
PTPRD and DCC Are Novel BACE1 Substrates Differentially Expressed in Alzheimer’s Disease: A Data Mining and Bioinformatics Study

 , ,
, ,
Abstract
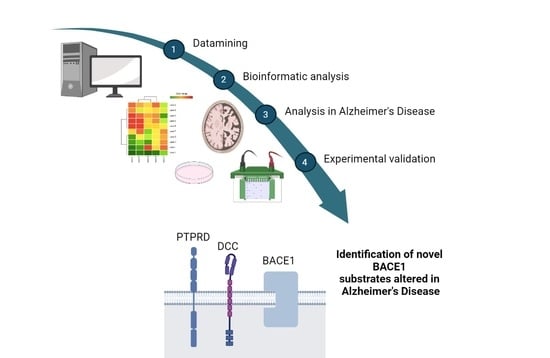
1. Introduction
1.1. The Role of BACE1 in Alzheimer’s Disease
1.2. Neuronal Roles and Substrates of BACE1
1.3. Non-Neuronal Roles and Substrates of BACE1
2. Results
2.1. BACE1-Regulated Proteins
2.2. BACE1 Substrates
2.3. BACE1 Substrates Altered in Alzheimer’s Disease
2.4. In Silico Modelling of Predicted Substrate Interactions
2.5. Experimental Validation of Novel Substrates
3. Discussion
3.1. Novel BACE1-Regulated Proteins and Potential Substrates
3.2. Pathways and Proteins Associated with BACE1 in AD
3.2.1. Receptor-Type Protein Tyrosine Phosphatases (PTPR)
3.2.2. Netrin Receptor DCC
3.2.3. Cell Adhesion
3.2.4. Ephrins and Ephrin Receptors
3.2.5. Axon Guidance
4. Materials and Methods
4.1. Computational Identification of BACE1 Substrates
4.2. Identification of Proteins Altered in Alzheimer’s Disease
4.3. Analysis of Protein Lists
4.4. In Silico Modelling of BACE1 Substrate Interactions
4.5. Mice
4.6. Primary Endothelial Cell Isolation and Protein Harvest
4.7. Cell Culture
4.8. Western Blotting
4.9. Quantitative RT-PCR
4.10. Statistical Tests
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vassar, R.; Kovacs, D.M.; Yan, R.; Wong, P.C. The β-Secretase Enzyme BACE in Health and Alzheimer’s Disease: Regulation, Cell Biology, Function, and Therapeutic Potential. J. Neurosci. 2009, 29, 12787–12794. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Di Fede, G.; Catania, M.; Morbin, M.; Rossi, G.; Suardi, S.; Mazzoleni, G.; Merlin, M.; Giovagnoli, A.R.; Prioni, S.; Erbetta, A.; et al. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 2009, 323, 1473–1477. [Google Scholar] [CrossRef]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef]
- Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat. Genet. 1992, 1, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, M.N.; Cummings, J. Open Peer Commentary to “Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE Trials as reported by Biogen December 2019”. Alzheimers Dement 2021, 17, 702–703. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Aisen, P.; Lemere, C.; Atri, A.; Sabbagh, M.; Salloway, S. Aducanumab produced a clinically meaningful benefit in association with amyloid lowering. Alzheimer’s Res. Ther. 2021, 13, 98. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Ou-Yang, M.H.; Kurz, J.E.; Nomura, T.; Popovic, J.; Rajapaksha, T.W.; Dong, H.; Contractor, A.; Chetkovich, D.M.; Tourtellotte, W.G.; Vassar, R. Axonal organization defects in the hippocampus of adult conditional BACE1 knockout mice. Sci. Transl. Med. 2018, 10, eaao5620. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Song, L.; Laird, F.; Wong, P.C.; Lee, H.K. BACE1 Knock-Outs Display Deficits in Activity-Dependent Potentiation of Synaptic Transmission at Mossy Fiber to CA3 Synapses in the Hippocampus. J. Neurosci. 2008, 28, 8677–8681. [Google Scholar] [CrossRef] [PubMed]
- Laird, F.M.; Cai, H.B.; Savonenko, A.V.; Farah, M.H.; He, K.W.; Melnikova, T.; Wen, H.J.; Chiang, H.C.; Xu, G.L.; Koliatsos, V.E.; et al. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J. Neurosci. 2005, 25, 11693–11709. [Google Scholar] [CrossRef]
- Egan, M.F.; Mukai, Y.; Voss, T.; Kost, J.; Stone, J.; Furtek, C.; Mahoney, E.; Cummings, J.L.; Tariot, P.N.; Aisen, P.S.; et al. Further analyses of the safety of verubecestat in the phase 3 EPOCH trial of mild-to-moderate Alzheimer’s disease. Alzheimers Res. 2019, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Singh, N.; Yao, A.Y.; Zhou, J.; He, W.; Hu, X.; Yan, R. BACE1 controls synaptic function through modulating release of synaptic vesicles. Mol. Psychiatry 2021, 26, 6394–6410. [Google Scholar] [CrossRef]
- Njavro, J.R.; Klotz, J.; Dislich, B.; Wanngren, J.; Shmueli, M.D.; Herber, J.; Kuhn, P.H.; Kumar, R.; Koeglsperger, T.; Conrad, M.; et al. Mouse brain proteomics establishes MDGA1 and CACHD1 as in vivo substrates of the Alzheimer protease BACE1. FASEB J. 2020, 34, 2465–2482. [Google Scholar] [CrossRef]
- Pigoni, M.; Wanngren, J.; Kuhn, P.H.; Munro, K.M.; Gunnersen, J.M.; Takeshima, H.; Feederle, R.; Voytyuk, I.; De Strooper, B.; Levasseur, M.D.; et al. Seizure protein 6 and its homolog seizure 6-like protein are physiological substrates of BACE1 in neurons. Mol. Neurodegener. 2016, 11, 67. [Google Scholar] [CrossRef]
- Lichtenthaler, S.F.; Dominguez, D.I.; Westmeyer, G.G.; Reiss, K.; Haass, C.; Saftig, P.; De Strooper, B.; Seed, B. The cell adhesion protein P-selectin glycoprotein ligand-1 is a substrate for the aspartyl protease BACE1. J. Biol. Chem. 2003, 278, 48713–48719. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.Q. Physiological Functions of the beta-Site Amyloid Precursor Protein Cleaving Enzyme 1 and 2. Front. Molec. Neurosci. 2017, 10, 12. [Google Scholar] [CrossRef]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The β-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef]
- Meakin, P.J.; Harper, A.J.; Hamilton, D.L.; Gallagher, J.; McNeilly, A.D.; Burgess, L.A.; Vaanholt, L.M.; Bannon, K.A.; Latcham, J.; Hussain, I.; et al. Reduction in BACE1 decreases body weight, protects against diet-induced obesity and enhances insulin sensitivity in mice. Biochem. J. 2012, 441, 285–296. [Google Scholar] [CrossRef]
- Meakin, P.J.; Coull, B.M.; Tuharska, Z.; McCaffery, C.; Akoumianakis, I.; Antoniades, C.; Brown, J.; Griffin, K.J.; Platt, F.; Ozber, C.H.; et al. Elevated circulating amyloid concentrations in obesity and diabetes promote vascular dysfunction. J. Clin. Investig. 2020, 130, 4104–4117. [Google Scholar] [CrossRef] [PubMed]
- Meakin, P.J.; Mezzapesa, A.; Benabou, E.; Haas, M.E.; Bonardo, B.; Grino, M.; Brunel, J.M.; Desbois-Mouthon, C.; Biddinger, S.B.; Govers, R.; et al. The beta secretase BACE1 regulates the expression of insulin receptor in the liver. Nat. Commun. 2018, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Plucinska, K.; Dekeryte, R.; Koss, D.; Shearer, K.; Mody, N.; Whitfield, P.D.; Doherty, M.K.; Mingarelli, M.; Welch, A.; Riedel, G.; et al. Neuronal human BACE1 knockin induces systemic diabetes in mice. Diabetologia 2016, 59, 1513–1523. [Google Scholar] [CrossRef]
- Grinberg, L.T.; Korczyn, A.D.; Heinsen, H. Cerebral amyloid angiopathy impact on endothelium. Exp. Gerontol. 2012, 47, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N. Vascular factors in Alzheimer’s disease. Int. Psychogeriatr. 2003, 15, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.H.; Marjaux, E.; Imhof, A.; De Strooper, B.; Haass, C.; Lichtenthaler, S.F. Regulated intramembrane proteolysis of the interleukin-1 receptor II by alpha-, beta-, and gamma-secretase. J. Biol. Chem. 2007, 282, 11982–11995. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.L.; Vassar, R. Linking vascular disorders and Alzheimer’s disease: Potential involvement of BACE1. Neurobiol Aging 2009, 30, 1535–1544. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Devraj, K.; Poznanovic, S.; Spahn, C.; Schwall, G.; Harter, P.N.; Mittelbronn, M.; Antoniello, K.; Paganetti, P.; Muhs, A.; Heilemann, M.; et al. BACE-1 is expressed in the blood-brain barrier endothelium and is upregulated in a murine model of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2016, 36, 1281–1294. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef]
- Farkas, E.; Luiten, P.G.M. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog. Neurobiol. 2001, 64, 575–611. [Google Scholar] [CrossRef]
- Greenberg, S.M. Cerebral amyloid angiopathy and vessel dysfunction. Cereb. Dis. 2002, 13 (Suppl. 2), 42–47. [Google Scholar] [CrossRef] [PubMed]
- Meakin, P.J.; Jalicy, S.M.; Montagut, G.; Allsop, D.J.P.; Cavellini, D.L.; Irvine, S.W.; McGinley, C.; Liddell, M.K.; McNeilly, A.D.; Parmionova, K.; et al. Bace1-dependent amyloid processing regulates hypothalamic leptin sensitivity in obese mice. Sci. Rep. 2018, 8, 55. [Google Scholar] [CrossRef]
- Marwarha, G.; Raza, S.; Meiers, C.; Ghribi, O. Leptin attenuates BACE1 expression and amyloid-β genesis via the activation of SIRT1 signaling pathway. Biochim. Biophys. Acta 2014, 1842, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.H.; Koroniak, K.; Hogl, S.; Colombo, A.; Zeitschel, U.; Willem, M.; Volbracht, C.; Schepers, U.; Imhof, A.; Hoffmeister, A.; et al. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012, 31, 3157–3168. [Google Scholar] [CrossRef]
- Stutzer, I.; Selevsek, N.; Esterhazy, D.; Schmidt, A.; Aebersold, R.; Stoffel, M. Systematic Proteomic Analysis Identifies beta-Site Amyloid Precursor Protein Cleaving Enzyme 2 and 1 (BACE2 and BACE1) Substrates in Pancreatic beta-Cells. J. Biol. Chem. 2013, 288, 10536–10547. [Google Scholar] [CrossRef]
- Dislich, B.; Wohlrab, F.; Bachhuber, T.; Muller, S.A.; Kuhn, P.H.; Hogl, S.; Meyer-Luehmann, M.; Lichtenthaler, S.F. Label-free Quantitative Proteomics of Mouse Cerebrospinal Fluid Detects beta-Site APP Cleaving Enzyme (BACE1) Protease Substrates In Vivo. Mol. Cell. Proteom. 2015, 14, 2550–2563. [Google Scholar] [CrossRef] [PubMed]
- Hemming, M.L.; Elias, J.E.; Gygi, S.P.; Selkoe, D.J. Identification of beta-Secretase (BACE1) Substrates Using Quantitative Proteomics. PLoS ONE 2009, 4, e8477. [Google Scholar] [CrossRef]
- Johnson, J.L.; Chambers, E.; Jayasundera, K. Application of a Bioinformatics-Based Approach to Identify Novel Putative in vivo BACE1 Substrates. Biomed. Eng. Comput. Biol. 2013, 5, 1–15. [Google Scholar] [CrossRef]
- Lau, S.F.; Cao, H.; Fu, A.K.Y.; Ip, N.Y. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 25800–25809. [Google Scholar] [CrossRef]
- Honorato, R.V.; Koukos, P.I.; Jiménez-García, B.; Tsaregorodtsev, A.; Verlato, M.; Giachetti, A.; Rosato, A.; Bonvin, A.M. Structural Biology in the Clouds: The WeNMR-EOSC Ecosystem. Front. Mol. Biosci. 2021, 8, 729513. [Google Scholar] [CrossRef]
- van Zundert, G.C.P.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; De Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- Kim, W.; Watanabe, H.; Lomoio, S.; Tesco, G. Spatiotemporal processing of neural cell adhesion molecules 1 and 2 by BACE1 in vivo. J. Biol. Chem. 2021, 296, 100372. [Google Scholar] [CrossRef]
- Turner, R.T.; Koelsch, G.; Hong, L.; Castenheira, P.; Ghosh, A.; Tang, J. Subsite specificity of memapsin 2 (beta-secretase): Implications for inhibitor design. Biochemistry 2001, 40, 10001–10006. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.J.; Barao, S.; Laga, M.; Bockstael, K.; Borgers, M.; Gijsen, H.; Annaert, W.; Moechars, D.; Mercken, M.; Gevaer, K.; et al. The Neural Cell Adhesion Molecules L1 and CHL1 Are Cleaved by BACE1 Protease In Vivo. J. Biol. Chem. 2012, 287, 25927–25940. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, D.; Lombino, F.; Liu, X.R.; D’Adamio, L. APP Is Cleaved by Bace1 in Pre-Synaptic Vesicles and Establishes a Pre-Synaptic Interactome, via Its Intracellular Domain, with Molecular Complexes that Regulate Pre-Synaptic Vesicles Functions. PLoS ONE 2014, 9, e108576. [Google Scholar] [CrossRef]
- Zou, L.; Wang, Z.; Shen, L.; Bao, G.B.; Wang, T.; Kang, J.H.; Pei, G. Receptor tyrosine kinases positively regulate BACE activity and Amyloid-beta production through enhancing BACE internalization. Cell Res. 2007, 17, 389–401. [Google Scholar] [CrossRef]
- Munji, R.N.; Soung, A.L.; Weiner, G.A.; Sohet, F.; Semple, B.D.; Trivedi, A.; Gimlin, K.; Kotoda, M.; Korai, M.; Aydin, S.; et al. Profiling the mouse brain endothelial transcriptome in health and disease models reveals a core blood-brain barrier dysfunction module. Nat. Neurosci. 2019, 22, 1892–1902. [Google Scholar] [CrossRef]
- Luo, X.; Prior, M.; He, W.; Hu, X.; Tang, X.; Shen, W.; Yadav, S.; Kiryu-Seo, S.; Miller, R.; Trapp, B.D.; et al. Cleavage of neuregulin-1 by BACE1 or ADAM10 protein produces differential effects on myelination. J. Biol. Chem. 2011, 286, 23967–23974. [Google Scholar] [CrossRef] [PubMed]
- Zabolotny, J.M.; Bence-Hanulec, K.K.; Stricker-Krongrad, A.; Haj, F.; Wang, Y.; Minokoshi, Y.; Kim, Y.B.; Elmquist, J.K.; Tartaglia, L.A.; Kahn, B.B.; et al. PTP1B regulates leptin signal transduction in vivo. Dev. Cell 2002, 2, 489–495. [Google Scholar] [CrossRef]
- Pulido, R.; Serra-Pagès, C.; Tang, M.; Streuli, M. The LAR/PTP delta/PTP sigma subfamily of transmembrane protein-tyrosine-phosphatases: Multiple human LAR, PTP delta, and PTP sigma isoforms are expressed in a tissue-specific manner and associate with the LAR-interacting protein LIP.1. Proc. Natl. Acad. Sci. USA 1995, 92, 11686–11690. [Google Scholar] [CrossRef]
- Uhl, G.R.; Martinez, M.J. PTPRD: Neurobiology, genetics, and initial pharmacology of a pleiotropic contributor to brain phenotypes. Ann. N. Y. Acad. Sci. 2019, 1451, 112–129. [Google Scholar] [CrossRef] [PubMed]
- Chibnik, L.B.; White, C.C.; Mukherjee, S.; Raj, T.; Yu, L.; Larson, E.B.; Montine, T.J.; Keene, C.D.; Sonnen, J.; Schneider, J.A.; et al. Susceptibility to neurofibrillary tangles: Role of the PTPRD locus and limited pleiotropy with other neuropathologies. Mol. Psychiatry 2018, 23, 1521–1529. [Google Scholar] [CrossRef]
- Tomita, H.; Cornejo, F.; Aranda-Pino, B.; Woodard, C.L.; Rioseco, C.C.; Neel, B.G.; Alvarez, A.R.; Kaplan, D.R.; Miller, F.D.; Cancino, G.I. The Protein Tyrosine Phosphatase Receptor Delta Regulates Developmental Neurogenesis. Cell Rep. 2020, 30, 215–228.e215. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Tian, M.; Zhang, X.; Xu, J.; Yang, B.; Yu, J.; Li, F.; Li, Y.; Li, S.; Li, X. Role of the JAK2/STAT3 signaling pathway in the pathogenesis of type 2 diabetes mellitus with macrovascular complications. Oncotarget 2017, 8, 96958–96969. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhang, F.; Mao, J.; Lu, Y.; Li, J.; Ma, W.; Fan, S.; Zhang, C.; Li, Q.; Wang, B.; et al. Protein tyrosine phosphatase receptor-type δ acts as a negative regulator suppressing breast cancer. Oncotarget 2017, 8, 98798–98811. [Google Scholar] [CrossRef]
- Nguyen, A.; Cai, H. Netrin-1 induces angiogenesis via a DCC-dependent ERK1/2-eNOS feed-forward mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 6530–6535. [Google Scholar] [CrossRef] [PubMed]
- Böhmer, F.D.; Friedrich, K. Protein tyrosine phosphatases as wardens of STAT signaling. Jakstat 2014, 3, e28087. [Google Scholar] [CrossRef]
- Chiba, T.; Yamada, M.; Sasabe, J.; Terashita, K.; Shimoda, M.; Matsuoka, M.; Aiso, S. Amyloid-beta causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons. Mol. Psychiatry 2009, 14, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Serafini, T.; Colamarino, S.A.; Leonardo, E.D.; Wang, H.; Beddington, R.; Skarnes, W.C.; Tessier-Lavigne, M. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell 1996, 87, 1001–1014. [Google Scholar] [CrossRef]
- Lourenço, F.C.; Galvan, V.; Fombonne, J.; Corset, V.; Llambi, F.; Müller, U.; Bredesen, D.E.; Mehlen, P. Netrin-1 interacts with amyloid precursor protein and regulates amyloid-beta production. Cell Death Differ. 2009, 16, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.; Wennström. Cell adhesion molecules in Alzheimer’s disease. Degener. Neurol. Neuromuscul. Dis. 2012, 2, 65–77. [Google Scholar] [CrossRef]
- Nagara, Y.; Hagiyama, M.; Hatano, N.; Futai, E.; Suo, S.; Takaoka, Y.; Murakami, Y.; Ito, A.; Ishiura, S. Tumor suppressor cell adhesion molecule 1 (CADM1) is cleaved by a disintegrin and metalloprotease 10 (ADAM10) and subsequently cleaved by γ-secretase complex. Biochem. Biophys. Res. Commun. 2012, 417, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Strekalova, H.; Buhmann, C.; Kleene, R.; Eggers, C.; Saffell, J.; Hemperly, J.; Weiller, C.; Müller-Thomsen, T.; Schachner, M. Elevated levels of neural recognition molecule L1 in the cerebrospinal fluid of patients with Alzheimer disease and other dementia syndromes. Neurobiol. Aging 2006, 27, 1–9. [Google Scholar] [CrossRef] [PubMed]
- van Abel, D.; Michel, O.; Veerhuis, R.; Jacobs, M.; van Dijk, M.; Oudejans, C.B. Direct downregulation of CNTNAP2 by STOX1A is associated with Alzheimer’s disease. J. Alzheimers Dis. 2012, 31, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Martin-de-Saavedra, M.D.; Santos, M.; Varea, O.; Spielman, B.; Gao, R.; Forrest, M.; Myczek, K.; Khalatyan, N.; Hall, E.; Sanz-Clemente, A.; et al. CNTNAP2 Ectodomain, Detected in Neuronal and CSF Sheddomes, Modulates Ca2+ Dynamics and Network Synchrony. bioRxiv 2019, 605378. [Google Scholar] [CrossRef]
- Wang, X.; Wang, C.; Pei, G. α-secretase ADAM10 physically interacts with β-secretase BACE1 in neurons and regulates CHL1 proteolysis. J. Mol. Cell. Biol. 2018, 10, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Hitt, B.; Riordan, S.M.; Kukreja, L.; Eimer, W.A.; Rajapaksha, T.W.; Vassar, R. beta-Site Amyloid Precursor Protein (APP)-cleaving Enzyme 1 (BACE1)-deficient Mice Exhibit a Close Homolog of L1 (CHL1) Loss-of-function Phenotype Involving Axon Guidance Defects. J. Biol. Chem. 2012, 287, 38408–38425. [Google Scholar] [CrossRef]
- Bot, N.; Schweizer, C.; Ben Halima, S.; Fraering, P.C. Processing of the synaptic cell adhesion molecule neurexin-3beta by Alzheimer disease alpha- and gamma-secretases. J. Biol. Chem. 2011, 286, 2762–2773. [Google Scholar] [CrossRef]
- Kania, A.; Klein, R. Mechanisms of ephrin–Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell. Biol. 2016, 17, 240–256. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.; Zou, Y.; Poo, M.; Tessier-Lavigne, M. Binding of DCC by netrin-1 to mediate axon guidance independent of adenosine A2B receptor activation. Science 2001, 291, 1976–1982. [Google Scholar] [CrossRef]
- Sabatier, C.; Plump, A.S.; Le, M.; Brose, K.; Tamada, A.; Murakami, F.; Lee, E.Y.; Tessier-Lavigne, M. The divergent Robo family protein rig-1/Robo3 is a negative regulator of slit responsiveness required for midline crossing by commissural axons. Cell 2004, 117, 157–169. [Google Scholar] [CrossRef]
- Borgius, L.; Nishimaru, H.; Caldeira, V.; Kunugise, Y.; Löw, P.; Reig, R.; Itohara, S.; Iwasato, T.; Kiehn, O. Spinal glutamatergic neurons defined by EphA4 signaling are essential components of normal locomotor circuits. J. Neurosci. 2014, 34, 3841–3853. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids. Res. 2021, 50, D439–D444. [Google Scholar] [CrossRef]
- Patel, S.; Vuillard, L.; Cleasby, A.; Murray, C.W.; Yon, J. Apo and inhibitor complex structures of BACE (beta-secretase). J. Mol. Biol. 2004, 343, 407–416. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell. Rep. Methods 2021, 1, 100014. [Google Scholar] [CrossRef] [PubMed]
- Assmann, J.C.; Müller, K.; Wenzel, J.; Walther, T.; Brands, J.; Thornton, P.; Allan, S.M.; Schwaninger, M. Isolation and Cultivation of Primary Brain Endothelial Cells from Adult Mice. Bio. Protoc. 2017, 7, e2294. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACE | CNTNAP4 | LINGO2 | NLGN4X | PCDHGC3 | SDK2 |
| ACP2 | CRIM1 | LMAN2 | NRCAM | PLXNB1 | SEMA4A |
| ADAM22 | CSF1R | LPL | NRP2 | PLXNB2 | SEMA4B |
| ALCAM | DCC | LRFN4 | NRXN3 | PODXL2 | SEMA4C |
| ALG2 | DNER | LRIG1 | NTM | PTPRD | SEMA6A |
| APLP2 | DPEP2 | LRIG2 | NTRK2 | PTPRK | SEMA7A |
| APP | DSG2 | LRIG3 | OPCML | PTPRM | SEZ6 |
| ATP6AP1 | EGFR | LRP11 | PCDH1 | PTPRN | SEZ6L |
| ATP6AP2 | EMB | LRP4 | PCDH10 | PTPRS | SLITRK1 |
| CACHD1 | EPHA2 | LRRN1 | PCDH17 | PTPRT | SORCS1 |
| CADM1 | EPHA4 | LYVE1 | PCDH19 | PTPRU | SORCS3 |
| CADM4 | EPHA7 | MBTPS1 | PCDH20 | PVR | SORL1 |
| CDH8 | GLG1 | MET | PCDH7 | QSOX2 | SSR1 |
| CEACAM1 | GPC3 | MMP17 | PCDH8 | RGMA | TLR9 |
| CHL1 | GPC4 | MPZL1 | PCDH9 | RGMB | TMEM132A |
| CLSTN2 | IL6ST | NCAM1 | PCDHGA11 | ROBO1 | TMEM132B |
| CLSTN3 | L1CAM | NCSTN | PCDHGA12 | ROBO2 | TMEM132E |
| CNST | LAMP1 | NEO1 | PCDHGA4 | RTN4RL1 | TMX3 |
| CNTNAP1 | LDLR | NFASC | PCDHGA5 | SDC4 | VCAM1 |
| CNTNAP2 | LIFR | NLGN1 | PCDHGA8 | SDK1 | VLDLR |
| Protein Name | Gene Symbol |
|---|---|
| Activated Leukocyte Cell Adhesion Molecule | ALCAM |
| Amyloid Beta Precursor Like Protein 2 | APLP2 |
| VWFA and cache domain-containing protein 1 | CACHD1 |
| Cell Adhesion Molecule 1 | CADM1 |
| Contactin Associated Protein 2 | CNTNAP2 |
| Contactin Associated Protein Family Member 4 | CNTNAP4 |
| Cysteine Rich Transmembrane BMP Regulator 1 | CRIM1 |
| Netrin receptor DCC | DCC |
| Delta and Notch-like epidermal growth factor-related receptor | DNER |
| Ephrin type-A receptor 4 | EPHA4 |
| Low-density lipoprotein receptor | LDLR |
| LIF Receptor Subunit Alpha | LIFR |
| Lymphatic Vessel Endothelial Hyaluronan Receptor 1 | LYVE1 |
| Neurexin-3 | NRXN3 |
| Neurotrimin | NTM |
| Neurotrophic Receptor Tyrosine Kinase 2 | NTRK2 |
| Protocadherin-10 | PCDH10 |
| Plexin B1 | PLXNB1 |
| Protein Tyrosine Phosphatase Receptor Type D | PTPRD |
| Protein Tyrosine Phosphatase Receptor Type M | PTPRM |
| Roundabout Guidance Receptor 1 | ROBO1 |
| Roundabout Guidance Receptor 2 | ROBO2 |
| Syndecan 4 | SDC4 |
| Sidekick Cell Adhesion Molecule 1 | SDK1 |
| Sortilin Related Receptor 1 | SORL1 |
| Reference | Study Type | Experimental Conditions | How Data Was Used | Statistical Analysis by Authors |
|---|---|---|---|---|
| [11] | SILAC proteomics | Brain membrane fraction from WT and BACE1 KO mice | Identify BACE1-regulated proteins | Log2 ratios of technical replicates were averaged and average protein log2 fold changes were calculated between BACE1 KO and WT samples. A two-sided Student’s t test evaluated the significance of proteins. Permutation-based FDR estimation was used. |
| [30] | SPECS proteomics | Primary neurons from BACE1 inhibitor treated, WT, and BACE1 KO mice | Identify BACE1-regulated proteins | A variance score (VS = absolute value of (standard error of the mean/(1 − mean))) was calculated for all proteins. Proteins with a vs. of ≤0.35 were considered as proteins with a consistent change upon BACE1 inhibition. |
| [31] | Loss/Gain-of-Function assays | MIN6 with knockdown of BACE1 and BACE2 | Identify BACE1-regulated proteins | Protein significance analysis was performed using SRMstats where a constant normalisation was performed for all runs to equalise the median peak intensities of the heavy transitions from all the peptides between runs. |
| [32] | Quantitative proteomics | CSF from WT and BACE1 −/− mice | Identify BACE1-regulated proteins | Using the mean, the average LFQ intensity within each biological group was calculated. A two-sided t test was used, and the p value was corrected using false discovery rate (FDR)-based multiple hypothesis testing. |
| [33] | SILAC proteomics | HeLa/HEK with BACE1 overexpression | Identify BACE1-regulated proteins | Proteins containing peptides with at least 65% of the total signal derived from the BACE1 condition were considered as putative substrates. This threshold value is equivalent to a 1.857-fold increase in peptide abundance. |
| [34] | BACE1 substrate prediction | Bioinformatic analysis | Identify potential BACE1 substrates | |
| [35] | Single nucleus RNA sequencing | Single nucleus prefrontal cortical samples of AD patients and normal control (NC) subjects | Used to identify genes differentially expressed in Alzheimer’s disease | Data were background corrected and quantile normalised. Differential expression was performed via limma using a block design to leverage technical replicates. Genes with a false discovery rate (FDR)-adjusted p < 0.05 were considered differentially expressed. An algorithm based on the hypergeometric distribution is used to calculate enrichment p values. |
| [36] | RNA sequencing | Endothelial and brain cells from mouse models of stroke, multiple sclerosis (EAE), traumatic brain injury (TBI), and epilepsy | Identify genes differentially expressed in endothelial cells, dependent on disease | Stratified samples according to classification of AD and NC samples and compared the transcriptome profiles of individual cell types between AD and NC samples by the Wilcoxon rank-sum test using the FindMarkers function with the parameters logfc.threshold = 0 and test.use = wilcox. The level of statistical significance for cell-type-specific transcriptomic changes was set at an adjusted p < 0.1 and a log2 fold change ≥0.1 or ≤−0.1. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor, H.A.; Simmons, K.J.; Clavane, E.M.; Trevelyan, C.J.; Brown, J.M.; Przemyłska, L.; Watt, N.T.; Matthews, L.C.; Meakin, P.J. PTPRD and DCC Are Novel BACE1 Substrates Differentially Expressed in Alzheimer’s Disease: A Data Mining and Bioinformatics Study. Int. J. Mol. Sci. 2022, 23, 4568. https://doi.org/10.3390/ijms23094568
Taylor HA, Simmons KJ, Clavane EM, Trevelyan CJ, Brown JM, Przemyłska L, Watt NT, Matthews LC, Meakin PJ. PTPRD and DCC Are Novel BACE1 Substrates Differentially Expressed in Alzheimer’s Disease: A Data Mining and Bioinformatics Study. International Journal of Molecular Sciences. 2022; 23(9):4568. https://doi.org/10.3390/ijms23094568
Chicago/Turabian StyleTaylor, Hannah A., Katie J. Simmons, Eva M. Clavane, Christopher J. Trevelyan, Jane M. Brown, Lena Przemyłska, Nicole T. Watt, Laura C. Matthews, and Paul J. Meakin. 2022. "PTPRD and DCC Are Novel BACE1 Substrates Differentially Expressed in Alzheimer’s Disease: A Data Mining and Bioinformatics Study" International Journal of Molecular Sciences 23, no. 9: 4568. https://doi.org/10.3390/ijms23094568
APA StyleTaylor, H. A., Simmons, K. J., Clavane, E. M., Trevelyan, C. J., Brown, J. M., Przemyłska, L., Watt, N. T., Matthews, L. C., & Meakin, P. J. (2022). PTPRD and DCC Are Novel BACE1 Substrates Differentially Expressed in Alzheimer’s Disease: A Data Mining and Bioinformatics Study. International Journal of Molecular Sciences, 23(9), 4568. https://doi.org/10.3390/ijms23094568