 1.85 | ||
| 5F9J | YLSPIASPL | 2.51 | ||
| 5FA3 | GLLPELPAV | 1.86 | ||
| A * 24:02 | 3I6L | QFKDNVILL | 2.4 | |
| 2BCK | VYGFVRACL | 2.8 |
| HLA-A Genotype | Score Value kal/mol | Ligand RMSD (Å) |
|---|---|---|
| A * 11:02 | −250.13 | 0.389 |
| A * 26:01 | −332.26 | 3.100 |
| A * 31:01 | −270.40 | 0.781 |
| A * 33:03 | −262.92 | 0.596 |
| HBV Peptide | HLA-A Genotype | PDB-ID | HLA Peptide | △Affinity kcal/mol | △Stability (Solvated) kcal/mol | △Hydropathy | △Prime Energy kcal/mol | Prime Affinity kcal/mol |
|---|---|---|---|---|---|---|---|---|
| FLWEWASVR | A * 02:01 | 1AO7 | LLFGYPVYV | 59.73 | 48.23 | 0.47 | 6.36 | −231.538 |
| 1BD2 | LLFGYPVYV | 100.45 | 57.18 | 0.31 | 56.03 | −186.495 | ||
| 1DUZ | LLFGYPVYV | −1 | 25.04 | 1.72 | −77.56 | −215.093 | ||
| 1QEW | FLWGPRALV | 29.19 | 3.38 | 0.76 | −44.56 | −187.811 | ||
| 1JHT | ALGIGILTV | −25.79 | 58.4 | −1.97 | −64.88 | −220.932 | ||
| 3I6G | GLMWLSYFV | 101.23 | 93.87 | −1.41 | 135.48 | −116.632 | ||
| 5ENW | GLKEGIPAL | 207.63 | 162.23 | 0.49 | 286.45 | −38.59 | ||
| 5F9J | YLSPIASPL | −10.79 | 19.31 | −0.59 | −127.65 | −245.795 | ||
| 6NCA | YVLDHLIVV | −15.94 | 18.86 | 0.1 | −54.54 | −209.7 | ||
| 5FA3 | GLLPELPAV | 111.81 | 62.17 | 1.55 | 51.95 | −112.188 | ||
| A * 33:03 | 5WJL template | GTSGSPIVNR | −19.43 | 21.53 | −0.70 | −56.20 | −244.411 | |
| A * 24:02 | 3I6L | QFKDNVILL | 11.43 | 13.94 | 0.82 | 57.69 | −195.086 | |
| 2BCK | VYGFVRACL | 23.14 | −4.06 | 1.14 | −24.61 | −189.799 | ||
| VWLSVIWMMW | A * 02:01 | 5F7D | GLKEGIPALD | 98.89 | 128.14 | 2.77 | 241.54 | −129.266 |
| 5D9S | FVLELEPEWTV | 109.48 | 101.41 | 4.06 | 244.59 | −132.558 | ||
| 5EOT | GLLPELPAVGG | 91.86 | 129.52 | 2.76 | 161.08 | −141.106 | ||
| 3I6K | TLACFVLAAV | 39.08 | 41.57 | -0.07 | 56.11 | −160.656 | ||
| 1I4F | GVYDGREHTV | 135.81 | 153.98 | 6.16 | 391.17 | −65.913 | ||
| A * 02:07 | 3OXS | FLPSDFFPSV | 96.17 | 49.52 | 4.83 | 103.16 | −142.360 | |
| A * 24:02 | 5WWI | LYKKLKREMTF | 6.77 | 3.73 | 6.62 | 90.22 | −240.244 | |
| 5WXD | LYKKLKREMTF | −9.87 | 12.91 | 7.73 | 82.75 | −243.243 | ||
| ETVLEYLVSV | A * 26:01 | 6AT9 template | AQDIYRASYY | 76.53 | −35.56 | 2.22 | 131.59 | −109.727 |
| A * 11:01 | 5WJL | GTSGSPIVNR | 86.11 | 38.35 | 0.58 | 121.02 | −125.356 | |
| 5WJN | GTSGSPIINR | 70.9 | 30.3 | 1.15 | 87.87 | −164.241 | ||
| 5WKF | GTSGSPIVNR | 372.07 | 168.26 | 0.43 | 536.89 | 115.8 | ||
| 5WKH | GTSGSPIINR | 57.75 | 25.51 | 2.3 | 69.94 | −165.814 | ||
| 1QVO | QVPLRPMTYK | 55.37 | −3.89 | 0.53 | 34.32 | −164.671 | ||
| A * 02:01 | 5YXU | KLVALGINAV | 109.1 | 66.08 | 0.18 | 162.51 | −159.462 | |
| 1I4F | GVYDGREHTV | 179.02 | 34.6 | 4.12 | 284.63 | −22.858 | ||
| 3I6K | TLACFVLAAV | 3.67 | −4.35 | −2.31 | −55.59 | −194.707 | ||
| 3UTQ | ALWGPDPAAA | −15.25 | 27.51 | 1.61 | −41.95 | −211.162 |
| HLA-Peptide | Number of Hydrogen Bonds | Number of Salt Bridge | Number of Pi-Pi Bonds |
|---|---|---|---|
| A * 02:01-1JHT- ALGIGILTV | 12 | 1 | 0 |
| A * 02:01-1JHT-FLWEWAFVR | 13 | 3 | 1 |
| A * 24:02-5WXD-LYKKLKREMTF | 10 | 1 | 2 |
| A * 24:02-5WXD-VWLSVIWMMW | 10 | 1 | 4 |
| A * 02:01-3UTQ-ALWGPDPAAA | 11 | 1 | 0 |
| A * 02:01-3UTQ-ETVLEYLVSV | 14 | 4 | 0 |
| HBV Peptide | HLA-A Genotype | PDB-ID | HLA Peptide | Binding Free Energy kcal/mol |
|---|---|---|---|---|
| SMYPSCCCTK | A * 11:01 | 5GRD | SSCSSCPLSK | −64.234 |
| A * 02:01 | 1I4F | GVYDGREHTV | −80.997 | |
| A * 03:01 | 3RL2 | QVPLRPMTYK | −76.071 | |
| ETVLEYLVSV | A * 26:01 | modeling | −90.805 | |
| A * 11:01 | 5GRD | SSCSSCPLSK | −93.877 | |
| A * 02:01 | 3UTQ | ALWGPDPAAA | −76.755 | |
| Epitopes | Origin Protein | HBV Genotype(s) | Position (Start–End) | Bioinformatics Prediction |
|---|---|---|---|---|
| FLPSDFFPSI | HBeAg | B/C | 47–56 | A * 02:07 > A * 02:01 > A * 11:01 |
| LLDTASALY | HBeAg | A/B/D | 59–67 | A * 01:01 > A * 11:02 |
| ILCWGELMNL | HBeAg | B/C | 88–97 | A * 02:07 > A * 24:02 |
| ASRELVVSY | HBeAg | B/C | 109–117 | A * 02:01 > A * 30:01 > A * 02:06 |
| SYVNVNMGL | HBeAg | A/C/D | 116–124 | A * 24:02 > A * 02:07 |
| WFHISCLTF | HBeAg | A/B/C/D | 131–139 | A * 24:02 > A * 02:07 > A * 02:01 |
| ETVLEYLVSV | HBeAg | C | 142–151 | A * 02:01 > A * 11:01 > A * 26:01 |
| ILSTLPETTV | HBeAg | A/B/C/D | 168–177 | A * 02:01 > A * 02:03 |
| STLPETTVVR | HBeAg | A/B/C/D | 170–179 | A * 11:01 > A * 02:07 |
| ASPISSIFSR | HBsAg | C | 158–167 | A * 11:01 |
| SPISSIFSR | HBsAg | C | 159–167 | A * 02:01 > A * 11:01 |
| SAISSISSK | HBsAg | B | 159–167 | A * 11:01>A * 02:03 |
| LQAGFFSLTK | HBsAg | B | 189–198 | A * 02:07 > A * 24:02 > A * 11:02 |
| QAGFFSLTK | HBsAg | B | 190–198 | A * 11:01 > A * 31:01 |
| QAGFFLLTR | HBsAg | C/D | 190–198 | A * 11:01 > A * 11:02 |
| CPGYRWMCLR | HBsAg | A/B/C/D | 243–252 | A * 33:03 |
| LFILLLCLI | HBsAg | A/C/D | 258–266 | A * 24:02 |
| LLDYQGMLPV | HBsAg | A/B/C/D | 271–280 | A * 02:03 > A * 24:02 |
| SMYPSCCCTK | HBsAg | C | 306–315 | A * 02:01 > A * 11:01 > A * 03:01 |
| FLWEWASVR | HBsAg | C | 335–343 | A * 02:01 > A * 33:03 > A * 24:02 |
| VWLSVIWMMW | HBsAg | A/B/C/D | 364–373 | A * 24:02 > A * 02:01 > A * 02:07 |
| MMWYWGPSL | HBsAg | A/C/D | 371–379 | A * 02:01 > A * 02:07 > A * 24:02 |
| MMWFWGPSL | HBsAg | B | 371–379 | A * 24:02 > A * 02:07 > A * 03:01 |
| MMWYWGPSLY | HBsAg | A/C/D | 371–380 | A * 24:02 > A * 03:01 > A * 02:07 |
| LYSILSPFL | HBsAg | C/D | 379–387 | A * 24:02 > A * 02:01 |
| TVNAHQILPK | HBx | A/D | 82–91 | A * 11:01 > A * 30:01 |
| TVNAHGNLPK | HBx | B | 82–91 | A * 02:01 > A * 11:01 > A * 03:01 |
| TVNAHQVLPK | HBx | C | 82–91 | A * 11:01 |
| STTDLEAYFK | HBx | A/B/C/D | 104–113 | A * 02:01 > A * 11:01 > A * 02:06 |
| RLKVFVLGG | HBx | A/B/C/D | 128–136 | A * 24:02 > A * 30:01 |
| KVFVLGGCR | HBx | A/B/C/D | 130–138 | A * 24:02 > A * 31:01 |
| VCAPAPCNF | HBx | D | 142–150 | A * 24:02 |
| LYSSTVPCF | HBpol | B | 62–70 | A * 24:02 > A * 33:03 > A * 02:01 |
| LYSSTVPVF | HBpol | C | 62–70 | A * 24:02 > A * 02:07 > A * 02:01 |
| FYPKVTKYL | HBpol | D | 115–123 | A * 24:02 > A * 11:01 |
| KVTKYLPLDK | HBpol | D | 118–127 | A * 11:01 |
| TLWKAGILYK | HBpol | A/B/C/D | 150–159 | A * 03:01 > A * 24:02 > A * 11:02 |
| FLLAQFTSA | HBpol | A/B/C/D | 524–532 | A * 02:03 > A * 02:07 |
| LLAQFTSAI | HBpol | A/B/C/D | 525–533 | A * 02:03 |
| PTYKAFLCK | HBpol | C/D | 671–679 | A * 11:01 > A * 02:06 |
| HTAELLAACF | HBpol | A/B/C/D | 726–735 | A * 24:02 > A * 26:01 |
| RSRSGAKLI | HBpol | B/C | 737–745 | A * 02:01 > A * 02:06 > A * 30:01 |
| RSRSGANIL | HBpol | B/C | 737–745 | A * 02:01 > A * 30:01 |
| KLIGTDNSV | HBpol | A/C | 743–751 | A * 02:01 > A * 02:03 |
| KLIGTHNSV | HBpol | B | 743–751 | A * 02:01 > A * 02:03 > A * 11:01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mei, X.; Li, X.; Zhao, C.; Liu, A.; Ding, Y.; Shen, C.; Li, J. The Use of Molecular Dynamics Simulation Method to Quantitatively Evaluate the Affinity between HBV Antigen T Cell Epitope Peptides and HLA-A Molecules. Int. J. Mol. Sci. 2022, 23, 4629. https://doi.org/10.3390/ijms23094629
Mei X, Li X, Zhao C, Liu A, Ding Y, Shen C, Li J. The Use of Molecular Dynamics Simulation Method to Quantitatively Evaluate the Affinity between HBV Antigen T Cell Epitope Peptides and HLA-A Molecules. International Journal of Molecular Sciences. 2022; 23(9):4629. https://doi.org/10.3390/ijms23094629
Chicago/Turabian StyleMei, Xueyin, Xingyu Li, Chen Zhao, Anna Liu, Yan Ding, Chuanlai Shen, and Jian Li. 2022. "The Use of Molecular Dynamics Simulation Method to Quantitatively Evaluate the Affinity between HBV Antigen T Cell Epitope Peptides and HLA-A Molecules" International Journal of Molecular Sciences 23, no. 9: 4629. https://doi.org/10.3390/ijms23094629
APA StyleMei, X., Li, X., Zhao, C., Liu, A., Ding, Y., Shen, C., & Li, J. (2022). The Use of Molecular Dynamics Simulation Method to Quantitatively Evaluate the Affinity between HBV Antigen T Cell Epitope Peptides and HLA-A Molecules. International Journal of Molecular Sciences, 23(9), 4629. https://doi.org/10.3390/ijms23094629