
CDDO-Me Attenuates Clasmatodendrosis in CA1 Astrocyte by Inhibiting HSP25-AKT Mediated DRP1-S637 Phosphorylation in Chronic Epilepsy Rats

Abstract
:1. Introduction
2. Results
2.1. CDDO-Me Attenuates HSP25-Mediated Autophagy in CA1 Astrocytes
2.2. CDDO-Me Reduces AKT S473 Phosphoprylation and Mitochondrial Length in CA1 Astrocytes
2.3. CDDO-Me Reduces DRP1 S637 Phosphorylation in CA1 Astrocytes without Affecting S616 Phosphorylation
2.4. HSP25 Knockdown Inhibits AKT S473 and DRP1 S637 Phosphorylations in CA1 Astrocytes
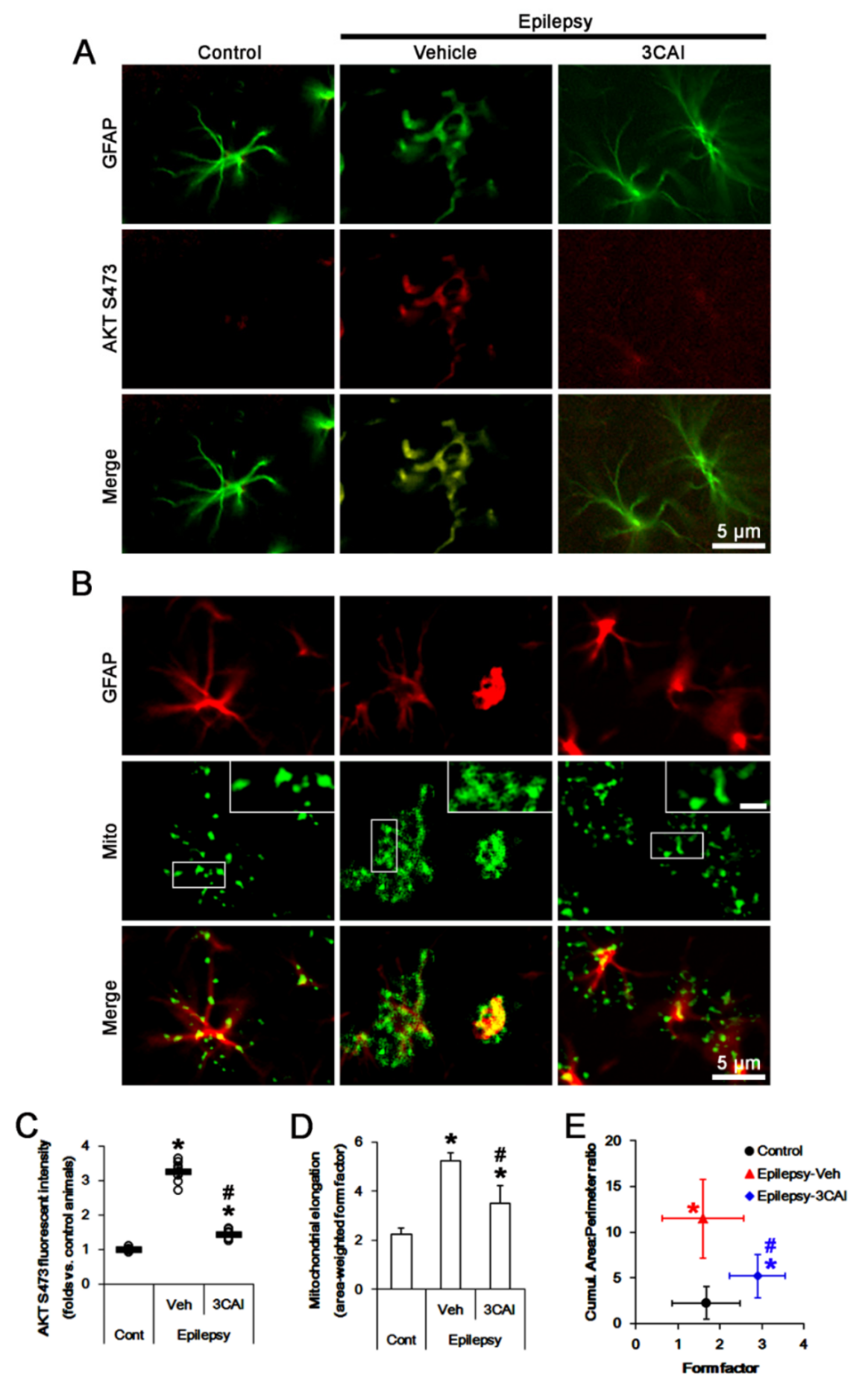
2.5. 3CAI Decreases AKT S473 and DRP1 S637 Phosphorylations in CA1 Astrocytes
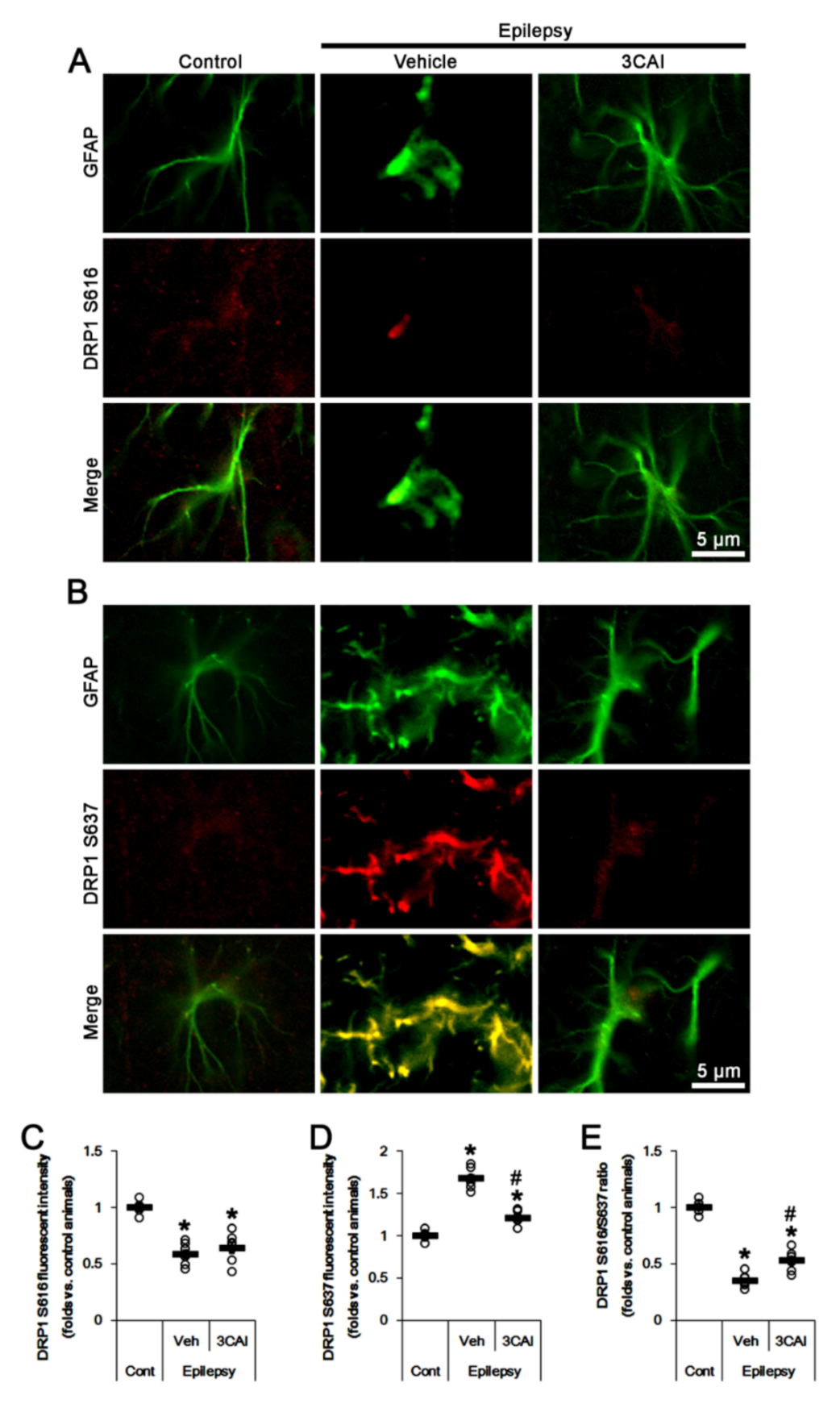
2.6. CDDO-Me and 3CAI Decrease DRP1 S637, but Not S616, Phosphorylations in CA1 Astrocytes
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Chemicals
4.2. Epilepsy Model
4.3. Surgery for CDDO-Me, HSP25 or 3CAI Infusion
4.4. Tissue Processing and Immunohistochemistry
4.5. Western Blots
4.6. Cell Count, Measurement of Fluorescent Intensity and Mitochondrial Morphometry
4.7. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3CAI | 3-chloroacetyl-indole |
| AMPAR | α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor |
| ARE | antioxidant-response element |
| ATP | adenosine triphosphate |
| Aβ | amyloid-β |
| BBB | blood brain barrier |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| CDDO-Me | 2-cyano-3,12-dioxo-oleana-1,9(11)-dien-28-oic acid methyl ester |
| CDK | cyclin dependent kinase |
| DRP1 | dynamin-related protein 1 |
| ER | endoplasmic reticulum |
| ERK1/2 | extracellular signal-related kinases 1/2 |
| Fis-1 | fission related protein-1 |
| GABA | γ-aminobutyric acid |
| GFAP | glial fibrillary acidic protein |
| HO-1 | heme oxygenase-1 |
| HSP25 | heat shock protein 25 |
| LAMP1 | lysosomal-associated membrane protein 1 |
| MFF | mitochondrial fission factor |
| MFN1 | mitofusin 1 |
| mTOR | mammalian target of rapamycin |
| Nrf2 | nuclear factor-erythroid 2-related factor 2 |
| OPA1 | optic atrophy 1 |
| PHLPP | pleckstrin homology domain and leucine-rich repeat protein phosphatase |
| PKA | protein kinase A |
| ROS | reactive oxygen species |
| SD | Sprague-Dawley |
| SP1 | specificity protein 1 |
| TLE | temporal lobe epilepsy |
| UPS | ubiquitin proteasome system. |
References
- Wittner, L.; Maglóczky, Z.; Borhegyi, Z.; Halász, P.; Tóth, S.; Erőss, L.; Szabó, Z.; Freund, T. Preservation of perisomatic inhibitory input of granule cells in the epileptic human dentate gyrus. Neuroscience 2001, 108, 587–600. [Google Scholar] [CrossRef]
- Malmgren, K.; Thom, M. Hippocampal sclerosis-Origins and imaging. Epilepsia 2012, 53, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Briellmann, R.S.; Kalnins, R.M.; Berkovic, S.F.; Jackson, G.D. Hippocampal pathology in refractory temporal lobe epilepsy: T2-weighted signal change reflects dentate gliosis. Neurology 2002, 58, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Vessal, M.; Dugani, C.B.; Solomon, D.A.; Burnham, W.M.; Ivy, G.O. Might astrocytes play a role in maintaining the seizure-prone state? Brain Res. 2005, 1044, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.-C.; Kim, D.-S.; Kwak, S.-E.; Kim, J.-E.; Won, M.H.; Kim, D.-W.; Choi, S.-Y.; Kwon, O.-S. Epileptogenic roles of astroglial death and regeneration in the dentate gyrus of experimental temporal lobe epilepsy. Glia 2006, 54, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Bedner, P.; Dupper, A.; Hüttmann, K.; Müller, J.; Herde, M.K.; Dublin, P.; Deshpande, T.; Schramm, J.; Häussler, U.; Haas, C.A.; et al. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 2015, 138, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.M.; Swanson, R.A. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia 2000, 32, 1–14. [Google Scholar] [CrossRef]
- Ransom, B.; Behar, T.; Nedergaard, M. New roles for astrocytes (stars at last). Trends Neurosci. 2003, 26, 520–522. [Google Scholar] [CrossRef]
- Sugawara, T.; Lewén, A.; Noshita, N.; Gasche, Y.; Chan, P.H. Effects of Global Ischemia Duration on Neuronal, Astroglial, Oligodendroglial, and Microglial Reactions in the Vulnerable Hippocampal CA1 Subregion in Rats. J. Neurotrauma 2002, 19, 85–98. [Google Scholar] [CrossRef]
- Revuelta, M.; Castaño, A.; Machado, A.; Cano, J.; Venero, J.L. Kainate-induced zinc translocation from presynaptic terminals causes neuronal and astroglial cell death and mRNA loss of BDNF receptors in the hippocampal formation and amygdala. J. Neurosci. Res. 2005, 82, 184–195. [Google Scholar] [CrossRef]
- Kim, D.-S.; Kim, J.-E.; Kwak, S.-E.; Choi, K.-C.; Kim, D.-W.; Kwon, O.-S.; Choi, S.-Y.; Kang, T.-C. Spatiotemporal characteristics of astroglial death in the rat hippocampo-entorhinal complex following pilocarpine-induced status epilepticus. J. Comp. Neurol. 2008, 511, 581–598. [Google Scholar] [CrossRef] [PubMed]
- Penfield, W. Neuroglia and microglia—The interstitial tissue of the central nervous system. In Special Cytology, the Form and Function of the Cell in Health and Disease; Cowdry, E.V., Ed.; Hoeber: New York, NY, USA, 1928; pp. 1033–1068. [Google Scholar]
- Ryu, H.; Kim, J.-E.; Yeo, S.-I.; Kim, D.-W.; Kwon, O.-S.; Choi, S.; Kang, T.-C. F-actin depolymerization accelerates clasmatodendrosis via activation of lysosome-derived autophagic astroglial death. Brain Res. Bull. 2011, 85, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.J.; Kim, J.E.; Yeo, S.I.; Kang, T.C. p65/RelA-Ser529 NF-κB subunit phosphorylation induces autophagic astroglial death (Clasmatodendrosis) following status epilepticus. Cell. Mol. Neurobiol. 2011, 31, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Fukuda, T.; Iwadate, K. Beading of the astrocytic processes (clasmatodendrosis) following head trauma is associated with protein degradation pathways. Brain Inj. 2013, 27, 1692–1697. [Google Scholar] [CrossRef]
- Bouchat, J.; Gilloteaux, J.; Suain, V.; Van Vlaender, D.; Brion, J.-P.; Nicaise, C. Ultrastructural Analysis of Thalamus Damages in a Mouse Model of Osmotic-Induced Demyelination. Neurotox. Res. 2019, 36, 144–162. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Kang, T.C. CDDO-Me attenuates astroglial autophagy via Nrf2-, ERK1/2-SP1- and Src-CK2-PTEN-PI3K/AKT-mediated signaling pathways in the hippocampus of chronic epilepsy rats. Antioxidants 2021, 10, 655. [Google Scholar] [CrossRef]
- Cuesta, R.; Laroia, G.; Schneider, R.J. Chaperone Hsp27 inhibits translation during heat shock by binding eIF4G and facilitating dissociation of cap-initiation complexes. Genes Dev. 2000, 14, 1460–1470. [Google Scholar] [CrossRef]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [Green Version]
- Jänen, S.B.; Chaachouay, H.; Richter-Landsberg, C. Autophagy is activated by proteasomal inhibition and involved in aggresome clearance in cultured astrocytes. Glia 2010, 58, 1766–1774. [Google Scholar] [CrossRef]
- Kim, J.E.; Ko, A.R.; Hyun, H.W.; Min, S.J.; Kang, T.C. P2RX7-MAPK1/2-SP1 axis inhibits MTOR independent HSPB1-mediated astroglial autophagy. Cell Death Dis. 2018, 9, 546. [Google Scholar] [CrossRef]
- Kim, J.-E.; Hyun, H.-W.; Min, S.-J.; Kang, T.-C. Sustained HSP25 Expression Induces Clasmatodendrosis via ER Stress in the Rat Hippocampus. Front. Cell. Neurosci. 2017, 11, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.S.; Kim, J.E. P2X7 receptor inhibits astroglial autophagy via regulating FAK- and PHLPP1/2-mediated AKT-S473 phosphorylation following kainic acid-induced seizures. Int. J. Mol. Sci. 2020, 21, 86476. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.-L.; Wang, S.-M.; Chen, C.-C.; Fong, T.-H.; Wu, M.-L. Mechanism of oxidative stress-induced intracellular acidosis in rat cerebellar astrocytes and C6 glioma cells. J. Physiol. 1997, 502, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Hulse, R.; Winterfield, J.; Kunkler, P.E.; Kraig, R.P. Astrocytic clasmatodendrosis in hippocampal organ culture. Glia 2001, 33, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Muranyi, M.; Ding, C.; He, Q.; Lin, Y.; Li, P.-A. Streptozotocin-Induced Diabetes Causes Astrocyte Death After Ischemia and Reperfusion Injury. Diabetes 2006, 55, 349–355. [Google Scholar] [CrossRef] [Green Version]
- Lewerenz, J.; Dargusch, R.; Maher, P. Lactacidosis modulates glutathione metabolism and oxidative glutamate toxicity. J. Neurochem. 2010, 113, 502–514. [Google Scholar] [CrossRef]
- Ko, A.-R.; Hyun, H.-W.; Min, S.-J.; Kim, J.-E. The Differential DRP1 Phosphorylation and Mitochondrial Dynamics in the Regional Specific Astroglial Death Induced by Status Epilepticus. Front. Cell. Neurosci. 2016, 10, 124. [Google Scholar] [CrossRef] [Green Version]
- Pandya, J.D.; Nukala, V.N.; Sullivan, P.G. Concentration dependent effect of calcium on brain mitochondrial bioenergetics and oxidative stress parameters. Front. Neuroenergetics 2013, 5, 10. [Google Scholar] [CrossRef]
- Lezi, E.; Swerdlow, R.H. Mitochondria in Neurodegeneration. Adv. Exp. Med. Biol. 2011, 942, 269–286. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.C. Mitochondria: Dynamic Organelles in Disease, Aging, and Development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Campello, S.; Scorrano, L. Mitochondrial shape changes: Orchestrating cell pathophysiology. EMBO Rep. 2010, 11, 678–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terman, A.; Gustafsson, B.; Brunk, U.T. Mitochondrial damage and intralysosomal degradation in cellular aging. Mol. Asp. Med. 2006, 27, 471–482. [Google Scholar] [CrossRef] [PubMed]
- DuBoff, B.; Götz, J.; Feany, M.B. Tau Promotes Neurodegeneration via DRP1 Mislocalization In Vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-E.; Ryu, H.J.; Kim, M.J.; Kang, T.-C. LIM kinase-2 induces programmed necrotic neuronal death via dysfunction of DRP1-mediated mitochondrial fission. Cell Death Differ. 2014, 21, 1036–1049. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, M.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira, C.R.; Rego, A.C. Insulin and IGF-1 improve mitochondrial function in a PI-3K/Akt-dependent manner and reduce mitochondrial generation of reactive oxygen species in Huntington’s disease knock-in striatal cells. Free Radic. Biol. Med. 2014, 74, 129–144. [Google Scholar] [CrossRef]
- Kang, T.-C. Nuclear Factor-Erythroid 2-Related Factor 2 (Nrf2) and Mitochondrial Dynamics/Mitophagy in Neurological Diseases. Antioxidants 2020, 9, 617. [Google Scholar] [CrossRef]
- Yang, Y.-L.; Li, J.; Liu, K.; Zhang, L.; Liu, Q.; Liu, B.; Qi, L.-W. Ginsenoside Rg5 increases cardiomyocyte resistance to ischemic injury through regulation of mitochondrial hexokinase-II and dynamin-related protein 1. Cell Death Dis. 2017, 8, e2625. [Google Scholar] [CrossRef] [Green Version]
- Tao, A.; Xu, X.; Kvietys, P.; Kao, R.; Martin, C.; Rui, T. Experimental diabetes mellitus exacerbates ischemia/reperfusion-induced myocardial injury by promoting mitochondrial fission: Role of down-regulation of myocardial Sirt1 and subsequent Akt/Drp1 interaction. Int. J. Biochem. Cell Biol. 2018, 105, 94–103. [Google Scholar] [CrossRef]
- Wu, B.; Luo, H.; Zhou, X.; Cheng, C.-Y.; Lin, L.; Liu, B.-L.; Liu, K.; Li, P.; Yang, H. Succinate-induced neuronal mitochondrial fission and hexokinase II malfunction in ischemic stroke: Therapeutical effects of kaempferol. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 2307–2318. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Yang, Y.-X.; Zhe, H.; He, Z.-X.; Zhou, S.-F. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Dev. Ther. 2014, 8, 2075–2088. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-E.; Lee, D.-S.; Park, H.; Kim, T.-H.; Kang, T.-C. Inhibition of AKT/GSK3β/CREB Pathway Improves the Responsiveness to AMPA Receptor Antagonists by Regulating GRIA1 Surface Expression in Chronic Epilepsy Rats. Biomedicines 2021, 9, 425. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, J.T.; Strack, S. Functional characterization of phosphorylation sites in dynamin-related protein 1. Methods Enzymol. 2009, 457, 231–253. [Google Scholar] [PubMed] [Green Version]
- Merrill, R.A.; Dagda, R.K.; Dickey, A.S.; Cribbs, J.T.; Green, S.H.; Usachev, Y.M.; Strack, S. Mechanism of Neuroprotective Mitochondrial Remodeling by PKA/AKAP1. PLoS Biol. 2011, 9, e1000612. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-E.; Park, H.; Kim, T.-H.; Kang, T.-C. LONP1 Regulates Mitochondrial Accumulations of HMGB1 and Caspase-3 in CA1 and PV Neurons Following Status Epilepticus. Int. J. Mol. Sci. 2021, 22, 2275. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12, 1528–1534. [Google Scholar] [CrossRef] [Green Version]
- Gozuacik, D.; Kimchi, A. Autophagy and cell death. Curr. Top. Dev. Biol. 2007, 78, 217–245. [Google Scholar]
- Tomimoto, H.; Akiguchi, I.; Wakita, H.; Suenaga, T.; Nakamura, S.; Kimura, J. Regressive changes of astroglia in white matter lesions in cerebrovascular disease and Alzheimer’s disease patients. Acta Neuropathol. 1997, 94, 146–152. [Google Scholar] [CrossRef]
- Sahlas, D.J.; Bilbao, J.M.; Swartz, R.H.; Black, S.E. Clasmatodendrosis correlating with periventricular hyperintensity in mixed dementia. Ann. Neurol. 2002, 52, 378–381. [Google Scholar] [CrossRef]
- Konishi, H.; Matsuzaki, H.; Tanaka, M.; Takemura, Y.; Kuroda, S.; Ono, Y.; Kikkawa, U. Activation of protein kinase B (Akt/RAC-protein kinase) by cellular stress and its association with heat shock protein Hsp27. FEBS Lett. 1997, 410, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Murashov, A.K.; Haq, I.; Hill, C.; Park, E.; Smith, M.; Wang, X.; Wang, X.; Goldberg, D.J.; Wolgemuth, D.J. Crosstalk between p38, Hsp25 and Akt in spinal motor neurons after sciatic nerve injury. Mol. Brain Res. 2001, 93, 199–208. [Google Scholar] [CrossRef]
- Bao, L.; Li, X.; Lin, Z. PTEN overexpression promotes glioblastoma death through triggering mitochondrial division and inactivating the Akt pathway. J. Recept. Signal Transduct. 2019, 39, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Park, H.; Choi, S.-H.; Kong, M.-J.; Kang, T.-C. CDDO-Me Selectively Attenuates CA1 Neuronal Death Induced by Status Epilepticus via Facilitating Mitochondrial Fission Independent of LONP1. Cells 2019, 8, 833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Park, H.; Kang, T.C. CDDO-Me distinctly regulates regional specific astroglial responses to status epilepticus via ERK1/2-Nrf2, PTEN-PI3K-AKT and NFκB signaling pathways. Antioxidants 2020, 9, 1026. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.P.; Huang, Y.L.; Chen, Y.J.; Juan, Y.H.; Hsu, C.L.; Nakahira, K.; Huang, Y.T.; Lin, M.W.; Wu, S.G.; Shih, J.Y.; et al. Multi-kinase framework promotes proliferation and invasion of lung adenocarcinoma through activation of dynamin-related protein 1. Mol. Oncol. 2021, 15, 560–578. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.I.; Lee, K.H.; Gabr, A.A.; Choi, G.E.; Kim, J.S.; Ko, S.H.; Han, H.J. Aβ-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim. Biophys. Acta 2016, 1863, 2820–2834. [Google Scholar] [CrossRef]
- Parone, P.A.; Da Cruz, S.; Tondera, D.; Mattenberger, Y.; James, D.I.; Maechler, P.; Barja, F.; Martinou, J.-C. Preventing Mitochondrial Fission Impairs Mitochondrial Function and Leads to Loss of Mitochondrial DNA. PLoS ONE 2008, 3, e3257. [Google Scholar] [CrossRef] [Green Version]
- Kageyama, Y.; Zhang, Z.; Roda, R.; Fukaya, M.; Wakabayashi, J.; Wakabayashi, N.; Kensler, T.W.; Reddy, P.H.; Iijima, M.; Sesaki, H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol. 2012, 197, 535–551. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.; Gibson, S.B. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J. Cell Sci. 2007, 120, 4155–4166. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-J.; Chen, T.-H.; Yang, L.-Y.; Shih, C.-M. Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Dis. 2014, 5, e1147. [Google Scholar] [CrossRef] [Green Version]
- Imai, T.; Takagi, T.; Kitashoji, A.; Yamauchi, K.; Shimazawa, M.; Hara, H. Nrf2 activator ameliorates hemorrhagic transformation in focal cerebral ischemia under warfarin anticoagulation. Neurobiol. Dis. 2016, 89, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Kitashoji, A.; Iwawaki, T.; Tsuruma, K.; Shimazawa, M.; Yoshimura, S.; Iwama, T.; Hara, H. Temporal activation of Nrf2 in the penumbra and Nrf2 activator-mediated neuroprotection in ischemia–reperfusion injury. Free Radic. Biol. Med. 2014, 72, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.-L.; Lin, C.-C.; Chen, Y.-W.; Hsiao, L.-D.; Yang, C.-M. CO Induces Nrf2-Dependent Heme Oxygenase-1 Transcription by Cooperating with Sp1 and c-Jun in Rat Brain Astrocytes. Mol. Neurobiol. 2015, 52, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Walz, W. Role of glial cells in the regulation of the brain ion microenvironment. Prog. Neurobiol. 1989, 33, 309–333. [Google Scholar] [CrossRef]
- Binder, D.K.; Yao, X.; Zador, Z.; Sick, T.J.; Verkman, A.S.; Manley, G.T. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia 2006, 53, 631–636. [Google Scholar] [CrossRef]
- D’Antuono, M.; Benini, R.; Biagini, G.; D’Arcangelo, G.; Barbarosie, M.; Tancredi, V.; Avoli, M. Limbic network interactions leading to hyperexcitability in a model of temporal lobe epilepsy. J. Neurophysiol. 2002, 87, 634–639. [Google Scholar] [CrossRef]
- Kim, J.-E.; Ryu, H.J.; Kim, M.-J.; Kim, D.-W.; Kwon, O.-S.; Choi, S.Y.; Kang, T.-C. Pyridoxal-5′-phosphate phosphatase/chronophin induces astroglial apoptosis via actin-depolymerizing factor/cofilin system in the rat brain following status epilepticus. Glia 2010, 58, 1937–1948. [Google Scholar] [CrossRef]
- Kim, J.E.; Park, H.; Lee, J.E.; Kang, T.C. Blockade of 67-kDa laminin receptor facilitates AQP4 down-regulation and BBB disruption via ERK1/2-and p38 MAPK-mediated PI3K/AKT activations. Cells 2020, 9, 1670. [Google Scholar] [CrossRef]
- Wang, X.Y.; Zhang, X.H.; Peng, L.; Liu, Z.; Yang, Y.X.; He, Z.X.; Dang, H.W.; Zhou, S.F. Bardoxolone methyl (CDDO-Me or RTA402) induces cell cycle arrest, apoptosis and autophagy via PI3K/Akt/mTOR and p38 MAPK/Erk1/2 signaling pathways in K562 cells. Am. J. Transl. Res. 2017, 9, 4652–4672. [Google Scholar]
- Kim, M.J.; Park, H.; Choi, S.H.; Kong, M.J.; Kim, J.E.; Kang, T.C. CDDO-Me attenuates vasogenic edema and astroglial death by regulating NF-κB p65 phosphorylations and Nrf2 expression following status epilepticus. Int. J. Mol. Sci. 2019, 20, 94862. [Google Scholar] [CrossRef] [Green Version]
- Borella, R.; Forti, L.; Gibellini, L.; De Gaetano, A.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Synthesis and anticancer activity of CDDO and CDDO-Me, two derivatives of natural triterpenoids. Molecules 2019, 24, 4097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeb, D.; Gao, X.; Jiang, H.; Dulchavsky, S.A.; Gautam, S.C. Oleanane Triterpenoid CDDO-Me inhibits growth and induces apoptosis in prostate cancer cells by independently targeting pro-survival Akt and mTOR. Prostate 2009, 69, 851–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-Y.; Zhe, H.; Zhao, R. Preclinical evidences toward the use of triterpenoid CDDO-Me for solid cancer prevention and treatment. Mol. Cancer 2014, 13, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volnova, A.; Tsytsarev, V.; Ganina, O.; Vélez-Crespo, G.E.; Alves, J.M.; Ignashchenkova, A.; Inyushin, M. The Anti-Epileptic Effects of Carbenoxolone In Vitro and In Vivo. Int. J. Mol. Sci. 2022, 23, 663. [Google Scholar] [CrossRef]
- Shu, Y.; Zhu, C.; Zeng, M.; Zhan, Q.; Hu, Z.; Wu, X. The protective effect of carbenoxolone on gap junction damage in the hippocampal CA1 area of a temporal lobe epilepsy rat model. Ann. Transl. Med. 2019, 7, 624. [Google Scholar] [CrossRef]
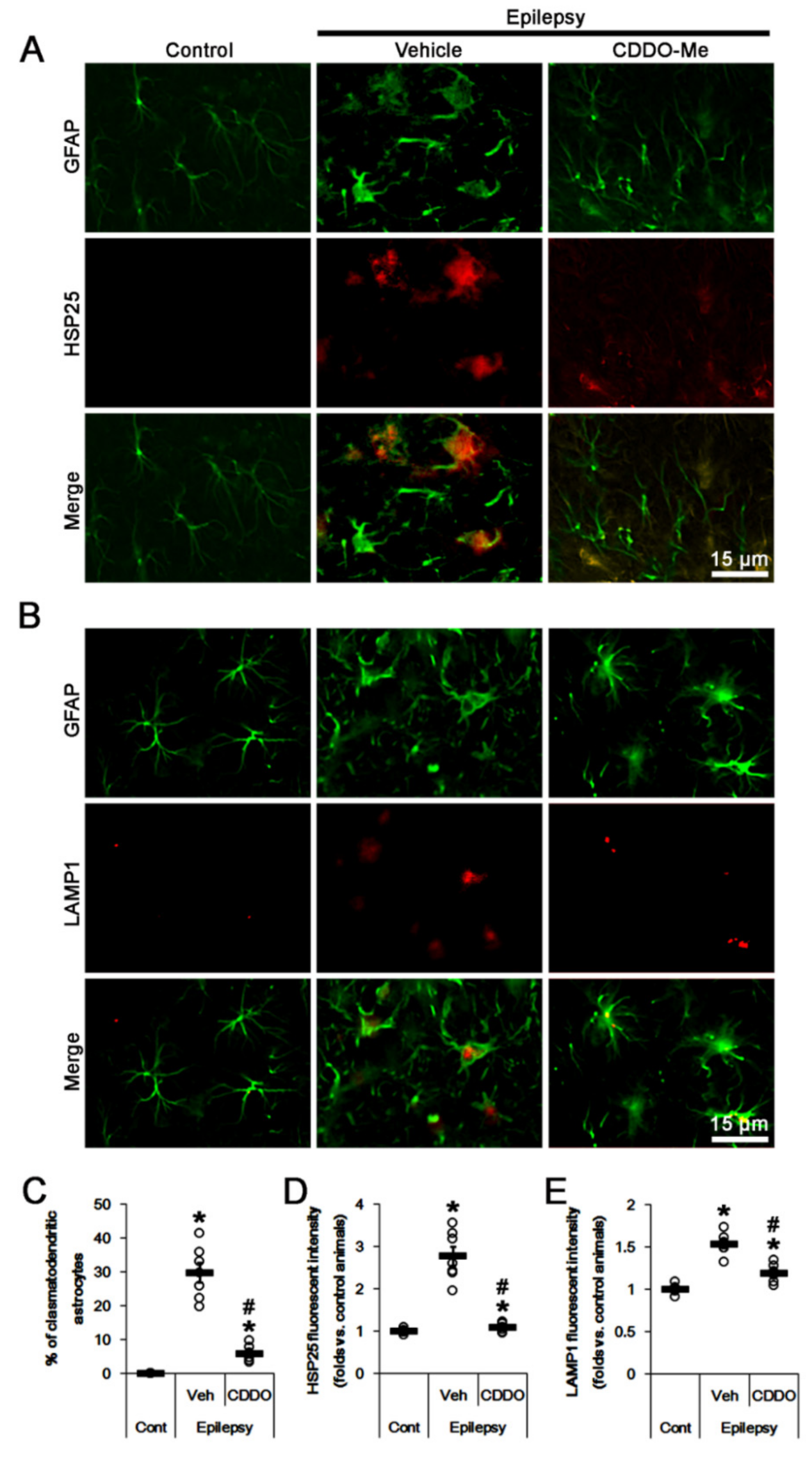
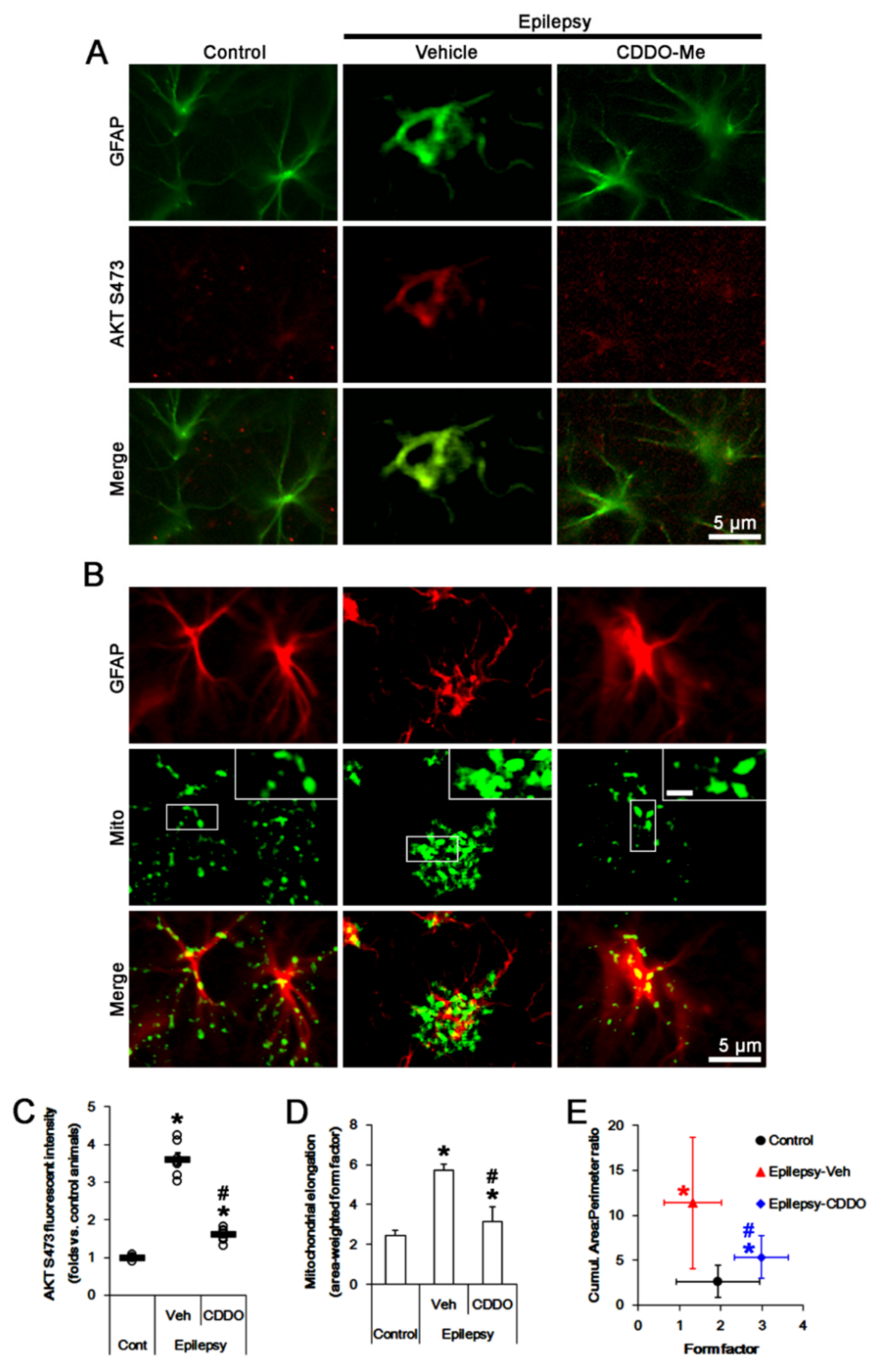

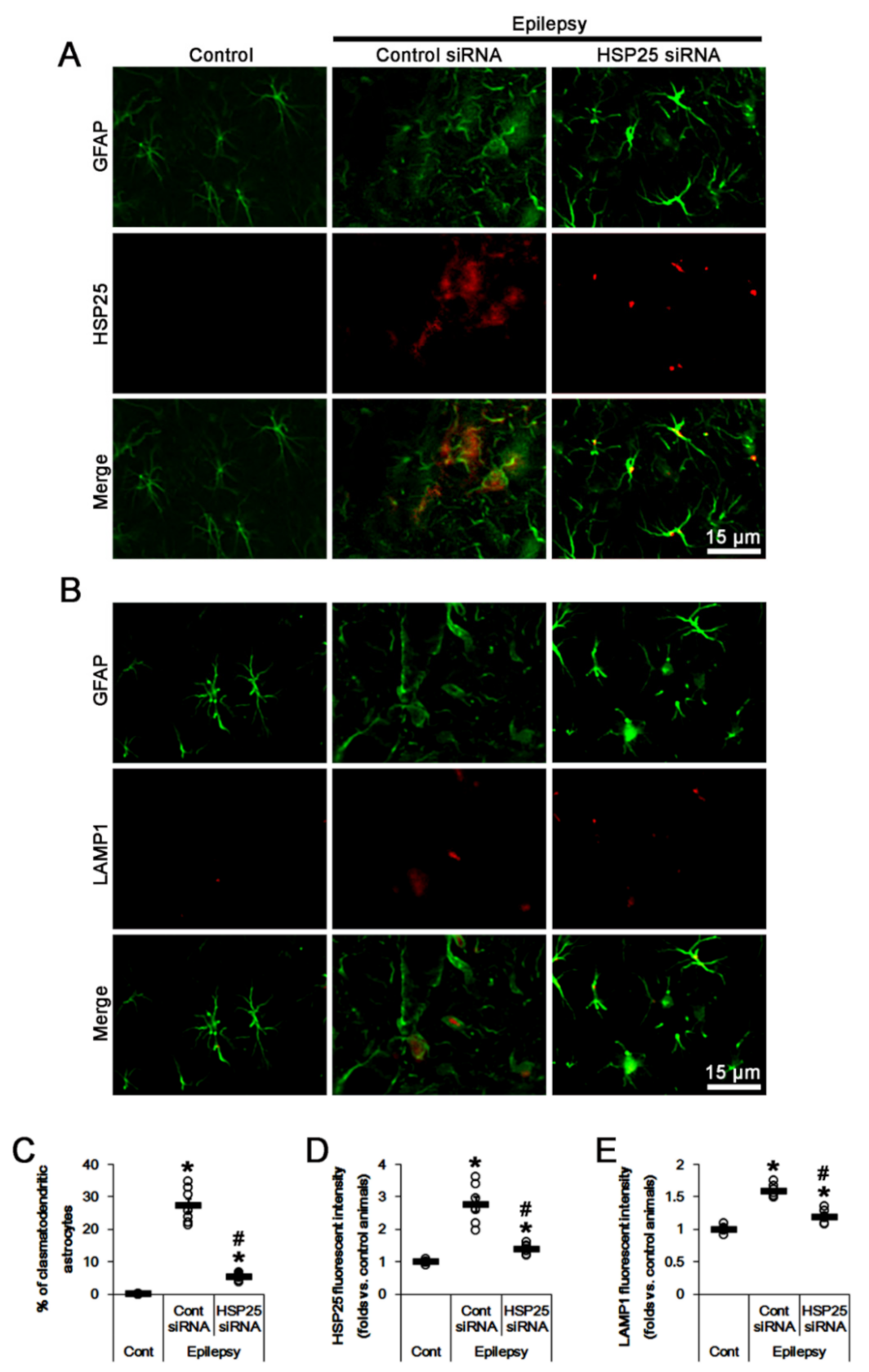

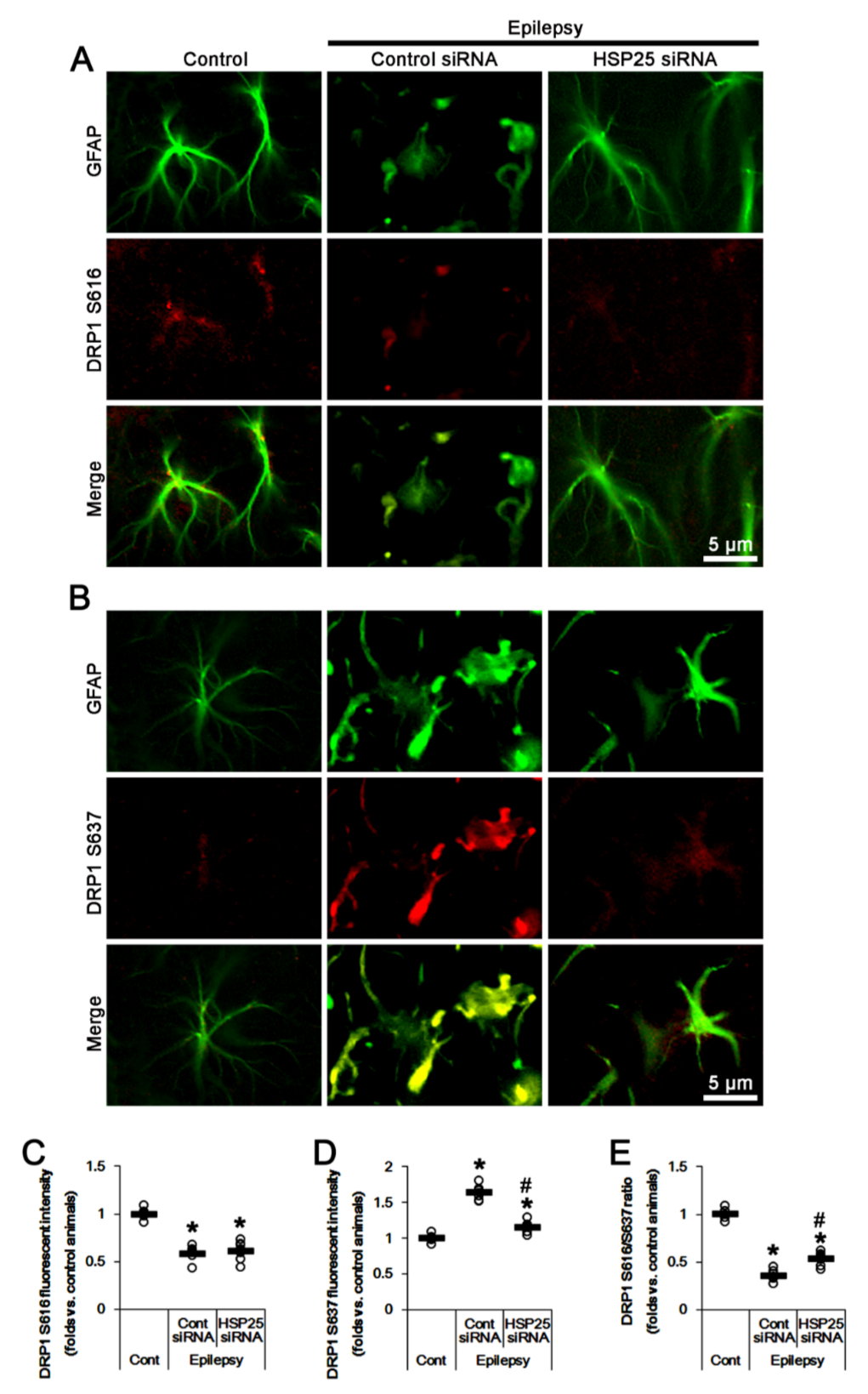



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Host | Manufacturer (Catalog Number) | Dilution Used |
|---|---|---|---|
| AKT | Rabbit | Cell signaling (#9272) | 1:1000 (WB) |
| AKT S473 | Rabbit | Cell Signaling (#4060) | 1:100 (IF) 1:1000 (WB) |
| DRP1 | Rabbit | Thermo (PA1-16987) | 1:1000 (WB) |
| DRP1 S616 | Rabbit | Cell Signaling (#4494) | 1:500 (IF) 1:1000 (WB) |
| DRP1 S637 | Rabbit | Cell Signaling (#4867) | 1:500 (IF) 1:1000 (WB) |
| GFAP | Rabbit Mouse | Abcam (#ab7260) Millipore (#MAB3402) | 1:500 (IF) 1:2000 (IF) |
| HSP25 | Rabbit | Enzo (#ADI-SPA-801) | 1:500 (IF) 1:1000 (WB) |
| LAMP1 | Rabbit | Lifespan (#LS-B580) | 1:200 (IF) |
| Mitochondrial marker (MTCO1) | Mouse | Abcam (#ab14705) | 1:500 (IF) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.-S.; Kim, T.-H.; Park, H.; Kim, J.-E. CDDO-Me Attenuates Clasmatodendrosis in CA1 Astrocyte by Inhibiting HSP25-AKT Mediated DRP1-S637 Phosphorylation in Chronic Epilepsy Rats. Int. J. Mol. Sci. 2022, 23, 4569. https://doi.org/10.3390/ijms23094569
Lee D-S, Kim T-H, Park H, Kim J-E. CDDO-Me Attenuates Clasmatodendrosis in CA1 Astrocyte by Inhibiting HSP25-AKT Mediated DRP1-S637 Phosphorylation in Chronic Epilepsy Rats. International Journal of Molecular Sciences. 2022; 23(9):4569. https://doi.org/10.3390/ijms23094569
Chicago/Turabian StyleLee, Duk-Shin, Tae-Hyun Kim, Hana Park, and Ji-Eun Kim. 2022. "CDDO-Me Attenuates Clasmatodendrosis in CA1 Astrocyte by Inhibiting HSP25-AKT Mediated DRP1-S637 Phosphorylation in Chronic Epilepsy Rats" International Journal of Molecular Sciences 23, no. 9: 4569. https://doi.org/10.3390/ijms23094569
APA StyleLee, D.-S., Kim, T.-H., Park, H., & Kim, J.-E. (2022). CDDO-Me Attenuates Clasmatodendrosis in CA1 Astrocyte by Inhibiting HSP25-AKT Mediated DRP1-S637 Phosphorylation in Chronic Epilepsy Rats. International Journal of Molecular Sciences, 23(9), 4569. https://doi.org/10.3390/ijms23094569