
p97/UBXD1 Generate Ubiquitylated Proteins That Are Sequestered into Nuclear Envelope Herniations in Torsin-Deficient Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
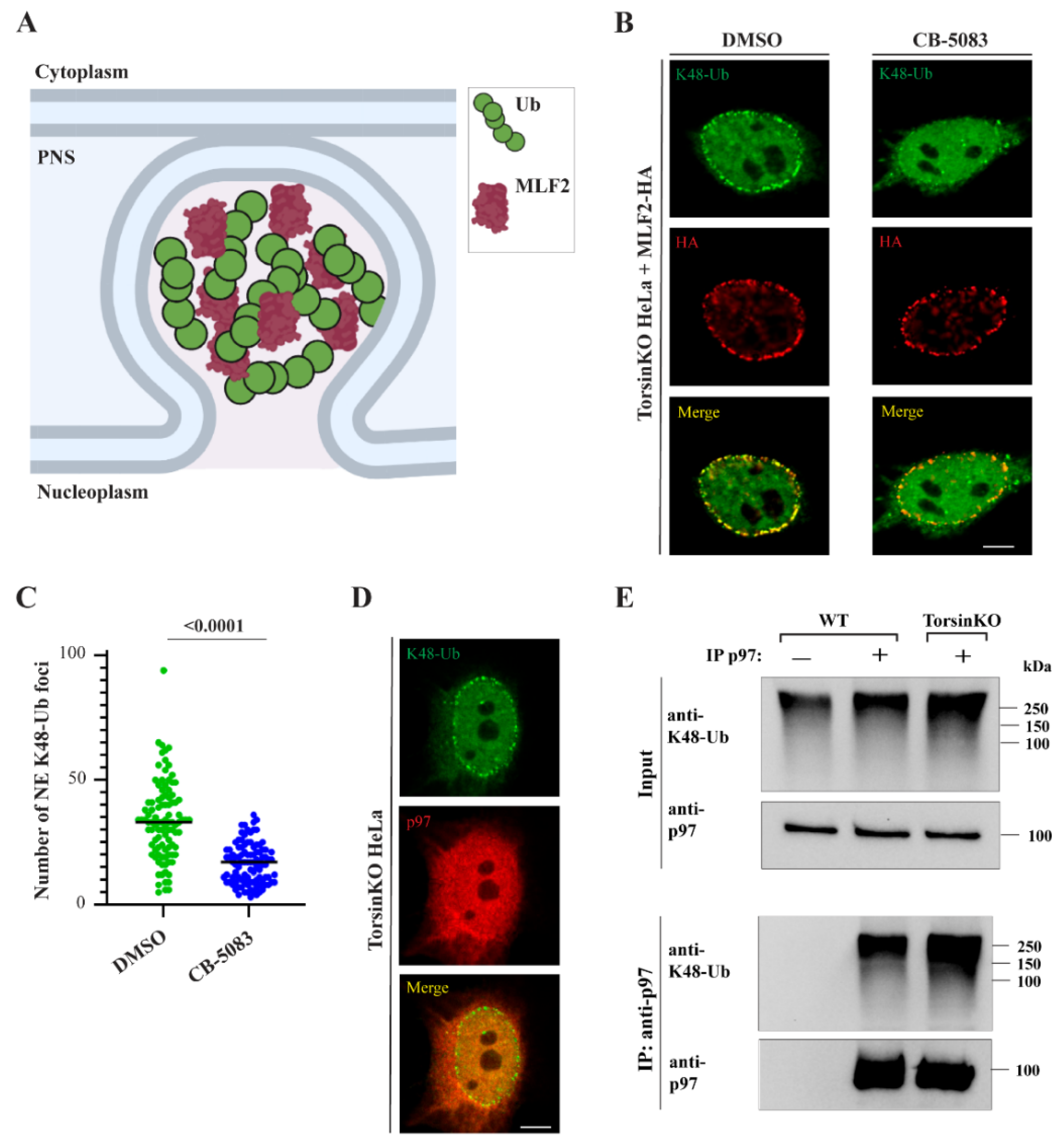
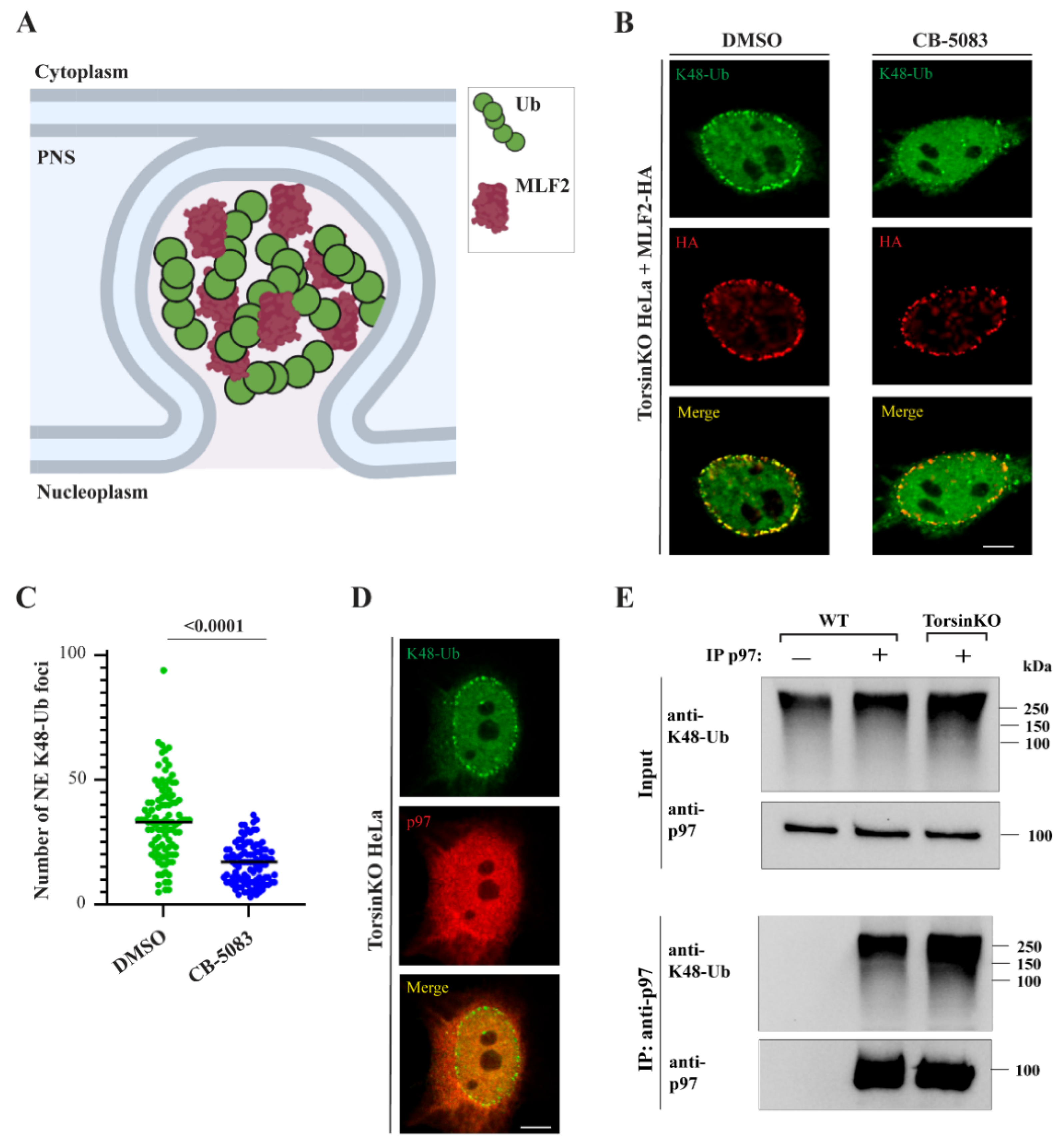
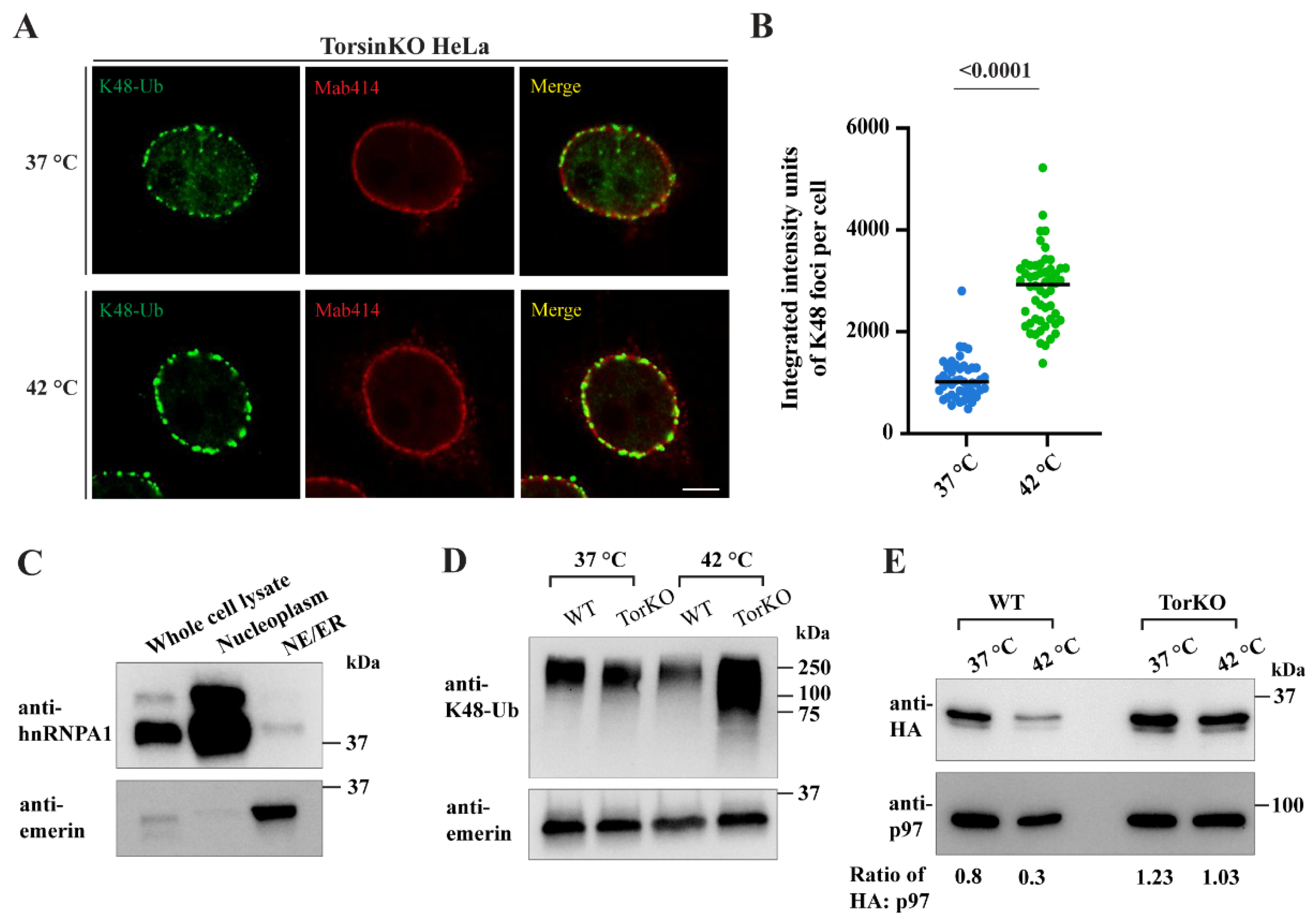
2.1. p97 Activity Is Required for K48-Ub Accumulation inside NE Blebs
2.2. p97 Associates with More K48-Ub in Torsin-Deficient Cells
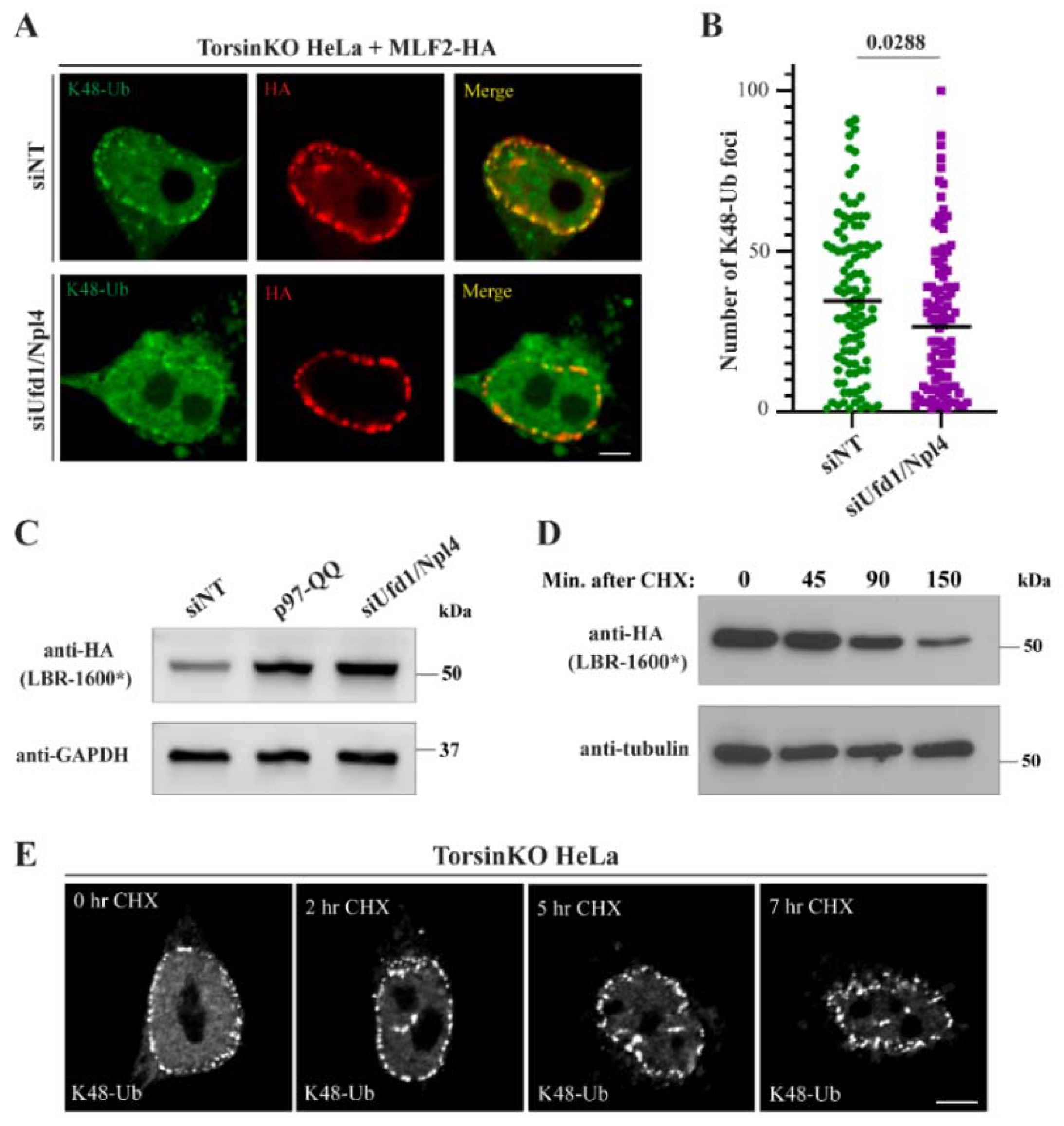
2.3. p97 Does Not Require the Ufd1/Npl4 Heterodimer for the Majority of the K48-Ub Protein Deposition to Blebs
2.4. Many Canonical ERAD-Associated E3 Ligases Do Not Significantly Contribute to the K48-Ub Protein inside Blebs
2.5. Newly Synthesized, Misfolded Proteins Do Not Account for the Majority of the K48-Ub Protein inside Blebs
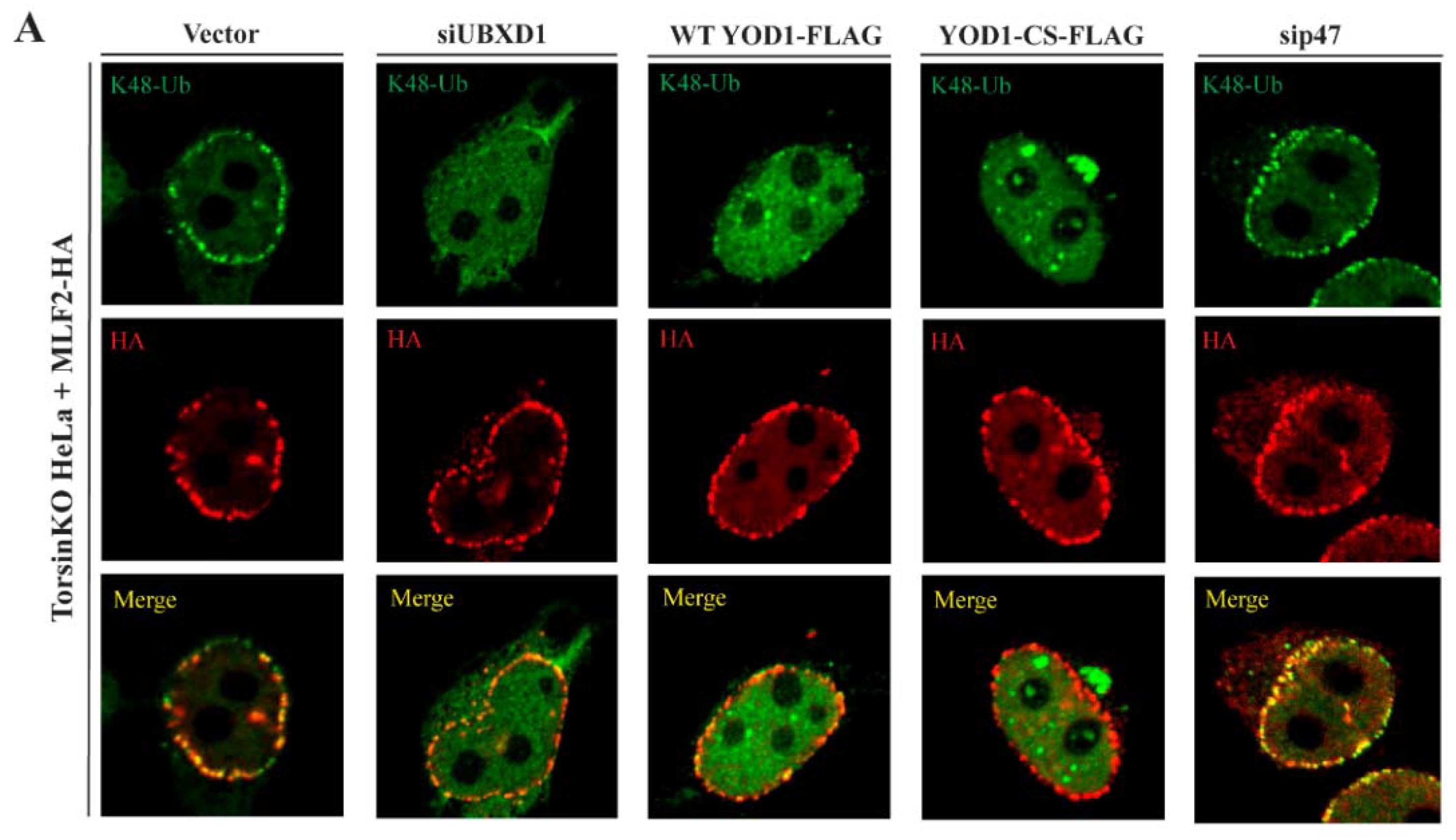
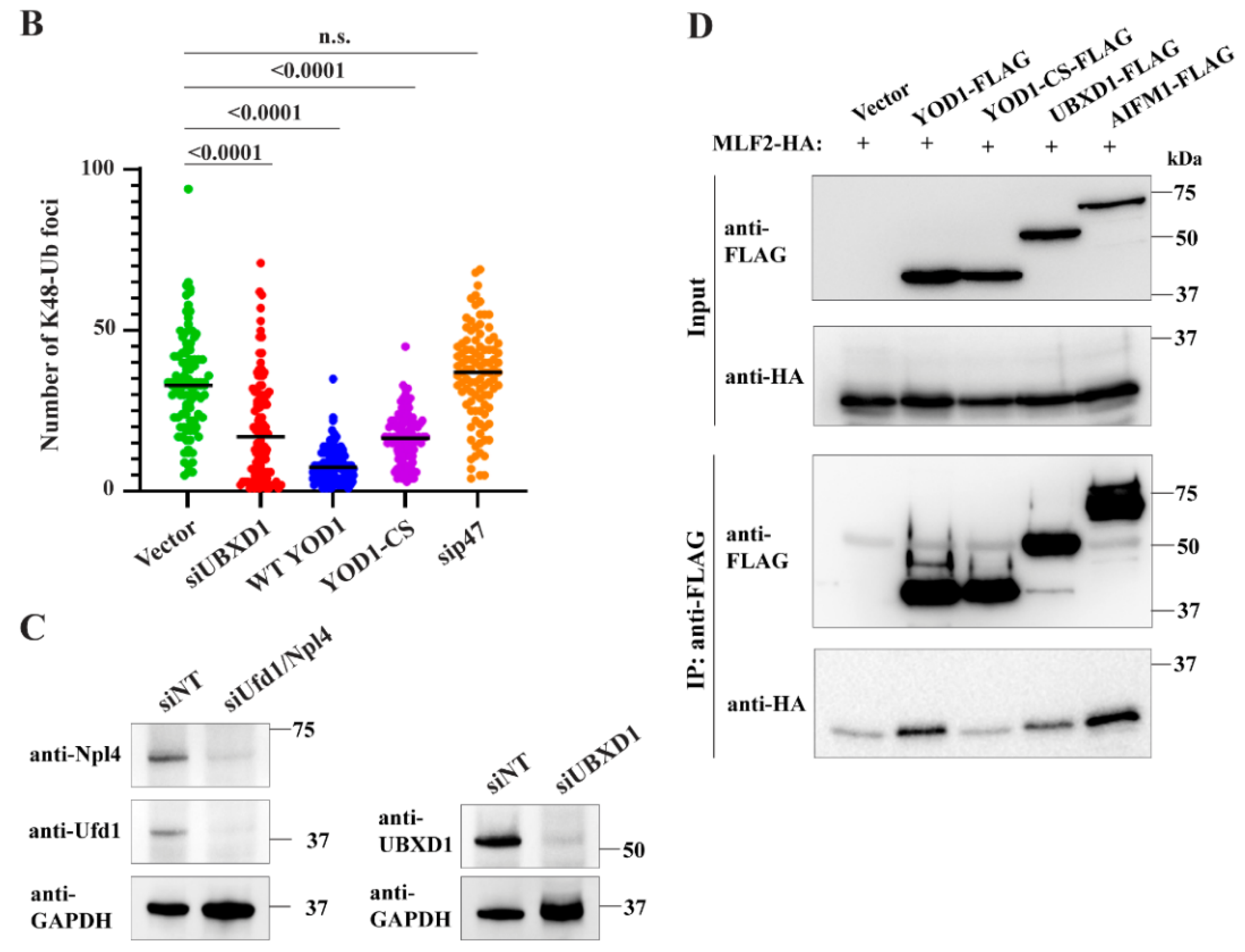
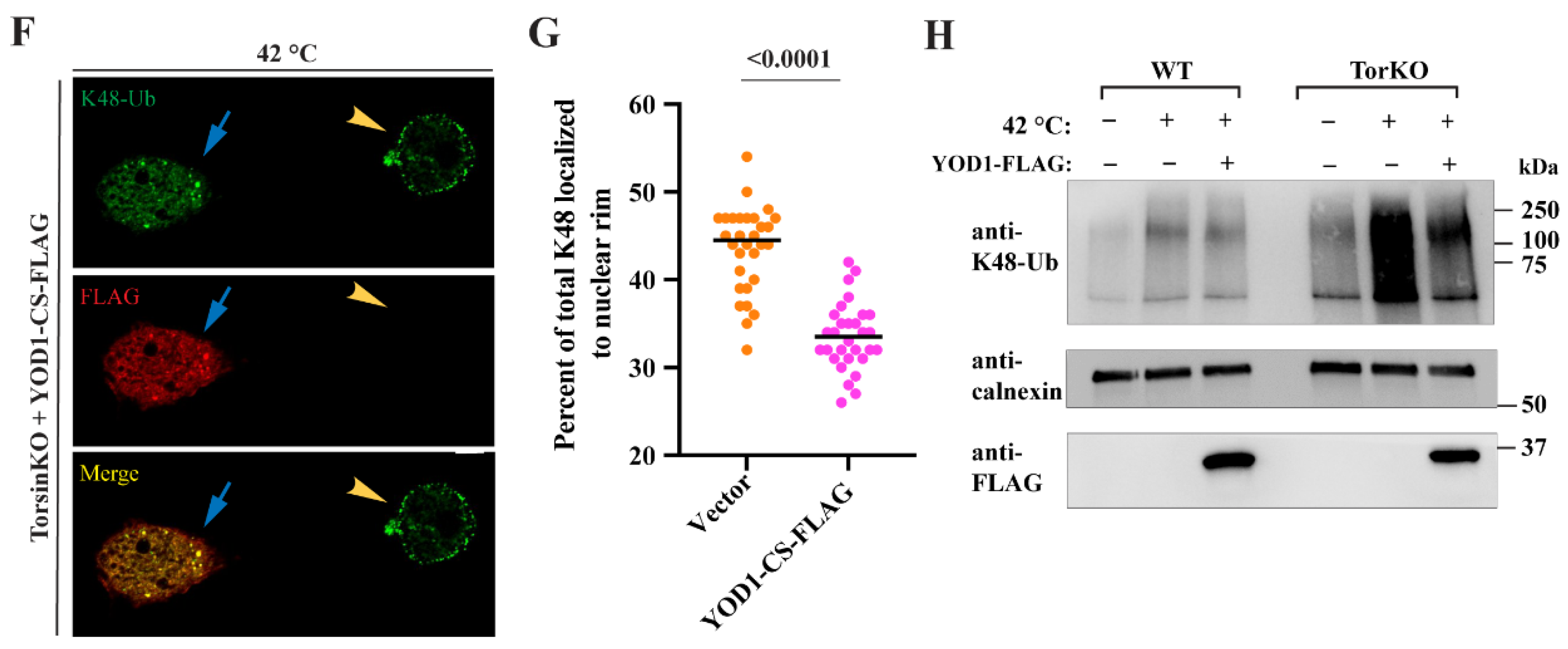
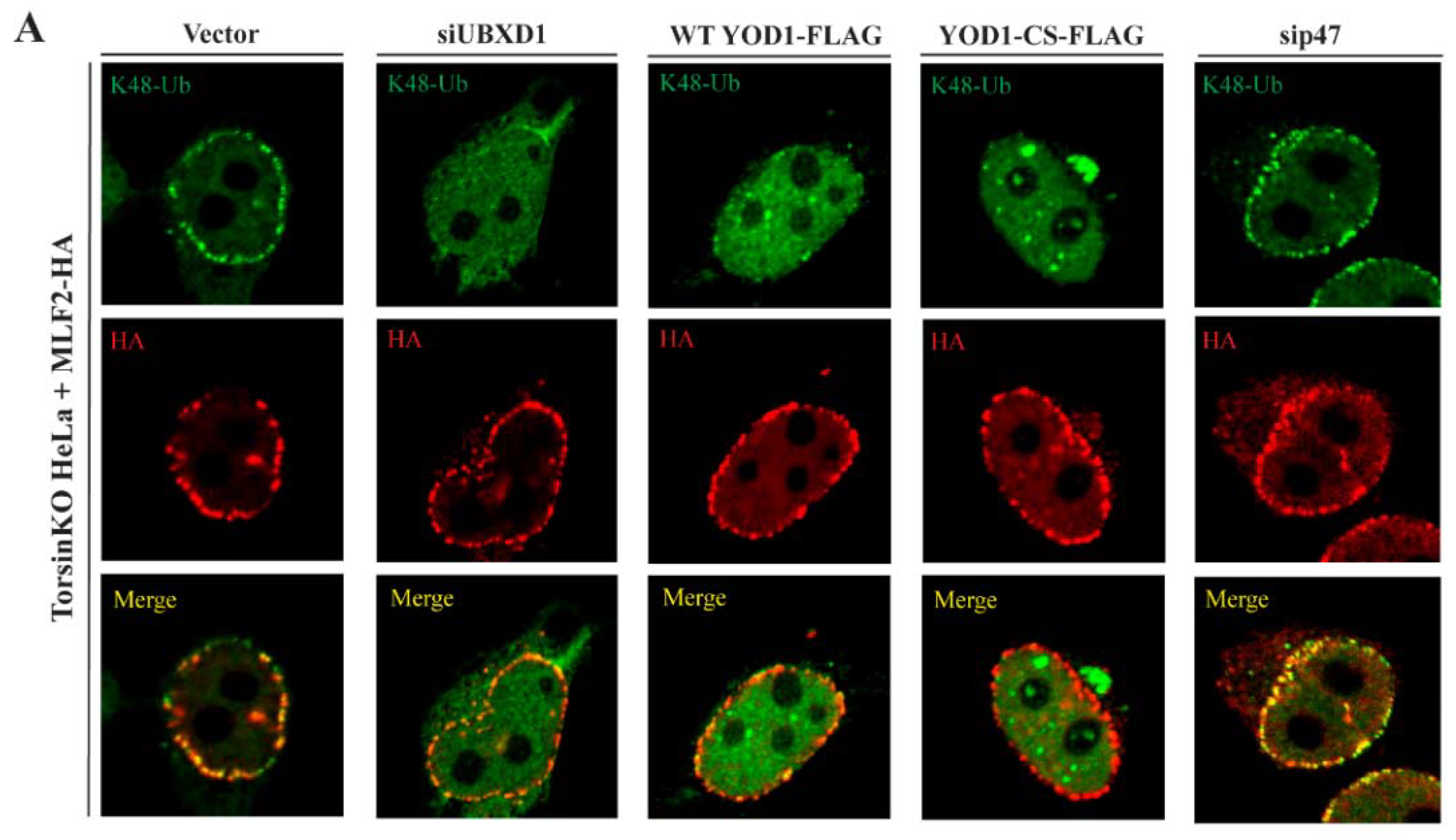
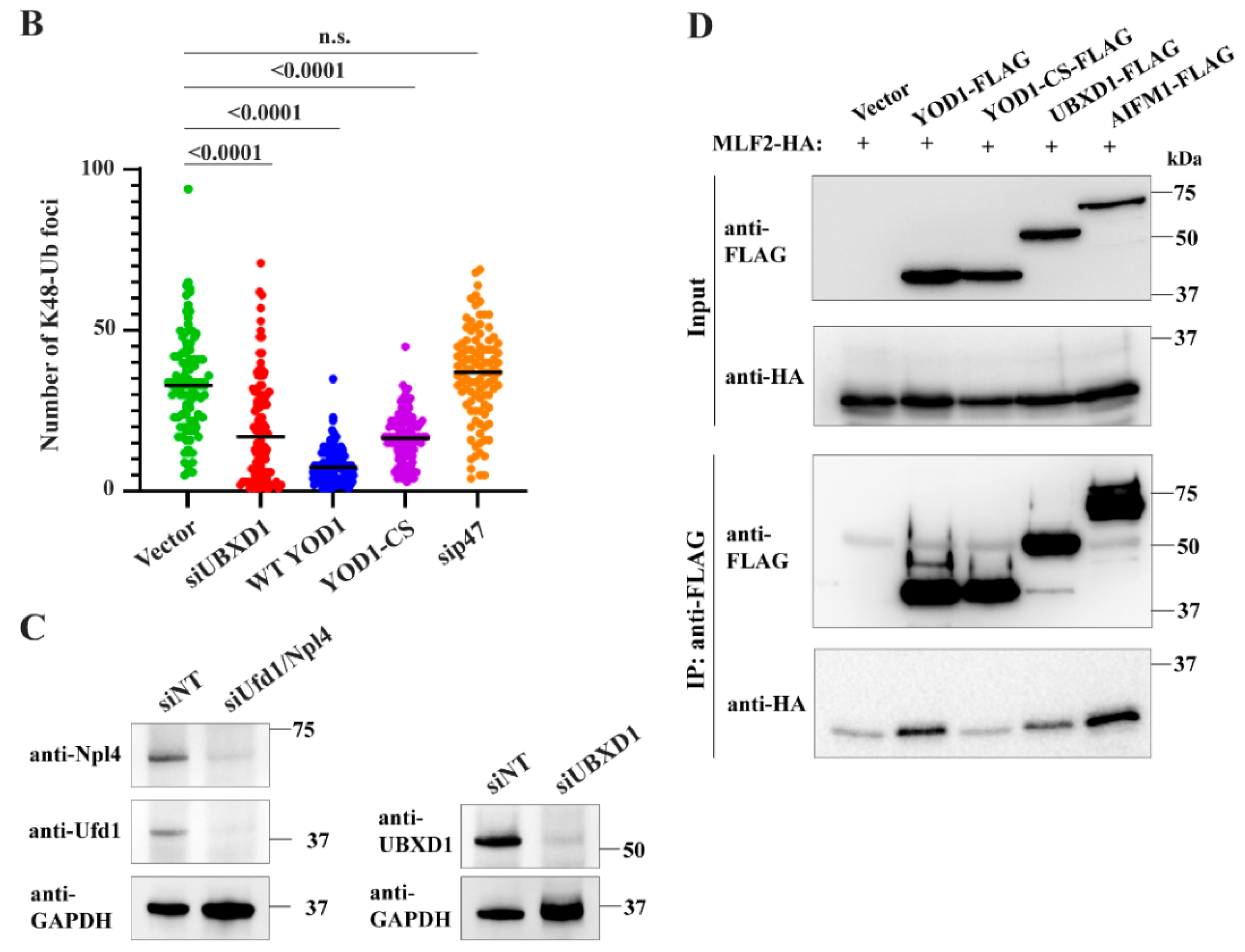
2.6. The Relevant p97 Activity for Accumulating K48-Ub inside Blebs Depends on the Cofactors YOD1 and UBXD1
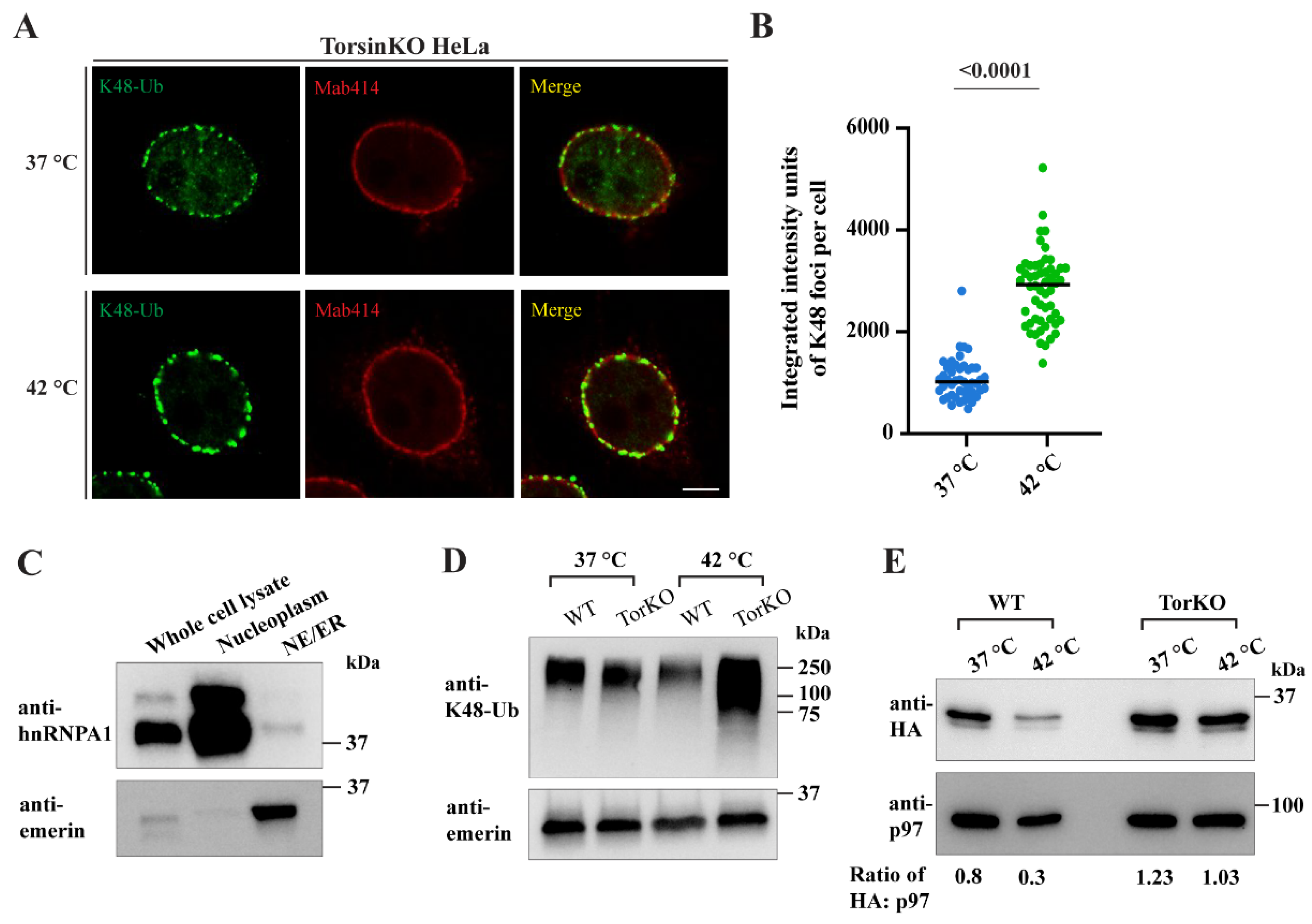
2.7. Provoking an Increase in Global Ubiquitylation Causes More K48-Ub Conjugates to Become Sequestered inside Blebs
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Cell Culture and Cell Lines
4.3. Small-Molecule Treatment, Plasmids, and Transient Transfections
4.4. siRNAs, Transient RNAi Knockdowns, and qPCR Validation
4.5. Immunofluorescence and Confocal Microscopy
4.6. Immunoprecipitation and Immunoblot Analysis
4.7. Cycloheximide Chase
4.8. NE Enrichment
4.9. Image Processing and Statical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goodchild, R.E.; Kim, C.E.; Dauer, W.T. Loss of the dystonia-associated protein torsina selectively disrupts the neuronal nuclear envelope. Neuron 2005, 48, 923–932. [Google Scholar] [CrossRef] [Green Version]
- Ozelius, L.J.; Hewett, J.W.; Page, C.E.; Bressman, S.B.; Kramer, P.L.; Shalish, C.; de Leon, D.; Brin, M.F.; Raymond, D.; Corey, D.P.; et al. The early-onset torsion dystonia gene (dyt1) encodes an atp-binding protein. Nat. Genet. 1997, 17, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Naismith, T.V.; Heuser, J.E.; Breakefield, X.O.; Hanson, P.I. Torsina in the nuclear envelope. Proc. Natl. Acad. Sci. USA 2004, 101, 7612–7617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Brown, R.S.; Chase, A.R.; Eisele, M.R.; Schlieker, C. Regulation of torsin atpases by lap1 and lull. Proc. Natl. Acad. Sci. USA 2013, 110, E1545–E1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.S.; Zhao, C.; Chase, A.R.; Wang, J.; Schlieker, C. The mechanism of torsin atpase activation. Proc. Natl. Acad. Sci. USA 2014, 111, E4822–E4831. [Google Scholar] [CrossRef] [Green Version]
- Sosa, B.A.; Demircioglu, F.E.; Chen, J.Z.; Ingram, J.; Ploegh, H.L.; Schwartz, T.U. How lamina-associated polypeptide 1 (lap1) activates torsin. eLife 2014, 3, e03239. [Google Scholar] [CrossRef]
- Goodchild, R.E.; Dauer, W.T. The aaa+ protein torsina interacts with a conserved domain present in lap1 and a novel er protein. J. Cell Biol. 2005, 168, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Demircioglu, F.E.; Sosa, B.A.; Ingram, J.; Ploegh, H.L.; Schwartz, T.U. Structures of torsina and its disease-mutant complexed with an activator reveal the molecular basis for primary dystonia. eLife 2016, 5, e17983. [Google Scholar] [CrossRef]
- Rose, A.E.; Brown, R.S.; Schlieker, C. Torsins: Not your typical aaa+ atpases. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 532–549. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Alegre, P. Advances in molecular and cell biology of dystonia: Focus on torsina. Neurobiol. Dis. 2019, 127, 233–241. [Google Scholar] [CrossRef]
- Rampello, A.J.; Prophet, S.M.; Schlieker, C. The role of torsin aaa+ proteins in preserving nuclear envelope integrity and safeguarding against disease. Biomolecules 2020, 10, 468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naismith, T.V.; Dalal, S.; Hanson, P.I. Interaction of torsina with its major binding partners is impaired by the dystonia-associated deltagag deletion. J. Biol. Chem. 2009, 284, 27866–27874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanGompel, M.J.; Nguyen, K.C.; Hall, D.H.; Dauer, W.T.; Rose, L.S. A novel function for the caenorhabditis elegans torsin ooc-5 in nucleoporin localization and nuclear import. Mol. Biol. Cell 2015, 26, 1752–1763. [Google Scholar] [CrossRef] [PubMed]
- Pappas, S.S.; Liang, C.C.; Kim, S.; Rivera, C.O.; Dauer, W.T. Torsina dysfunction causes persistent neuronal nuclear pore defects. Hum. Mol. Genet. 2018, 27, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Rampello, A.J.; Laudermilch, E.; Vishnoi, N.; Prophet, S.M.; Shao, L.; Zhao, C.; Lusk, C.P.; Schlieker, C. Torsin atpase deficiency leads to defects in nuclear pore biogenesis and sequestration of mlf. J. Cell Biol. 2020, 219, e201910185. [Google Scholar] [CrossRef]
- Jacquemyn, J.; Foroozandeh, J.; Vints, K.; Swerts, J.; Verstreken, P.; Gounko, N.V.; Gallego, S.F.; Goodchild, R. Torsin and nep1r1-ctdnep1 phosphatase affect interphase nuclear pore complex insertion by lipid-dependent and lipid-independent mechanisms. EMBO J. 2021, 40, e106914. [Google Scholar] [CrossRef]
- Laudermilch, E.; Tsai, P.L.; Graham, M.; Turner, E.; Zhao, C.; Schlieker, C. Dissecting torsin/cofactor function at the nuclear envelope: A genetic study. Mol. Biol. Cell 2016, 27, 3964–3971. [Google Scholar] [CrossRef]
- Prophet, S.M.; Rampello, A.J.; Niescier, R.F.; Shaw, J.E.; Koleske, A.J.; Schlieker, C. Mlf2 modulates phase separated nuclear envelope condensates that provoke dual proteotoxicity. bioRxiv 2021. [Google Scholar] [CrossRef]
- Liang, C.C.; Tanabe, L.M.; Jou, S.; Chi, F.; Dauer, W.T. Torsina hypofunction causes abnormal twisting movements and sensorimotor circuit neurodegeneration. J. Clin. Investig. 2014, 124, 3080–3092. [Google Scholar] [CrossRef] [Green Version]
- Lam, Y.A.; Lawson, T.G.; Velayutham, M.; Zweier, J.L.; Pickart, C.M. A proteasomal atpase subunit recognizes the polyubiquitin degradation signal. Nature 2002, 416, 763–767. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef] [PubMed]
- Huryn, D.M.; Kornfilt, D.J.P.; Wipf, P. P97: An emerging target for cancer, neurodegenerative diseases, and viral infections. J. Med. Chem. 2020, 63, 1892–1907. [Google Scholar] [CrossRef] [PubMed]
- Van den Boom, J.; Meyer, H. Vcp/p97-mediated unfolding as a principle in protein homeostasis and signaling. Mol. Cell 2018, 69, 182–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.J.; Wang, J.; Yao, B.; Wong, S.; Djakovic, S.; Kumar, B.; Rice, J.; Valle, E.; Soriano, F.; Menon, M.K.; et al. Discovery of a first-in-class, potent, selective, and orally bioavailable inhibitor of the p97 aaa atpase (cb-5083). J. Med. Chem. 2015, 58, 9480–9497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, D.J.; Le Moigne, R.; Djakovic, S.; Kumar, B.; Rice, J.; Wong, S.; Wang, J.; Yao, B.; Valle, E.; Kiss von Soly, S.; et al. Targeting the aaa atpase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell 2015, 28, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Rapoport, T.A. Mechanistic insights into er-associated protein degradation. Curr. Opin. Cell Biol. 2018, 53, 22–28. [Google Scholar] [CrossRef]
- Ji, Z.; Li, H.; Peterle, D.; Paulo, J.A.; Ficarro, S.B.; Wales, T.E.; Marto, J.A.; Gygi, S.P.; Engen, J.R.; Rapoport, T.A. Translocation of polyubiquitinated protein substrates by the hexameric cdc48 atpase. Mol. Cell 2022, 82, 570–584.e578. [Google Scholar] [CrossRef]
- Tsai, P.L.; Zhao, C.; Turner, E.; Schlieker, C. The lamin b receptor is essential for cholesterol synthesis and perturbed by disease-causing mutations. eLife 2016, 5, e16011. [Google Scholar] [CrossRef]
- Tsai, P.L.; Zhao, C.; Schlieker, C. Methodologies to monitor protein turnover at the inner nuclear membrane. Methods Enzymol. 2019, 619, 47–69. [Google Scholar]
- Wu, X.; Siggel, M.; Ovchinnikov, S.; Mi, W.; Svetlov, V.; Nudler, E.; Liao, M.; Hummer, G.; Rapoport, T.A. Structural basis of er-associated protein degradation mediated by the hrd1 ubiquitin ligase complex. Science 2020, 368, eaaz2449. [Google Scholar] [CrossRef]
- Schoebel, S.; Mi, W.; Stein, A.; Ovchinnikov, S.; Pavlovicz, R.; DiMaio, F.; Baker, D.; Chambers, M.G.; Su, H.; Li, D.; et al. Cryo-em structure of the protein-conducting erad channel hrd1 in complex with hrd. Nature 2017, 548, 352–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, C.C.; Vasic, V.; Stein, A. Doa10 is a membrane protein retrotranslocase in er-associated protein degradation. eLife 2020, 9, e56945. [Google Scholar] [CrossRef] [PubMed]
- Twomey, E.C.; Ji, Z.; Wales, T.E.; Bodnar, N.O.; Ficarro, S.B.; Marto, J.A.; Engen, J.R.; Rapoport, T.A. Substrate processing by the cdc48 atpase complex is initiated by ubiquitin unfolding. Science 2019, 365, eaax1033. [Google Scholar] [CrossRef] [PubMed]
- Mehrtash, A.B.; Hochstrasser, M. Ubiquitin-dependent protein degradation at the endoplasmic reticulum and nuclear envelope. Semin. Cell Dev. Biol. 2019, 93, 111–124. [Google Scholar] [CrossRef]
- Wu, X.; Rapoport, T.A. Translocation of proteins through a distorted lipid bilayer. Trends Cell Biol. 2021, 31, 473–484. [Google Scholar] [CrossRef]
- Ferro-Novick, S.; Reggiori, F.; Brodsky, J.L. Er-phagy, er homeostasis, and er quality control: Implications for disease. Trends Biochem. Sci. 2021, 46, 630–639. [Google Scholar] [CrossRef]
- Vasic, V.; Denkert, N.; Schmidt, C.C.; Riedel, D.; Stein, A.; Meinecke, M. Hrd1 forms the retrotranslocation pore regulated by auto-ubiquitination and binding of misfolded proteins. Nat. Cell Biol. 2020, 22, 274–281. [Google Scholar] [CrossRef]
- Schubert, U.; Anton, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef]
- Wolff, S.; Weissman, J.S.; Dillin, A. Differential scales of protein quality control. Cell 2014, 157, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Gloge, F.; Becker, A.H.; Kramer, G.; Bukau, B. Co-translational mechanisms of protein maturation. Curr. Opin. Struct. Biol. 2014, 24, 24–33. [Google Scholar] [CrossRef]
- Buchberger, A.; Schindelin, H.; Hanzelmann, P. Control of p97 function by cofactor binding. FEBS Lett. 2015, 589, 2578–2589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Tang, W.K.; Zhang, T.; Xia, D. A mighty “protein extractor” of the cell: Structure and function of the p97/cdc48 atpase. Front. Mol. Biosci. 2017, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. Function of the p97-ufd1-npl4 complex in retrotranslocation from the er to the cytosol: Dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 2003, 162, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Rabouille, C.; Newman, R.; Levine, T.P.; Pappin, D.; Freemont, P.; Warren, G. P47 is a cofactor for p97-mediated membrane fusion. Nature 1997, 388, 75–78. [Google Scholar] [CrossRef]
- Haines, D.S.; Lee, J.E.; Beauparlant, S.L.; Kyle, D.B.; den Besten, W.; Sweredoski, M.J.; Graham, R.L.; Hess, S.; Deshaies, R.J. Protein interaction profiling of the p97 adaptor ubxd1 points to a role for the complex in modulating ergic-53 trafficking. Mol. Cell. Proteomics 2012, 11, M111.016444. [Google Scholar] [CrossRef] [Green Version]
- Ritz, D.; Vuk, M.; Kirchner, P.; Bug, M.; Schutz, S.; Hayer, A.; Bremer, S.; Lusk, C.; Baloh, R.H.; Lee, H.; et al. Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by vcp and ubxd1 and impaired by vcp disease mutations. Nat. Cell Biol. 2011, 13, 1116–1123. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, C.; Kirchner, P.; Bug, M.; Grum, D.; Koerver, L.; Schulze, N.; Poehler, R.; Dressler, A.; Fengler, S.; Arhzaouy, K.; et al. Vcp/p97 cooperates with yod1, ubxd1 and plaa to drive clearance of ruptured lysosomes by autophagy. EMBO J. 2017, 36, 135–150. [Google Scholar] [CrossRef] [Green Version]
- Bento, A.C.; Bippes, C.C.; Kohler, C.; Hemion, C.; Frank, S.; Neutzner, A. Ubxd1 is a mitochondrial recruitment factor for p97/vcp and promotes mitophagy. Sci. Rep. 2018, 8, 12415. [Google Scholar] [CrossRef] [Green Version]
- Nagahama, M.; Ohnishi, M.; Kawate, Y.; Matsui, T.; Miyake, H.; Yuasa, K.; Tani, K.; Tagaya, M.; Tsuji, A. Ubxd1 is a vcp-interacting protein that is involved in er-associated degradation. Biochem. Biophys. Res. Commun. 2009, 382, 303–308. [Google Scholar] [CrossRef]
- Ernst, R.; Mueller, B.; Ploegh, H.L.; Schlieker, C. The otubain yod1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the er. Mol. Cell 2009, 36, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Ewing, R.M.; Chu, P.; Elisma, F.; Li, H.; Taylor, P.; Climie, S.; McBroom-Cerajewski, L.; Robinson, M.D.; O’Connor, L.; Li, M.; et al. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol. Syst. Biol. 2007, 3, 89. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Salokas, K.; Tamene, F.; Jiu, Y.; Weldatsadik, R.G.; Ohman, T.; Varjosalo, M. An ap-ms- and bioid-compatible mac-tag enables comprehensive mapping of protein interactions and subcellular localizations. Nat. Commun. 2018, 9, 1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parag, H.A.; Raboy, B.; Kulka, R.G. Effect of heat shock on protein degradation in mammalian cells: Involvement of the ubiquitin system. EMBO J. 1987, 6, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Fujimuro, M.; Sawada, H.; Yokosawa, H. Dynamics of ubiquitin conjugation during heat-shock response revealed by using a monoclonal antibody specific to multi-ubiquitin chains. Eur. J. Biochem. 1997, 249, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, B.A.; Gwon, Y.; Mishra, A.; Peng, J.; Nakamura, H.; Zhang, K.; Kim, H.J.; Taylor, J.P. Ubiquitination is essential for recovery of cellular activities after heat shock. Science 2021, 372, eabc3593. [Google Scholar] [CrossRef]
- Friant, S.; Meier, K.D.; Riezman, H. Increased ubiquitin-dependent degradation can replace the essential requirement for heat shock protein induction. EMBO J. 2003, 22, 3783–3791. [Google Scholar] [CrossRef]
- Elsiena Kuiper, E.F.; Gallardo, P.; Bergsma, T.; Mari, M.; Musskopf, M.K.; Kuipers, J.; Giepmans, B.N.G.; Steen, A.; Veenhoff, L.M.; Kampinga, H.H.; et al. The molecular chaperone dnajb6 provides surveillance of fg-nups and is required for interphase nuclear pore complex biogenesis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Weisheit, C.E.; Dauer, W.T. A novel conditional knock-in approach defines molecular and circuit effects of the dyt1 dystonia mutation. Hum. Mol. Genet. 2015, 24, 6459–6472. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liang, C.-C.; Pappas, S.S.; Dauer, W.T. Torsinb overexpression prevents abnormal twisting in dyt1 dystonia mouse models. eLife 2019, 9, e54285. [Google Scholar] [CrossRef]
- Goodchild, R.E.; Dauer, W.T. Mislocalization to the nuclear envelope: An effect of the dystonia-causing torsina mutation. Proc. Natl. Acad. Sci. USA 2004, 101, 847–852. [Google Scholar] [CrossRef] [Green Version]
- Vander Heyden, A.B.; Naismith, T.V.; Snapp, E.L.; Hanson, P.I. Static retention of the lumenal monotopic membrane protein torsina in the endoplasmic reticulum. EMBO J. 2011, 30, 3217–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nery, F.C.; Armata, I.A.; Farley, J.E.; Cho, J.A.; Yaqub, U.; Chen, P.; da Hora, C.C.; Wang, Q.; Tagaya, M.; Klein, C.; et al. Torsina participates in endoplasmic reticulum-associated degradation. Nat. Commun. 2011, 2, 393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.Y.; Hernandez-Ono, A.; Fedotova, T.; Ostlund, C.; Lee, M.J.; Gibeley, S.B.; Liang, C.C.; Dauer, W.T.; Ginsberg, H.N.; Worman, H.J. Nuclear envelope-localized torsina-lap1 complex regulates hepatic vldl secretion and steatosis. J. Clin. Investig. 2019, 130, 4885–4900. [Google Scholar] [CrossRef] [PubMed]
- Sasset, L.; Petris, G.; Cesaratto, F.; Burrone, O.R. The vcp/p97 and yod1 proteins have different substrate-dependent activities in endoplasmic reticulum-associated degradation (erad). J. Biol. Chem. 2015, 290, 28175–28188. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Gutierrez, G.J.; Ronai, Z.A. Ubiquitin-recognition protein ufd1 couples the endoplasmic reticulum (er) stress response to cell cycle control. Proc. Natl. Acad. Sci. USA 2011, 108, 9119–9124. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.M.; Wang, H.J.; Chen, F.; Guo, W.H.; Wang, Y.Y.; Li, H.Y.; Tang, J.H.; Ding, Y.; Shen, Y.C.; Li, M.; et al. Hrd1 suppresses the growth and metastasis of breast cancer cells by promoting igf-1r degradation. Oncotarget 2015, 6, 42854–42867. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Stirling, D.R.; Swain-Bowden, M.J.; Lucas, A.M.; Carpenter, A.E.; Cimini, B.A.; Goodman, A. Cellprofiler 4: Improvements in speed, utility and usability. BMC Bioinform. 2021, 22, 433. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prophet, S.M.; Naughton, B.S.; Schlieker, C. p97/UBXD1 Generate Ubiquitylated Proteins That Are Sequestered into Nuclear Envelope Herniations in Torsin-Deficient Cells. Int. J. Mol. Sci. 2022, 23, 4627. https://doi.org/10.3390/ijms23094627
Prophet SM, Naughton BS, Schlieker C. p97/UBXD1 Generate Ubiquitylated Proteins That Are Sequestered into Nuclear Envelope Herniations in Torsin-Deficient Cells. International Journal of Molecular Sciences. 2022; 23(9):4627. https://doi.org/10.3390/ijms23094627
Chicago/Turabian StyleProphet, Sarah M., Brigitte S. Naughton, and Christian Schlieker. 2022. "p97/UBXD1 Generate Ubiquitylated Proteins That Are Sequestered into Nuclear Envelope Herniations in Torsin-Deficient Cells" International Journal of Molecular Sciences 23, no. 9: 4627. https://doi.org/10.3390/ijms23094627
APA StyleProphet, S. M., Naughton, B. S., & Schlieker, C. (2022). p97/UBXD1 Generate Ubiquitylated Proteins That Are Sequestered into Nuclear Envelope Herniations in Torsin-Deficient Cells. International Journal of Molecular Sciences, 23(9), 4627. https://doi.org/10.3390/ijms23094627