
High SOX9 Maintains Glioma Stem Cell Activity through a Regulatory Loop Involving STAT3 and PML

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
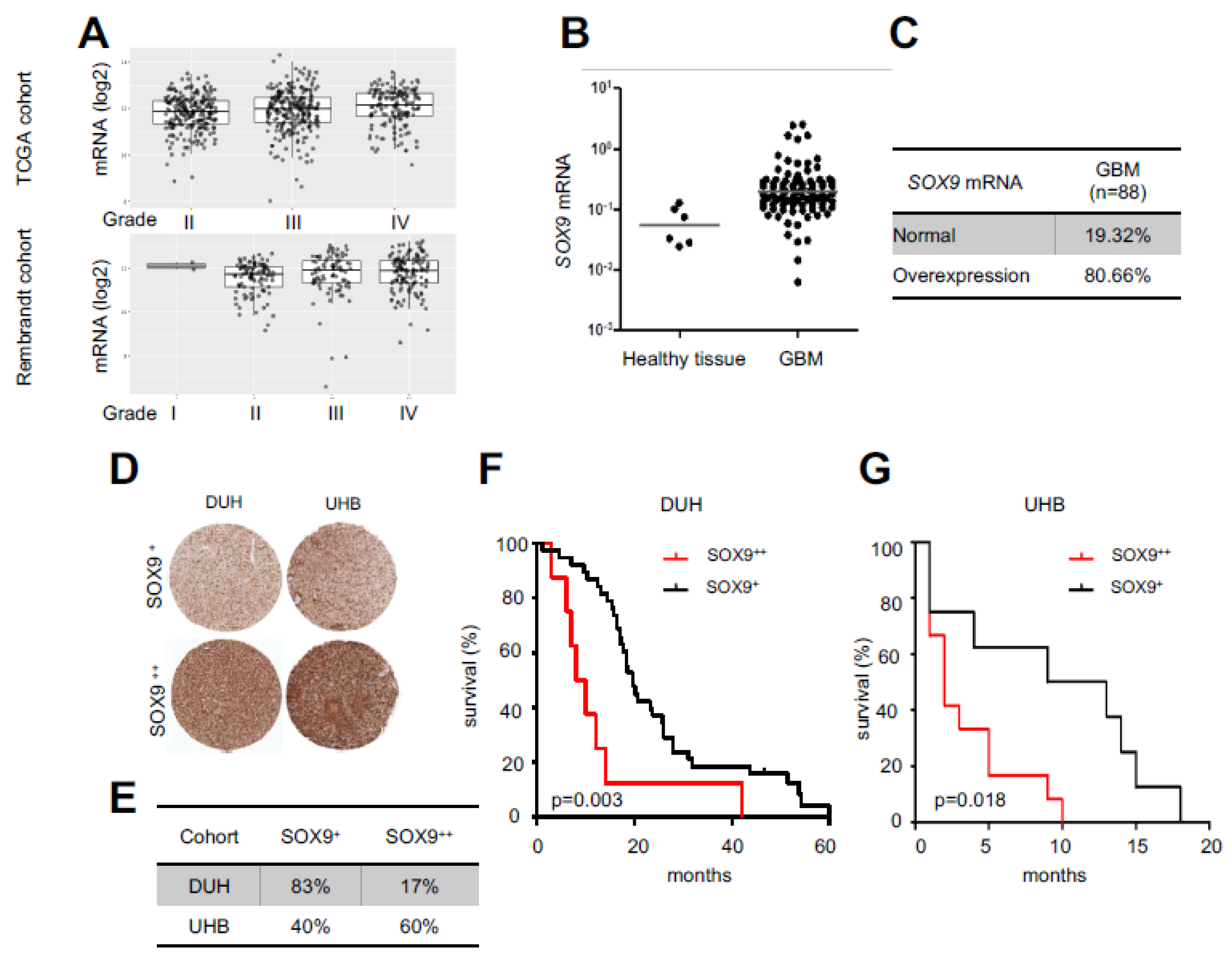
2.1. High SOX9 Levels Correlate with Lower Patient Survival
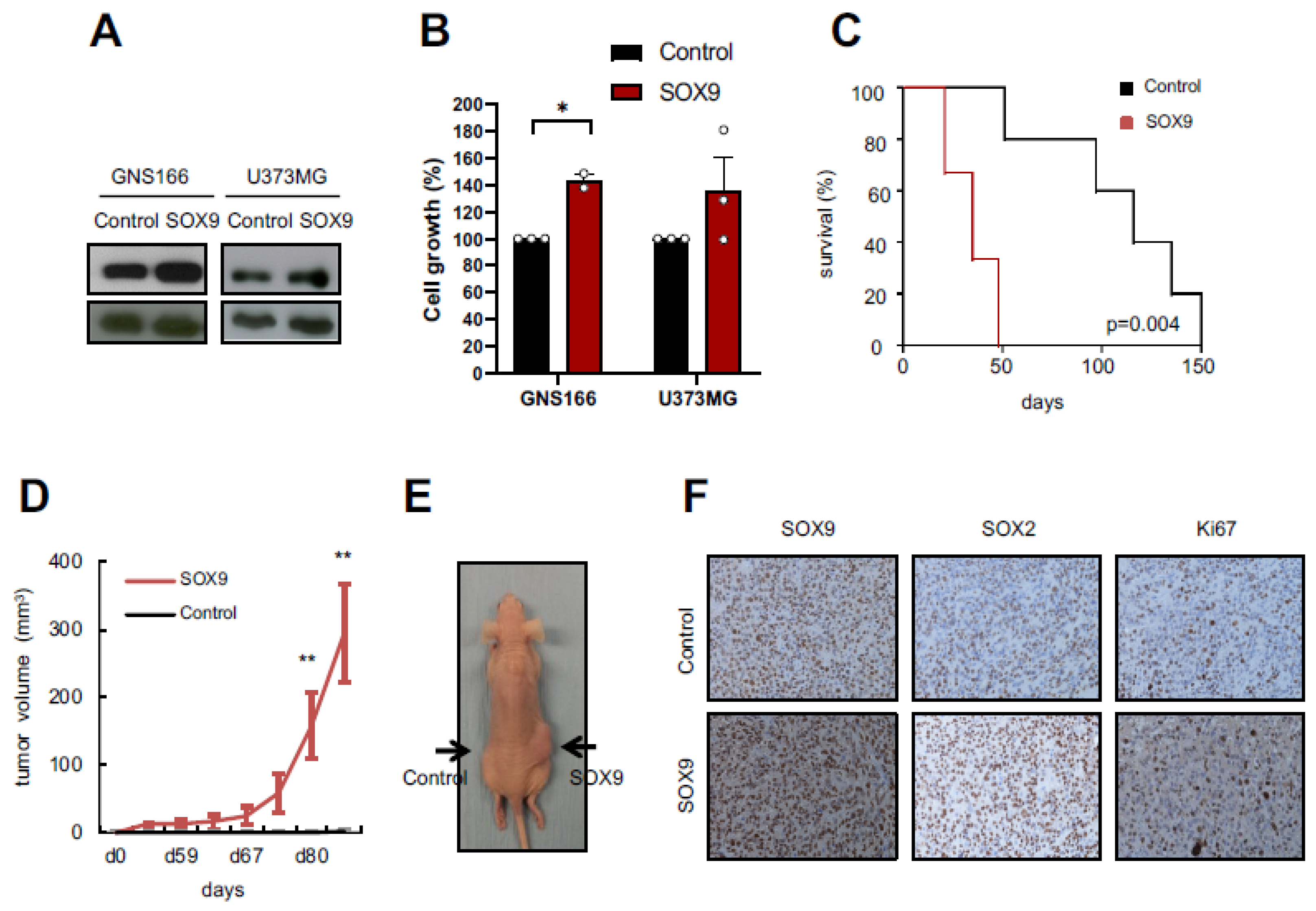
2.2. SOX9 Upregulation Increases Tumorigenic Capacity of GSCs
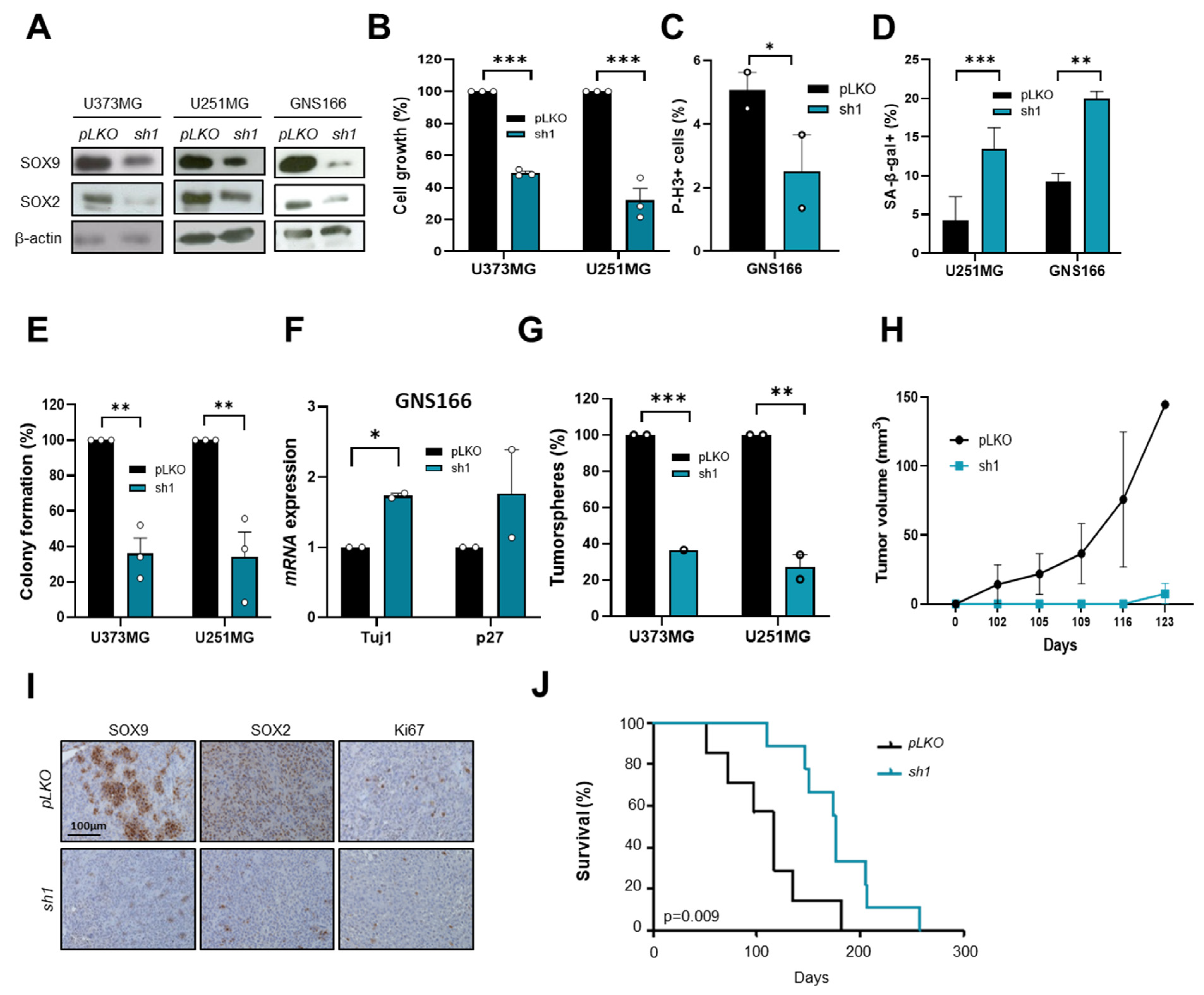
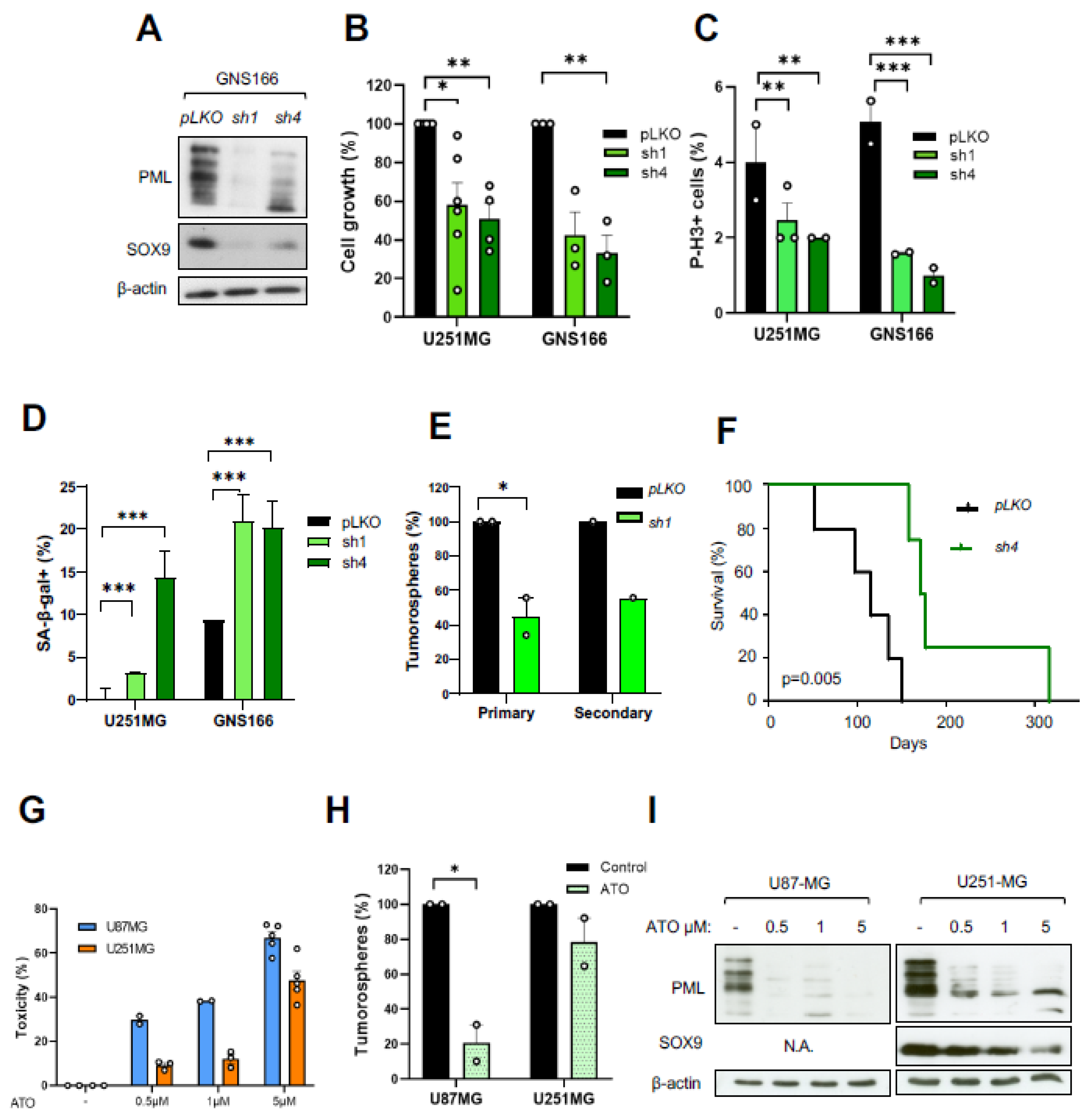
2.3. SOX9 Downregulation Decreases Stemness and Tumorigenic Capacity in GSCs
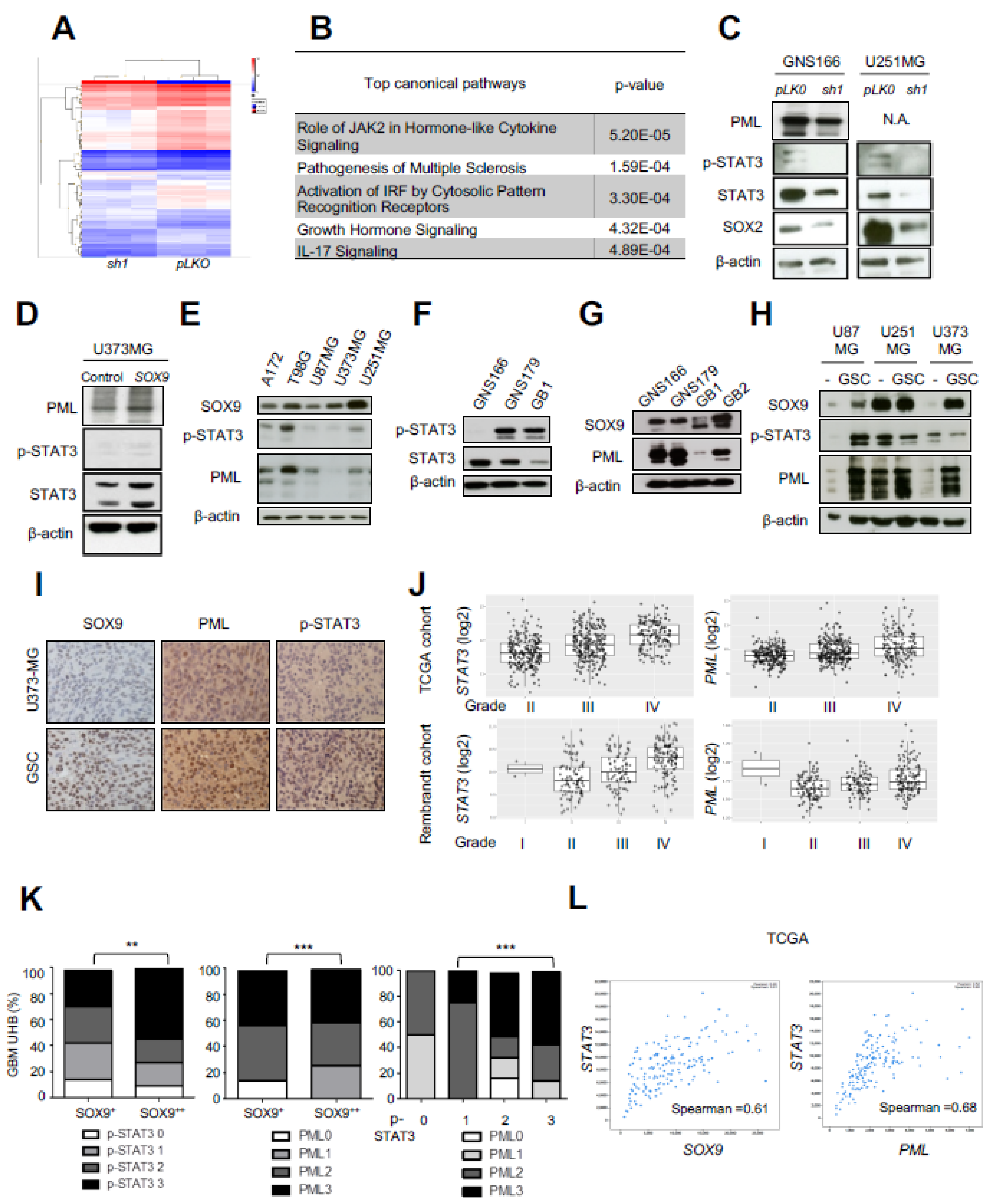
2.4. Transcriptomic Analysis Reveals STAT3 and PML as Mediators of SOX9 Activity in GSCs
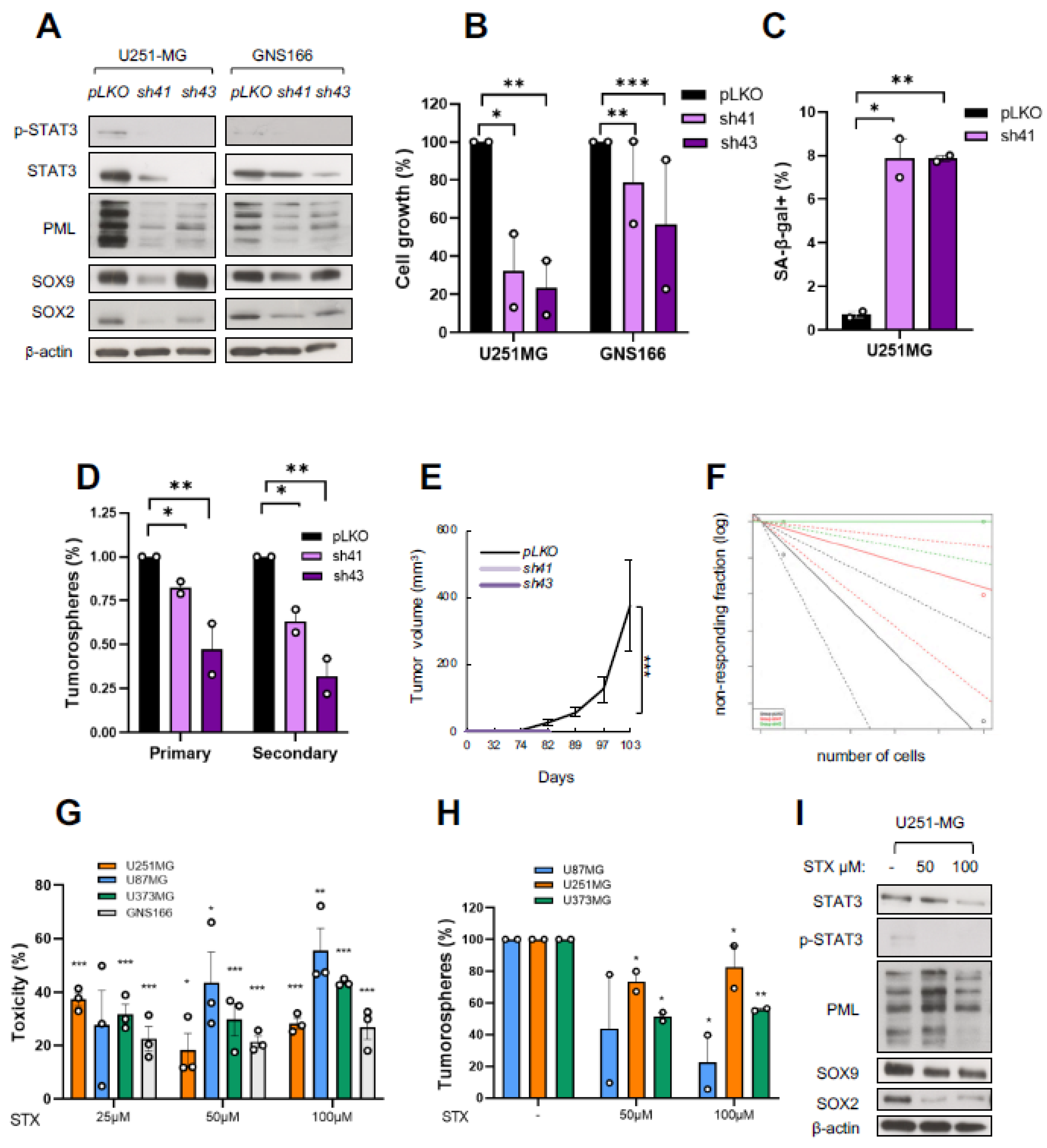
2.5. STAT3 Regulates GSC Activity and Its Pharmacological Inhibition Reduces Tumorigenicity
2.6. PML Regulates GSC Activity and Its Pharmacological Inhibition Reduces Tumorigenicity
2.7. The SOX9–STAT3–PML Axis Constitutes a Regulatory Loop That Modulates GSCs
3. Discussion
4. Materials and Methods
4.1. Human Subjects
4.2. Tissue Microarrays
4.3. Cell Culture
4.4. Gene Silencing and Overexpression
4.5. Cell Proliferation Assay
4.6. Oncosphere Formation Assay
4.7. Senescence-Associated β-Galactosidase Staining
4.8. Cell Viability Assay
4.9. Quantitative Real-Time PCR
4.10. Immunoblot
4.11. Immunofluorescence
4.12. In Vivo Carcinogenesis Assays
4.13. Immunohistochemistry
4.14. Transcriptome Analysis
4.15. Chromatin Immunoprecipitation Assay
4.16. Data Availability Statement
4.17. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Lan, X.; Joerg, D.; Cavalli, F.; Richards, L.; Nguyen, L.; Vanner, R.; Guilhamon, P.; Lee, L.; Kushida, M.; Pellacani, D.; et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Alcantara Llaguno, S.; Chen, J.; Kwon, C.H.; Jackson, E.L.; Li, Y.; Burns, D.K.; Parada, L.F. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 2009, 15, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Alcantara Llaguno, S.R.; Wang, Z.; Sun, D.; Chen, J.; Xu, J.; Kim, E.; Parada, L.F. Adult Lineage-Restricted CNS Progenitors Specify Distinct Glioblastoma Subtypes. Cancer Cell 2015, 28, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Lee, J.H. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suvà, M.L.; Riggi, N.; Bernstein, B.E. Epigenetic reprogramming in cancer. Science 2013, 339, 1567–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suva, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Bernstein, B.E. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Rocha, A.M.; Sampron, N.; Alonso, M.M.; Matheu, A. Role of SOX family of transcription factors in central nervous system tumors. Am. J. Cancer Res. 2014, 4, 312–324. [Google Scholar]
- Caren, H.; Stricker, S.H.; Bulstrode, H.; Gagrica, S.; Johnstone, E.; Bartlett, T.E.; Pollard, S.M. Glioblastoma Stem Cells Respond to Differentiation Cues but Fail to Undergo Commitment and Terminal Cell-Cycle Arrest. Stem Cell Rep. 2015, 5, 829–842. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986.e9. [Google Scholar] [CrossRef] [Green Version]
- Pritchett, J.; Athwal, V.; Roberts, N.; Hanley, N.A.; Hanley, K.P. Understanding the role of SOX9 in acquired diseases: Lessons from development. Trends Mol. Med. 2011, 17, 166–174. [Google Scholar] [CrossRef]
- Cheng, L.-C.; Pastrana, E.; Tavazoie, M.; Doetsch, F. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat. Neurosci. 2009, 12, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.E.; Wynn, S.L.; Sesay, A.; Cruz, C.; Cheung, M.; Gomez-Gaviro, M.V.; Booth, S.; Gao, B.; Cheah, K.S.E.; Lovell-Badge, R.; et al. SOX9 induces and maintains neural stem cells. Nat. Neurosci. 2010, 13, 1181–1189. [Google Scholar] [CrossRef]
- Swartling, F.J.; Savov, V.; Persson, A.I.; Chen, J.; Hackett, C.S.; Northcott, P.A.; Grimmer, M.R.; Lau, J.; Chesler, L.; Perry, A.; et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell 2012, 21, 601–613. [Google Scholar] [CrossRef] [Green Version]
- Matheu, A.; Collado, M.; Wise, C.; Manterola, L.; Cekaite, L.; Tye, A.J.; Canamero, M.; Bujanda, L.; Schedl, A.; Cheah, K.S.; et al. Oncogenicity of the developmental transcription factor Sox9. Cancer Res. 2012, 72, 1301–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Weinberg, R.A. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garros-Regulez, L.; Aldaz, P.; Arrizabalaga, O.; Moncho-Amor, V.; Carrasco-Garcia, E.; Manterola, L.; Matheu, A. mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert Opin. Ther. Targets 2016, 20, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, S.; Yuan, J.; Mao, X.-G.; Cao, Y.; Zong, J.; Tu, Y.; Zhang, Y. Oncogenic role of SOX9 expression in human malignant glioma. Med. Oncol. 2012, 29, 3484–3490. [Google Scholar] [CrossRef]
- Aldaz, P.; Otaegi-Ugartemendia, M.; Saenz-Antoñanzas, A.; García-Puga, M.; Moreno-Valladares, M.; Flores, J.M.; Gerovska, D.; Arauzo-Bravo, M.J.; Samprón, N.; Matheu, A.; et al. SOX9 promotes tumor progression through the axis BMI1-p21(CIP). Sci. Rep. 2020, 10, 357. [Google Scholar] [CrossRef] [Green Version]
- Sherry, M.M.; Reeves, A.; Wu, J.K.; Cochran, B.H. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009, 27, 2383–2392. [Google Scholar] [CrossRef] [Green Version]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. Interleukin 6 signaling regulates promyelocytic leukemia protein gene expression in human normal and cancer cells. J. Biol. Chem. 2012, 287, 26702–26714. [Google Scholar] [CrossRef] [Green Version]
- Martin-Martin, N.; Piva, M.; Urosevic, J.; Aldaz, P.; Sutherland, J.D.; Fernandez-Ruiz, S.; Carracedo, A. Stratification and therapeutic potential of PML in metastatic breast cancer. Nat. Commun. 2016, 7, 12595. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Davidsen, T. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Jeanne, M.; Benhenda, S.; Nasr, R.; Lei, M.; Peres, L.; de The, H. Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol. 2008, 10, 547–555. [Google Scholar] [CrossRef]
- Adam, R.C.; Yang, H.; Rockowitz, S.; Larsen, S.; Nikolova, M.; Oristian, D.S.; Polak, L.; Kadaja, M.; Asare, A.; Zheng, D.; et al. Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature 2015, 521, 366–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Xu, X.; Liu, N.; Cheng, Y.; Jin, W.; Zhang, P.; Wang, X.; Yang, H.; Liu, H.; Tu, Y. SOX9-PDK1 axis is essential for glioma stem cell self-renewal and temozolomide resistance. Oncotarget 2017, 9, 192–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Zhang, J.-Y.; Li, Y.-H.; Ren, F. Decreased expression of SOX9 indicates a better prognosis and inhibits the growth of glioma cells by inducing cell cycle arrest. Int. J. Clin. Exp. Pathol. 2015, 8, 10130–10138. [Google Scholar] [PubMed]
- Rahaman, S.O.; Harbor, P.C.; Chernova, O.; Barnett, G.H.; Vogelbaum, M.A.; Haque, S.J. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene 2002, 21, 8404–8413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.S.Y.; Sandanaraj, E.; Chong, Y.K.; Lim, S.W.; Koh, L.W.H.; Ng, W.H.; Tang, C. A STAT3-based gene signature stratifies glioma patients for targeted therapy. Nat. Commun. 2019, 10, 3601. [Google Scholar] [CrossRef] [PubMed]
- Birner, P.; Toumangelova-Uzeir, K.; Natchev, S.; Guentchev, M. STAT3 tyrosine phosphorylation influences survival in glioblastoma. J. Neurooncol. 2010, 100, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Bernardi, R.; Morotti, A.; Matsuoka, S.; Saglio, G.; Ikeda, Y.; Rosenblatt, J.; Avigan, D.E.; Teruya-Feldstein, J.; Pandolfi, P.P. PML targeting eradicates quiescent leukaemia-initiating cells. Nature 2008, 453, 1072–1078. [Google Scholar] [CrossRef]
- Carracedo, A.; Weiss, D.; Leliaert, A.K.; Bhasin, M.; de Boer, V.; Laurent, G.; Adams, A.C.; Sundvall, M.; Song, S.J.; Ito, K.; et al. A metabolic prosurvival role for PML in breast cancer. J. Clin. Investig. 2012, 122, 3088–3100. [Google Scholar] [CrossRef]
- Amodeo, V.; Deli, A.; Betts, J.; Bartesaghi, S.; Zhang, Y.; Richard-Londt, A.; Ellis, M.; Roshani, R.; Vouri, M.; Galavotti, S.; et al. A PML/Slit Axis Controls Physiological Cell Migration and Cancer Invasion in the CNS. Cell Rep. 2017, 20, 411–426. [Google Scholar] [CrossRef] [Green Version]
- Ashizawa, T.; Miyata, H.; Iizuka, A.; Komiyama, M.; Oshita, C.; Kume, A.; Akiyama, Y. Effect of the STAT3 inhibitor STX-0119 on the proliferation of cancer stem-like cells derived from recurrent glioblastoma. Int. J. Oncol. 2013, 43, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, Y.; Nonomura, C.; Ashizawa, T.; Iizuka, A.; Kondou, R.; Miyata, H.; Sugino, T.; Mitsuya, K.; Hayashi, N.; Nakasu, Y.; et al. The anti-tumor activity of the STAT3 inhibitor STX-0119 occurs via promotion of tumor-infiltrating lymphocyte accumulation in temozolomide-resistant glioblastoma cell line. Immunol. Lett. 2017, 190, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Fuh, B.; Sobo, M.; Cen, L.; Josiah, D.; Hutzen, B.; Cisek, K.; Bhasin, D.; Regan, N.; Lin, L.; Chan, C.; et al. LLL-3 inhibits STAT3 activity, suppresses glioblastoma cell growth and prolongs survival in a mouse glioblastoma model. Br. J. Cancer 2009, 100, 106–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, K.V.; Cseh, O.; Aman, A.; Weiss, S.; Luchman, H.A. The JAK2/STAT3 inhibitor pacritinib effectively inhibits patient-derived GBM brain tumor initiating cells in vitro and when used in combination with temozolomide increases survival in an orthotopic xenograft model. PLoS ONE 2017, 12, e0189670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Cheng, L.; Shi, Y.; Ke, S.Q.; Huang, Z.; Fang, X.; Chu, C.-W.; Xie, Q.; Bian, X.-W.; Rich, J.N.; et al. Arsenic trioxide disrupts glioma stem cells via promoting PML degradation to inhibit tumor growth. Oncotarget 2015, 6, 37300–37315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Jill, P.; Alexe, G.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Sun, D.; Chen, Y.-J.; Xie, X.; Shi, Y.; Tabar, V.; Brennan, C.W.; Bale, T.A.; Jayewickreme, C.D.; Laks, D.R.; et al. Cell Lineage-Based Stratification for Glioblastoma. Cancer Cell 2020, 38, 366–379.e8. [Google Scholar] [CrossRef] [PubMed]
- Gangoso, E.; Southgate, B.; Bradley, L.; Rus, S.; Galvez-Cancino, F.; McGivern, N.; Güç, E.; Kapourani, C.-A.; Byron, A.; Ferguson, K.M.; et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 2021, 184, 2454–2470.e26. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [Green Version]
- Matheu, A.; Klatt, P.; Serrano, M. Regulation of the INK4a/ARF locus by histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 42433–42441. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldaz, P.; Martín-Martín, N.; Saenz-Antoñanzas, A.; Carrasco-Garcia, E.; Álvarez-Satta, M.; Elúa-Pinin, A.; Pollard, S.M.; Lawrie, C.H.; Moreno-Valladares, M.; Samprón, N.; et al. High SOX9 Maintains Glioma Stem Cell Activity through a Regulatory Loop Involving STAT3 and PML. Int. J. Mol. Sci. 2022, 23, 4511. https://doi.org/10.3390/ijms23094511
Aldaz P, Martín-Martín N, Saenz-Antoñanzas A, Carrasco-Garcia E, Álvarez-Satta M, Elúa-Pinin A, Pollard SM, Lawrie CH, Moreno-Valladares M, Samprón N, et al. High SOX9 Maintains Glioma Stem Cell Activity through a Regulatory Loop Involving STAT3 and PML. International Journal of Molecular Sciences. 2022; 23(9):4511. https://doi.org/10.3390/ijms23094511
Chicago/Turabian StyleAldaz, Paula, Natalia Martín-Martín, Ander Saenz-Antoñanzas, Estefania Carrasco-Garcia, María Álvarez-Satta, Alejandro Elúa-Pinin, Steven M. Pollard, Charles H. Lawrie, Manuel Moreno-Valladares, Nicolás Samprón, and et al. 2022. "High SOX9 Maintains Glioma Stem Cell Activity through a Regulatory Loop Involving STAT3 and PML" International Journal of Molecular Sciences 23, no. 9: 4511. https://doi.org/10.3390/ijms23094511
APA StyleAldaz, P., Martín-Martín, N., Saenz-Antoñanzas, A., Carrasco-Garcia, E., Álvarez-Satta, M., Elúa-Pinin, A., Pollard, S. M., Lawrie, C. H., Moreno-Valladares, M., Samprón, N., Hench, J., Lovell-Badge, R., Carracedo, A., & Matheu, A. (2022). High SOX9 Maintains Glioma Stem Cell Activity through a Regulatory Loop Involving STAT3 and PML. International Journal of Molecular Sciences, 23(9), 4511. https://doi.org/10.3390/ijms23094511