
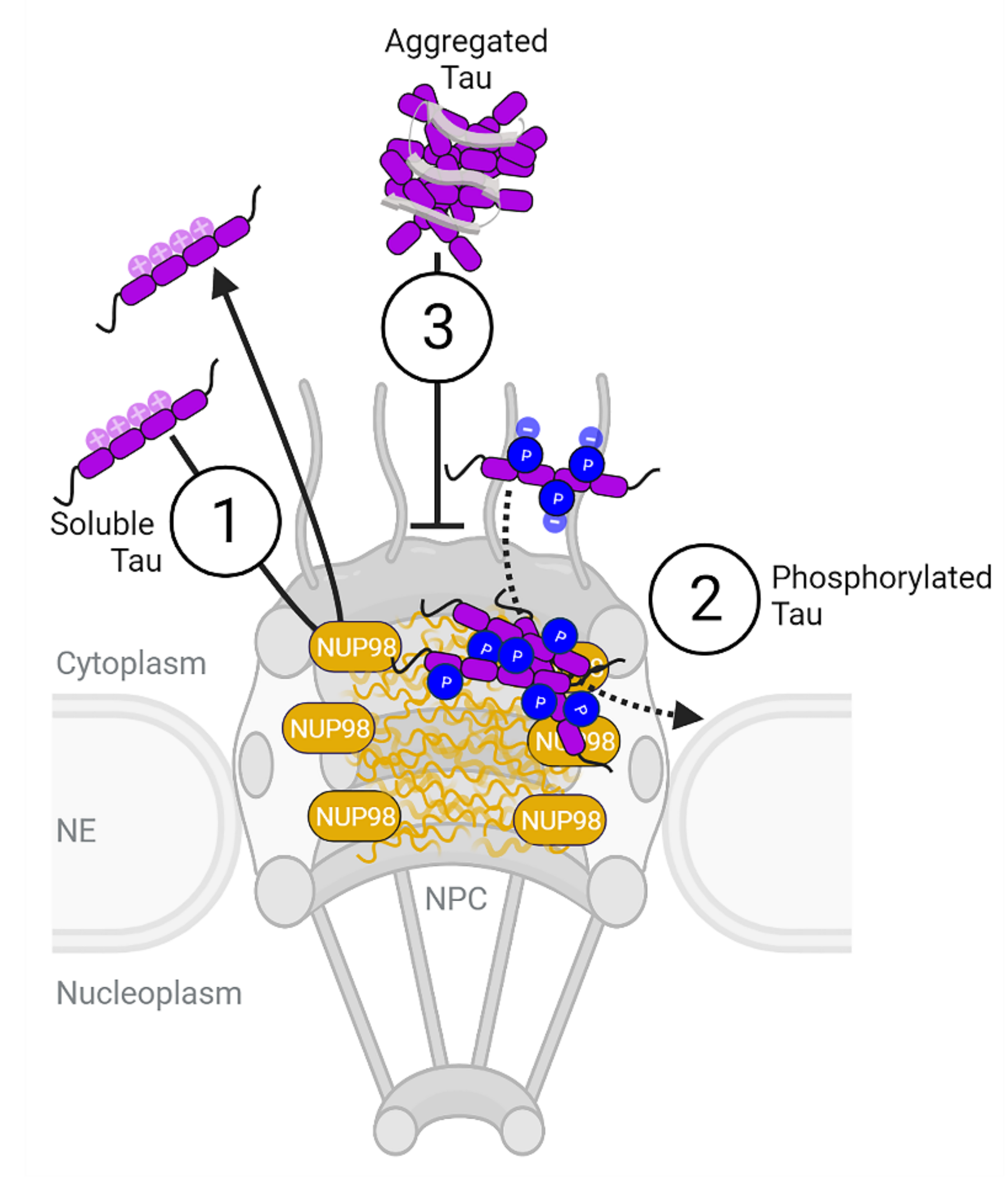
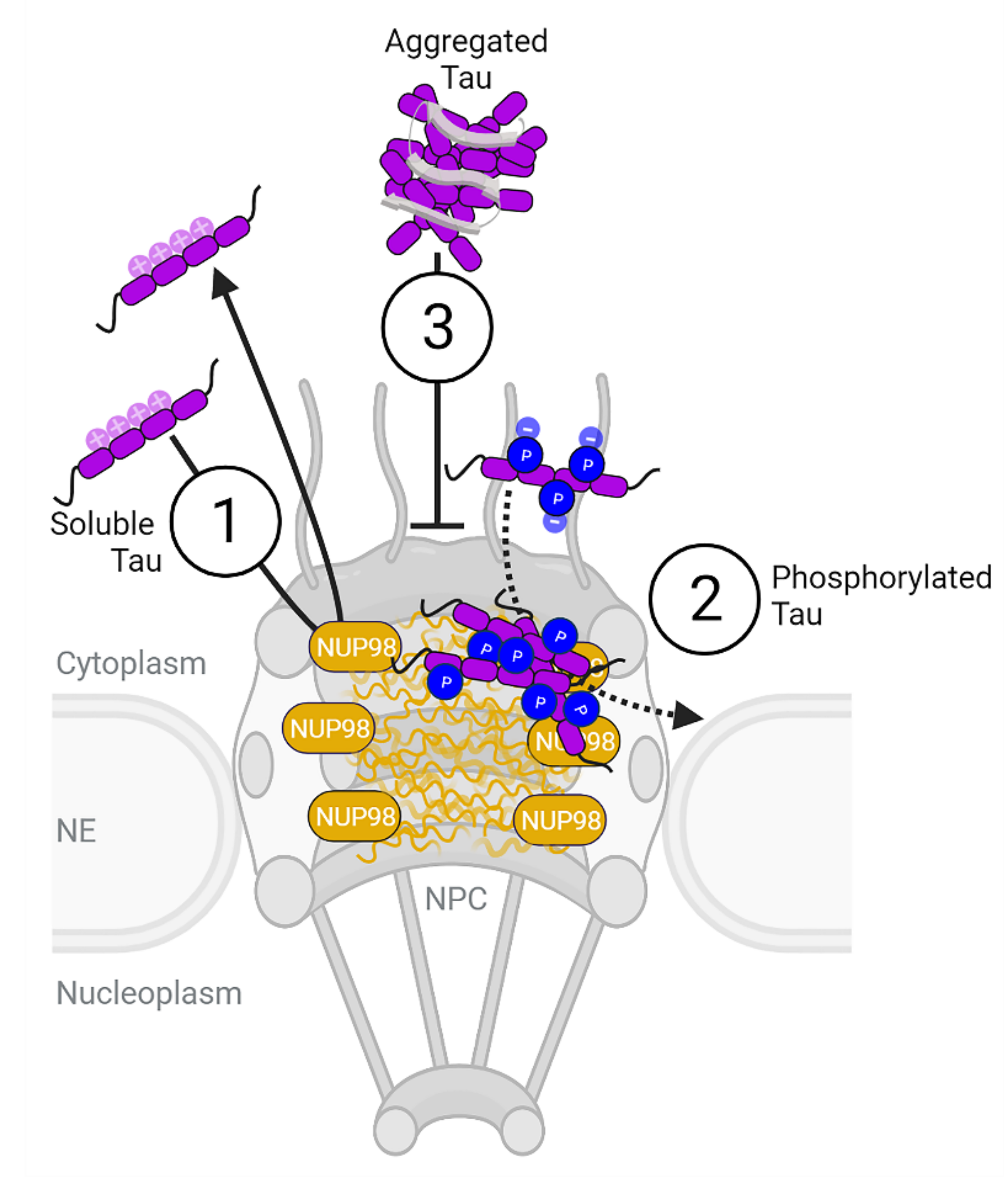
Phosphorylation but Not Oligomerization Drives the Accumulation of Tau with Nucleoporin Nup98

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
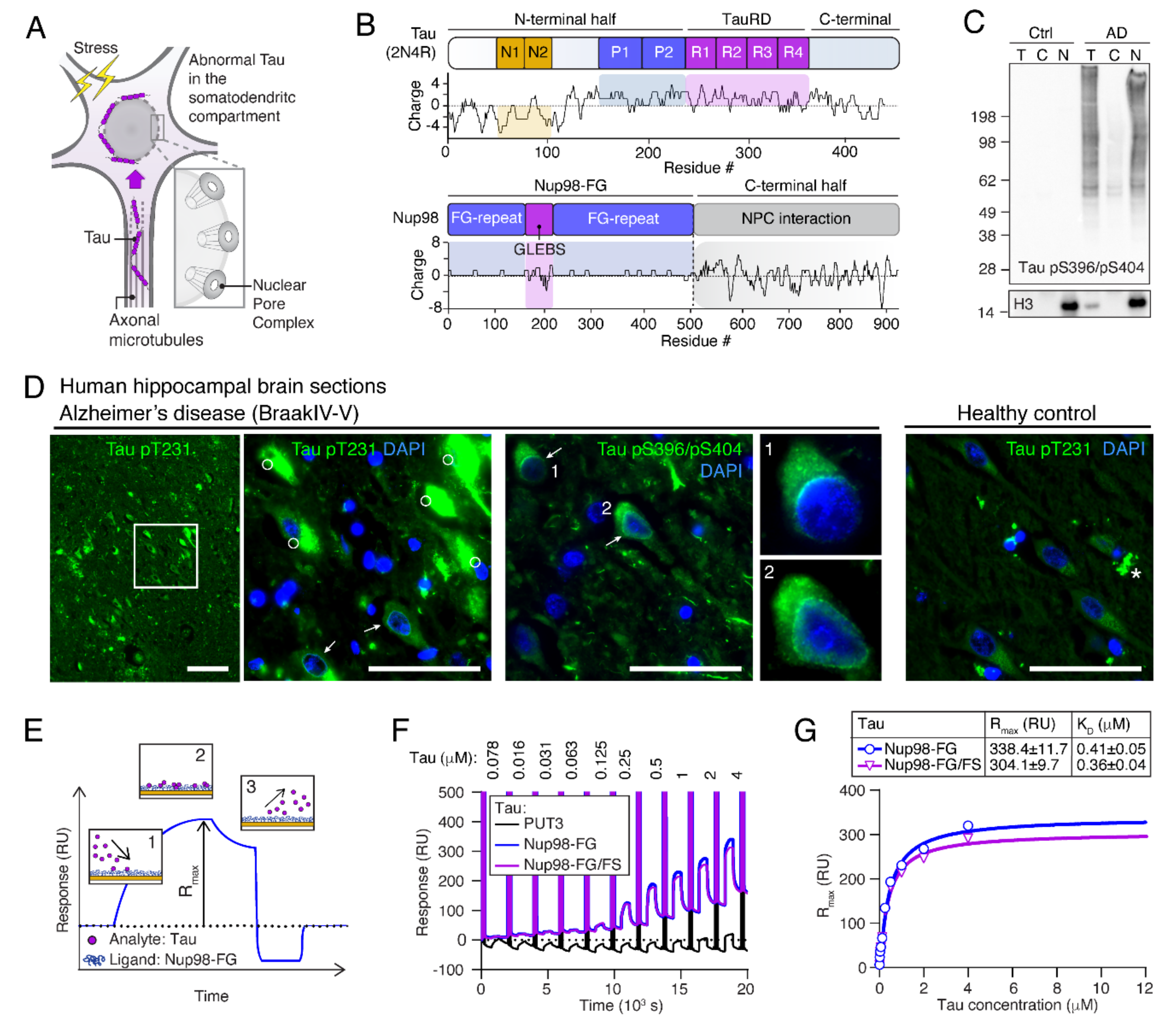
2.1. Tau Accumulates at Neuronal NEs in AD Brains
2.2. Human Tau Binds to Nup98-FG
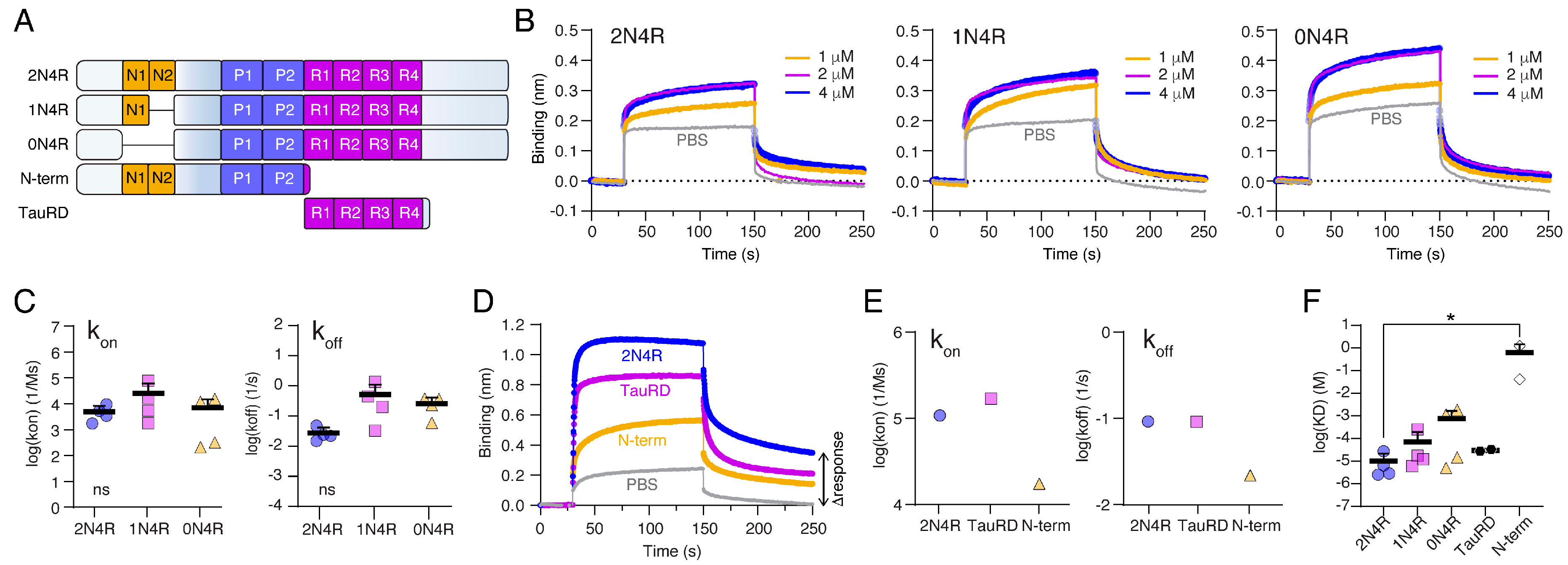
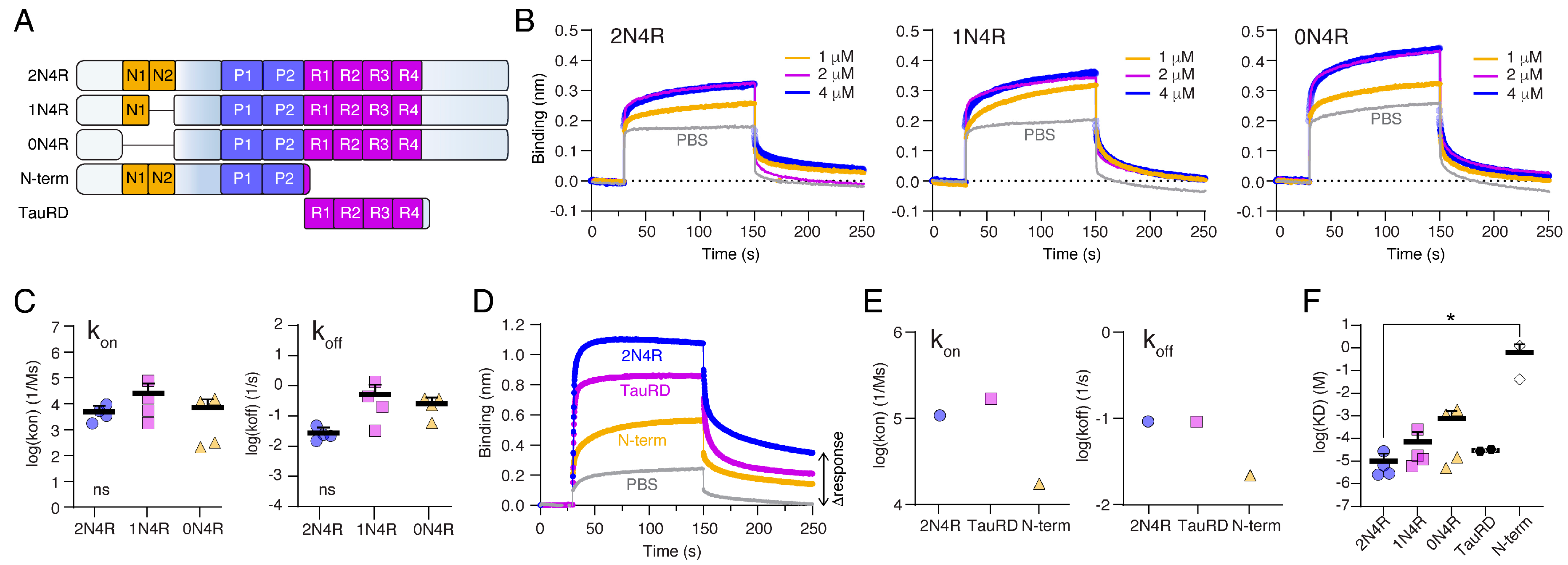
2.3. The Tau Repeat Domain Mediates the Binding to Nup98
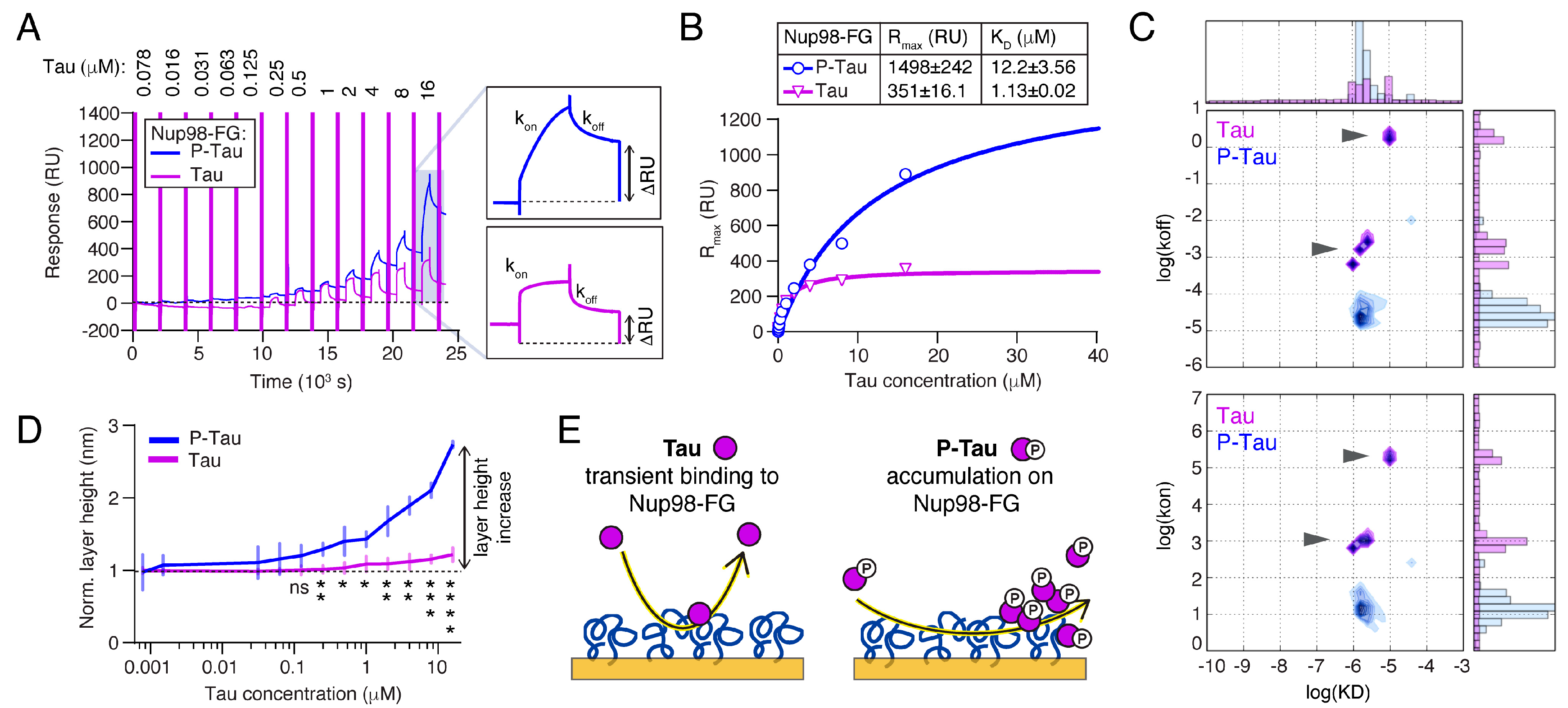
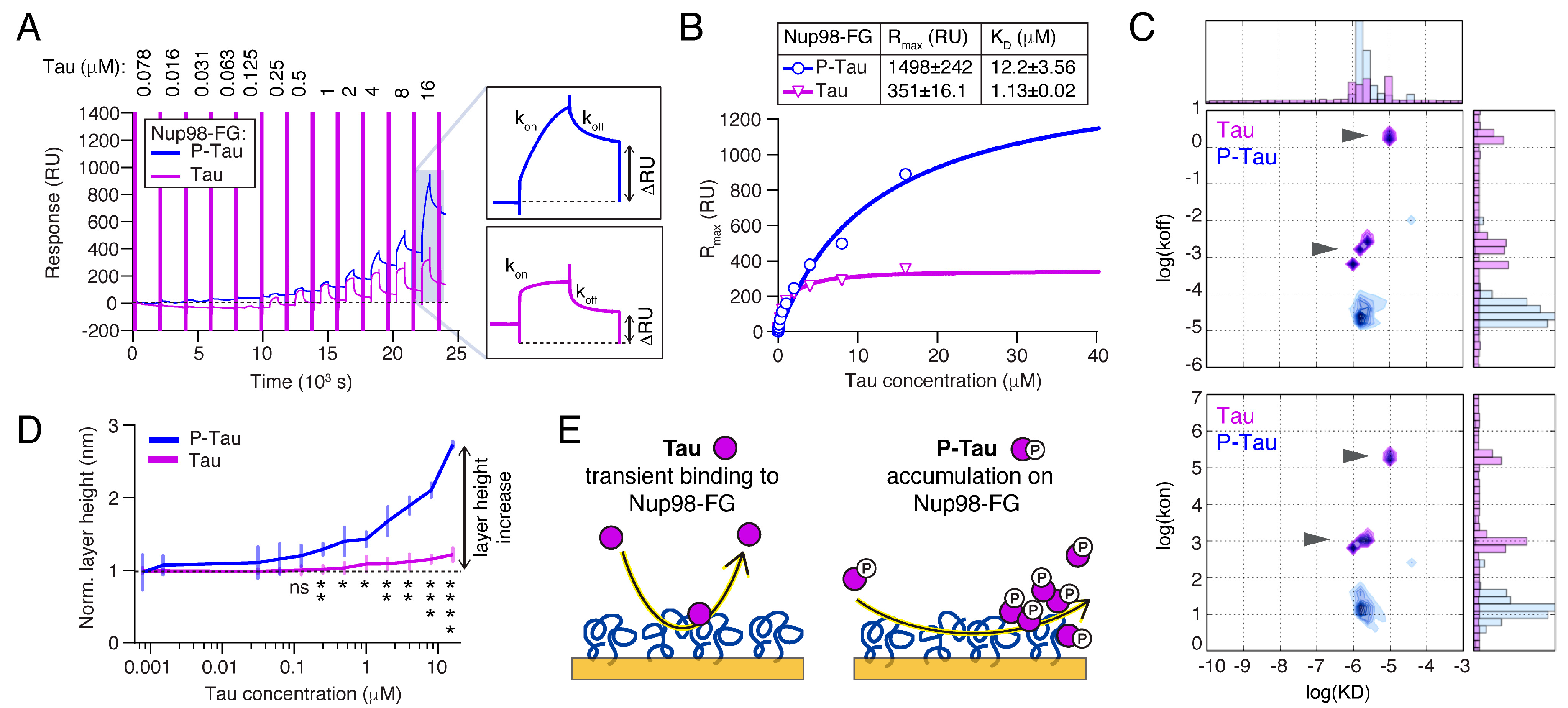
2.4. Phosphorylation Increases the Binding and Accumulation of Tau to Nup98-FG
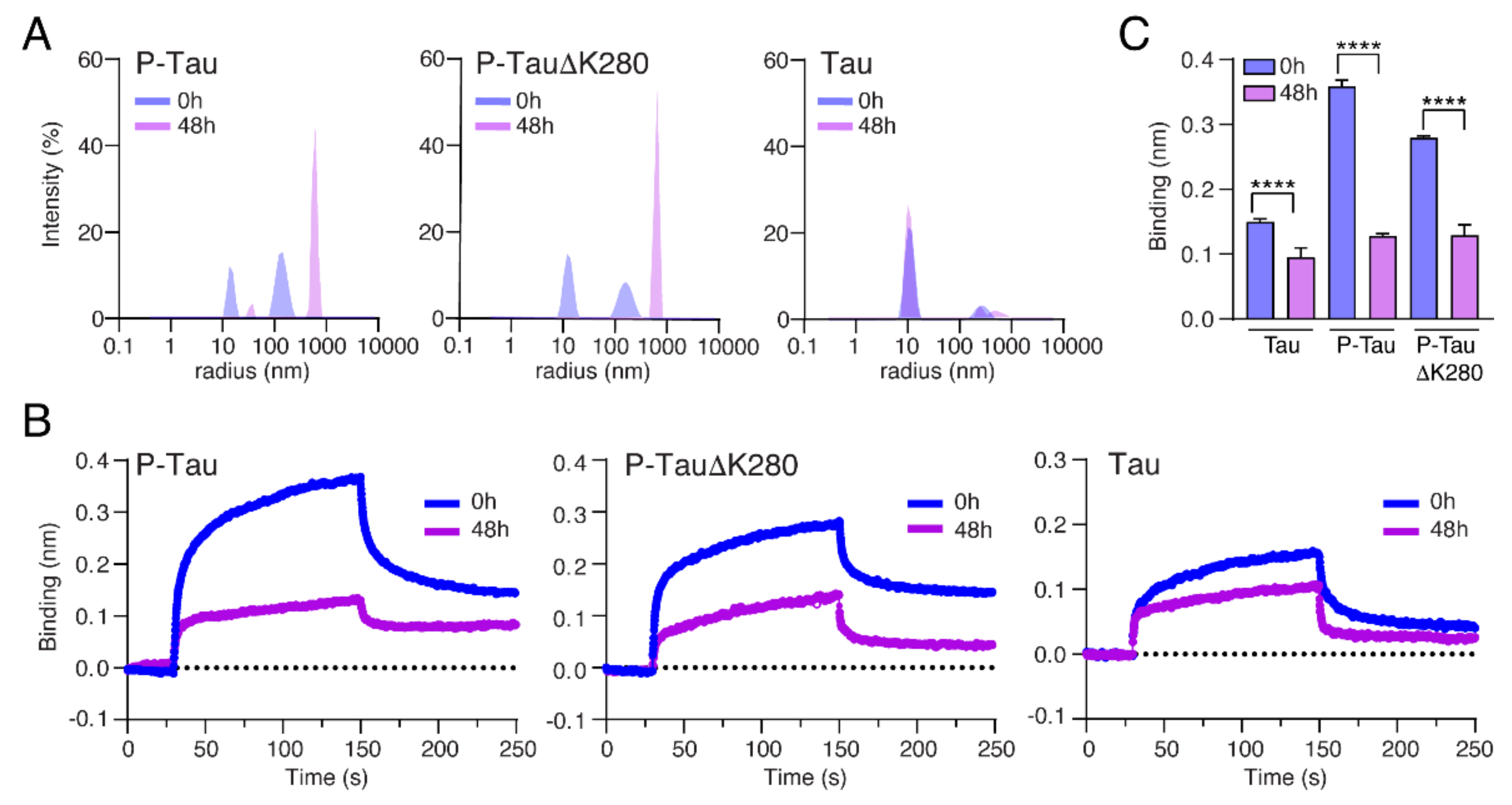
2.5. Oligomerization Reduces the Binding of Tau to Nup98-FG
3. Discussion
4. Materials and Methods
4.1. Human Samples
4.2. Tau Detection in AD Brain Sections (IHC)
4.3. Expression and Purification of Recombinant Proteins (E. coli and SF9 Tau)
4.4. Tau in Vitro Acetylation
4.5. Surface Plasmon Resonance
4.6. Bio-Layer Interferomertry
4.7. Dynamic Light Scattering (DLS) Measurements
4.8. Human Brain Nuclei Preparation
4.9. Coomassie, SDS-PAGE, and Western Blot
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mandelkow, E.M.; Biernat, J.; Drewes, G.; Gustke, N.; Trinczek, B.; Mandelkow, E. Tau Domains, Phosphorylation, and Interactions with Microtubules. Neurobiol. Aging 1995, 16, 355–362. [Google Scholar] [CrossRef]
- Ballatore, C.; Lee, V.M.Y.; Trojanowski, J.Q. Tau-Mediated Neurodegeneration in Alzheimer’s Disease and Related Disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G. Pathogenesis of the Tauopathies. J. Mol. Neurosci. 2011, 45, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.M.; Mudher, A. Are Tau Aggregates Toxic or Protective in Tauopathies? Front. Neurol. 2013, 4, 114. [Google Scholar] [CrossRef] [Green Version]
- Kopeikina, K.J.; Hyman, B.J.; Spires-Jones, T.L. Soluble Forms of Tau Are Toxic in Alzheimer’s Disease. Transl. Neurosci. 2012, 3, 223–233. [Google Scholar] [CrossRef]
- Zempel, H.; Mandelkow, E. Lost after Translation: Missorting of Tau Protein and Consequences for Alzheimer Disease. Trends Neurosci. 2014, 37, 721–732. [Google Scholar] [CrossRef]
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear Tau, a Key Player in Neuronal DNA Protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Kumar, Y.; Zempel, H.; Mandelkow, E.M.; Biernat, J.; Mandelkow, E. Novel Diffusion Barrier for Axonal Retention of Tau in Neurons and Its Failure in Neurodegeneration. EMBO J. 2011, 30, 4825–4837. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.Z.; Xia, Y.Y.; Grundke-Iqbal, I.; Iqbal, K. Abnormal Hyperphosphorylation of Tau: Sites, Regulation, and Molecular Mechanism of Neurofibrillary Degeneration. J. Alzheimer’s Dis. 2013, 33, S123–S139. [Google Scholar] [CrossRef]
- Sohn, P.D.; Tracy, T.E.; Son, H.I.; Zhou, Y.; Leite, R.E.P.; Miller, B.L.; Seeley, W.W.; Grinberg, L.T.; Gan, L. Acetylated Tau Destabilizes the Cytoskeleton in the Axon Initial Segment and Is Mislocalized to the Somatodendritic Compartment. Mol. Neurodegener. 2016, 11, 47. [Google Scholar] [CrossRef] [Green Version]
- Sotiropoulos, I.; Galas, M.C.; Silva, J.M.; Skoulakis, E.; Wegmann, S.; Maina, M.B.; Blum, D.; Sayas, C.L.; Mandelkow, E.M.; Mandelkow, E.; et al. Atypical, Non-Standard Functions of the Microtubule Associated Tau Protein. Acta Neuropathol. Commun. 2017, 5, 91. [Google Scholar] [CrossRef] [Green Version]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S.; et al. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron 2018, 99, 925–940.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochmair, J.; Exner, C.; Franck, M.; Dominguez-baquero, A.; Kraushar, M.L.; Mielke, T.; Radbruch, H.; Diez, L.; Kaniyappan, S.; Falke, S.; et al. Molecular Crowding and RNA Synergize to Promote Phase Separation, Microtubule Interaction, and Seeding of Tau Condensates. EMBO J. 2022, e108882. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, L.G.; Miskiewicz, H.B.; Tannenbaum, L.B.; Mirra, S.S. Nuclear Pore Complex Proteins in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2006, 65, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, B.; Bardai, F.H.; Feany, M.B. Lamin Dysfunction Mediates Neurodegeneration in Tauopathies. Curr. Biol. 2016, 26, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paonessa, F.; Evans, L.D.; Solanki, R.; Larrieu, D.; Wray, S.; Hardy, J.; Jackson, S.P.; Livesey, F.J. Microtubules Deform the Nuclear Membrane and Disrupt Nucleocytoplasmic Transport in Tau-Mediated Frontotemporal Dementia. Cell Rep. 2019, 26, 582–593.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, F.; Sousa, J.; Pereira, C.D.; Da Cruz e Silva, O.A.B.; Rebelo, S. Nuclear Envelope Dysfunction and Its Contribution to the Aging Process. Aging Cell 2020, 19, e13143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terry, L.J.; Shows, E.B.; Wente, S.R. Crossing the Nuclear Envelope: Hierarchical Regulation of Nucleocytoplasmic Transport. Science 2007, 318, 1412–1416. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Liang, C.; Li, F.; Wang, L.; Wu, X.; Lu, A.; Xiao, G.; Zhang, G. The Rules and Functions of Nucleocytoplasmic Shuttling Proteins. Int. J. Mol. Sci. 2018, 19, 1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakiyama, Y.; Mazur, A.; Kapinos, L.E.; Lim, R.Y.H. Spatiotemporal Dynamics of the Nuclear Pore Complex Transport Barrier Resolved by High-Speed Atomic Force Microscopy. Nat. Nanotechnol. 2016, 11, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Cornelison, G.L.; Levy, S.A.; Jenson, T.; Frost, B. Tau-Induced Nuclear Envelope Invagination Causes a Toxic Accumulation of MRNA in Drosophila. Aging Cell 2019, 18, e12847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butner, K.A.; Kirschner, M.W. Tau Protein Binds to Microtubules through a Flexible Array of Distributed Weak Sites. J. Cell Biol. 1991, 115, 717–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustke, N.; Trinczek, B.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Domains of τ Protein and Interactions with Microtubules. Biochemistry 1994, 33, 9511–9522. [Google Scholar] [CrossRef] [PubMed]
- Kadavath, H.; Hofele, R.V.; Biernat, J.; Kumar, S.; Tepper, K.; Urlaub, H.; Mandelkow, E.; Zweckstetter, M. Tau Stabilizes Microtubules by Binding at the Interface between Tubulin Heterodimers. Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506. [Google Scholar] [CrossRef] [Green Version]
- Mukrasch, M.D.; Von Bergen, M.; Biernat, J.; Fischer, D.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. The “Jaws” of the Tau-Microtubule Interaction. J. Biol. Chem. 2007, 282, 12230–12239. [Google Scholar] [CrossRef] [Green Version]
- Jeganathan, S.; Von Bergen, M.; Mandelkow, E.M.; Mandelkow, E. The Natively Unfolded Character of Tau and Its Aggregation to Alzheimer-like Paired Helical Filaments. Biochemistry 2008, 47, 10526–10539. [Google Scholar] [CrossRef]
- Daebel, V.; Chinnathambi, S.; Biernat, J.; Schwalbe, M.; Habenstein, B.; Loquet, A.; Akoury, E.; Tepper, K.; Müller, H.; Baldus, M.; et al. β-Sheet Core of Tau Paired Helical Filaments Revealed by Solid-State NMR. J. Am. Chem. Soc. 2012, 134, 13982–13989. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Tepper, K.; Kaniyappan, S.; Biernat, J.; Wegmann, S.; Mandelkow, E.M.; Müller, D.J.; Mandelkow, E. Stages and Conformations of the Tau Repeat Domain during Aggregation and Its Effect on Neuronal Toxicity. J. Biol. Chem. 2014, 289, 20318–20332. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM Structures of Tau Filaments from Alzheimer’s Disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Zhang, W.; Yang, Y.; Murzin, A.G.; Falcon, B.; Kotecha, A.; Van Beers, M.; Tarutani, A.; Kametani, F.; Garringer, H.J.; et al. Structure-Based Classification of Tauopathies. Nature 2021, 598, 359–363. [Google Scholar] [CrossRef]
- Falcon, B.; Zhang, W.; Schweighauser, M.; Murzin, A.G.; Vidal, R.; Garringer, H.J.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Tau Filaments from Multiple Cases of Sporadic and Inherited Alzheimer’s Disease Adopt a Common Fold. Acta Neuropathol. 2018, 136, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, D.; Zandi, R.; Kim, Y.W.; Colvin, M.; Rexach, M.; Gopinathan, A. Nuclear Pore Complex Protein Sequences Determine Overall Copolymer Brush Structure and Function. Biophys. J. 2014, 106, 1997–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayliss, R.; Littlewood, T.; Stewart, M. Structural Basis for the Interaction between FxFG Nucleoporin Repeats and Importin-β in Nuclear Trafficking. Cell 2000, 102, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Bednenko, J.; Cingolani, G.; Gerace, L. Nucleocytoplasmic Transport: Navigating the Channel. Traffic 2003, 4, 127–135. [Google Scholar] [CrossRef]
- Isgro, T.A.; Schulten, K. Binding Dynamics of Isolated Nucleoporin Repeat Regions to Importin-β. Structure 2005, 13, 1869–1879. [Google Scholar] [CrossRef] [PubMed]
- Jeganathan, S.; Von Bergen, M.; Brutlach, H.; Steinhoff, H.J.; Mandelkow, E. Global Hairpin Folding of Tau in Solution. Biochemistry 2006, 45, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Barghorn, S.; Biernat, J.; Mandelkow, E. Purification of Recombinant Tau Protein and Preparation of Alzheimer-Paired Helical Filaments in Vitro. Methods Mol. Biol. 2005, 299, 35–51. [Google Scholar] [CrossRef]
- Drepper, F.; Biernat, J.; Kaniyappan, S.; Meyer, H.E.; Mandelkow, E.M.; Warscheid, B.; Mandelkow, E. A Combinatorial Native MS and LC-MS/MS Approach Reveals High Intrinsic Phosphorylation of Human Tau but Minimal Levels of Other Key Modifications. J. Biol. Chem. 2020, 295, 18213–18225. [Google Scholar] [CrossRef]
- Schoch, R.L.; Kapinos, L.E.; Lim, R.Y.H. Nuclear Transport Receptor Binding Avidity Triggers a Self-Healing Collapse Transition in FG-Nucleoporin Molecular Brushes. Proc. Natl. Acad. Sci. USA 2012, 109, 16911–16916. [Google Scholar] [CrossRef] [Green Version]
- Trzeciakiewicz, H.; Tseng, J.H.; Wander, C.M.; Madden, V.; Tripathy, A.; Yuan, C.X.; Cohen, T.J. A Dual Pathogenic Mechanism Links Tau Acetylation to Sporadic Tauopathy. Sci. Rep. 2017, 7, 44102. [Google Scholar] [CrossRef]
- Ukmar-Godec, T.; Hutten, S.; Grieshop, M.P.; Rezaei-Ghaleh, N.; Cima-Omori, M.S.; Biernat, J.; Mandelkow, E.; Söding, J.; Dormann, D.; Zweckstetter, M. Lysine/RNA-Interactions Drive and Regulate Biomolecular Condensation. Nat. Commun. 2019, 10, 2909. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Sarmiento, J.; Troncoso, J.; Jackson, G.R.; Kayed, R. Identification of Oligomers at Early Stages of Tau Aggregation in Alzheimer’s Disease. FASEB J. 2012, 26, 1946–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, S.M.; Himmelstein, D.S.; Lancia, J.K.; Binder, L.I. Tau Oligomers and Tau Toxicity in Neurodegenerative Disease. Biochem. Soc. Trans. 2012, 40, 667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A Current View on Tau Protein Phosphorylation in Alzheimer’s Disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, H.; Mair, W.; Kumar, M.; Schlaffner, C.N.; Tang, S.; Beerepoot, P.; Fatou, B.; Guise, A.J.; Cheng, L.; Takeda, S.; et al. Tau PTM Profiles Identify Patient Heterogeneity and Stages of Alzheimer’s Disease. Cell 2020, 183, 1699–1713.e13. [Google Scholar] [CrossRef]
- Ren, Y.; Seo, H.S.; Blobel, G.; Hoelz, A. Structural and Functional Analysis of the Interaction between the Nucleoporin Nup98 and the MRNA Export Factor Rae1. Proc. Natl. Acad. Sci. USA 2010, 107, 10406–10411. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, C.E.J.; Fornerod, M.; Kasper, L.H.; Van Deursen, J.M.A. RAE1 Is a Shuttling MRNA Export Factor That Binds to a GLEBS-like NUP98 Motif at the Nuclear Pore Complex through Multiple Domains. J. Cell Biol. 2000, 145, 237–253. [Google Scholar] [CrossRef]
- Barghorn, S.; Mandelkow, E. Toward a Unified Scheme for the Aggregation of Tau into Alzheimer Paired Helical Filaments. Biochemistry 2002, 41, 14885–14896. [Google Scholar] [CrossRef]
- Meraz-Ríos, M.A.; Lira-De León, K.I.; Campos-Peña, V.; De Anda-Hernández, M.A.; Mena-López, R. Tau Oligomers and Aggregation in Alzheimer’s Disease. J. Neurochem. 2010, 112, 1353–1367. [Google Scholar] [CrossRef]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau Protein Liquid–Liquid Phase Separation Can Initiate Tau Aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef]
- Schwefel, D.; Groom, H.C.T.; Boucherit, V.C.; Christodoulou, E.; Walker, P.A.; Stoye, J.P.; Bishop, K.N.; Taylor, I.A. Structural Basis of Lentiviral Subversion of a Cellular Protein Degradation Pathway. Nature 2014, 505, 234–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapinos, L.E.; Schoch, R.L.; Wagner, R.S.; Schleicher, K.D.; Lim, R.Y.H. Karyopherin-Centric Control of Nuclear Pores Based on Molecular Occupancy and Kinetic Analysis of Multivalent Binding with FG Nucleoporins. Biophys. J. 2014, 106, 1751–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shannessy, D.J.; Brigham-Burke, M.; Karl Soneson, K.; Hensley, P.; Brooks, I. Determination of Rate and Equilibrium Binding Constants for Macromolecular Interactions Using Surface Plasmon Resonance: Use of Nonlinear Least Squares Analysis Methods. Anal. Biochem. 1993, 212, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Diez, L.; Kapinos, L.E.; Lim, R.Y.H.; Wegmann, S. Analysis of Tau: Nucleoporin Interactions by Surface Plasmon Resonance Spectroscopy. Methods Mol. Biol. 2020, in press. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diez, L.; Kapinos, L.E.; Hochmair, J.; Huebschmann, S.; Dominguez-Baquero, A.; Vogt, A.; Rankovic, M.; Zweckstetter, M.; Lim, R.Y.H.; Wegmann, S. Phosphorylation but Not Oligomerization Drives the Accumulation of Tau with Nucleoporin Nup98. Int. J. Mol. Sci. 2022, 23, 3495. https://doi.org/10.3390/ijms23073495
Diez L, Kapinos LE, Hochmair J, Huebschmann S, Dominguez-Baquero A, Vogt A, Rankovic M, Zweckstetter M, Lim RYH, Wegmann S. Phosphorylation but Not Oligomerization Drives the Accumulation of Tau with Nucleoporin Nup98. International Journal of Molecular Sciences. 2022; 23(7):3495. https://doi.org/10.3390/ijms23073495
Chicago/Turabian StyleDiez, Lisa, Larisa E. Kapinos, Janine Hochmair, Sabrina Huebschmann, Alvaro Dominguez-Baquero, Amelie Vogt, Marija Rankovic, Markus Zweckstetter, Roderick Y. H. Lim, and Susanne Wegmann. 2022. "Phosphorylation but Not Oligomerization Drives the Accumulation of Tau with Nucleoporin Nup98" International Journal of Molecular Sciences 23, no. 7: 3495. https://doi.org/10.3390/ijms23073495
APA StyleDiez, L., Kapinos, L. E., Hochmair, J., Huebschmann, S., Dominguez-Baquero, A., Vogt, A., Rankovic, M., Zweckstetter, M., Lim, R. Y. H., & Wegmann, S. (2022). Phosphorylation but Not Oligomerization Drives the Accumulation of Tau with Nucleoporin Nup98. International Journal of Molecular Sciences, 23(7), 3495. https://doi.org/10.3390/ijms23073495