
Chaperone-Mediated Autophagy in Pericytes: A Key Target for the Development of New Treatments against Glioblastoma Progression



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Glioblastoma Multiforme
1.2. CMA in Cancer
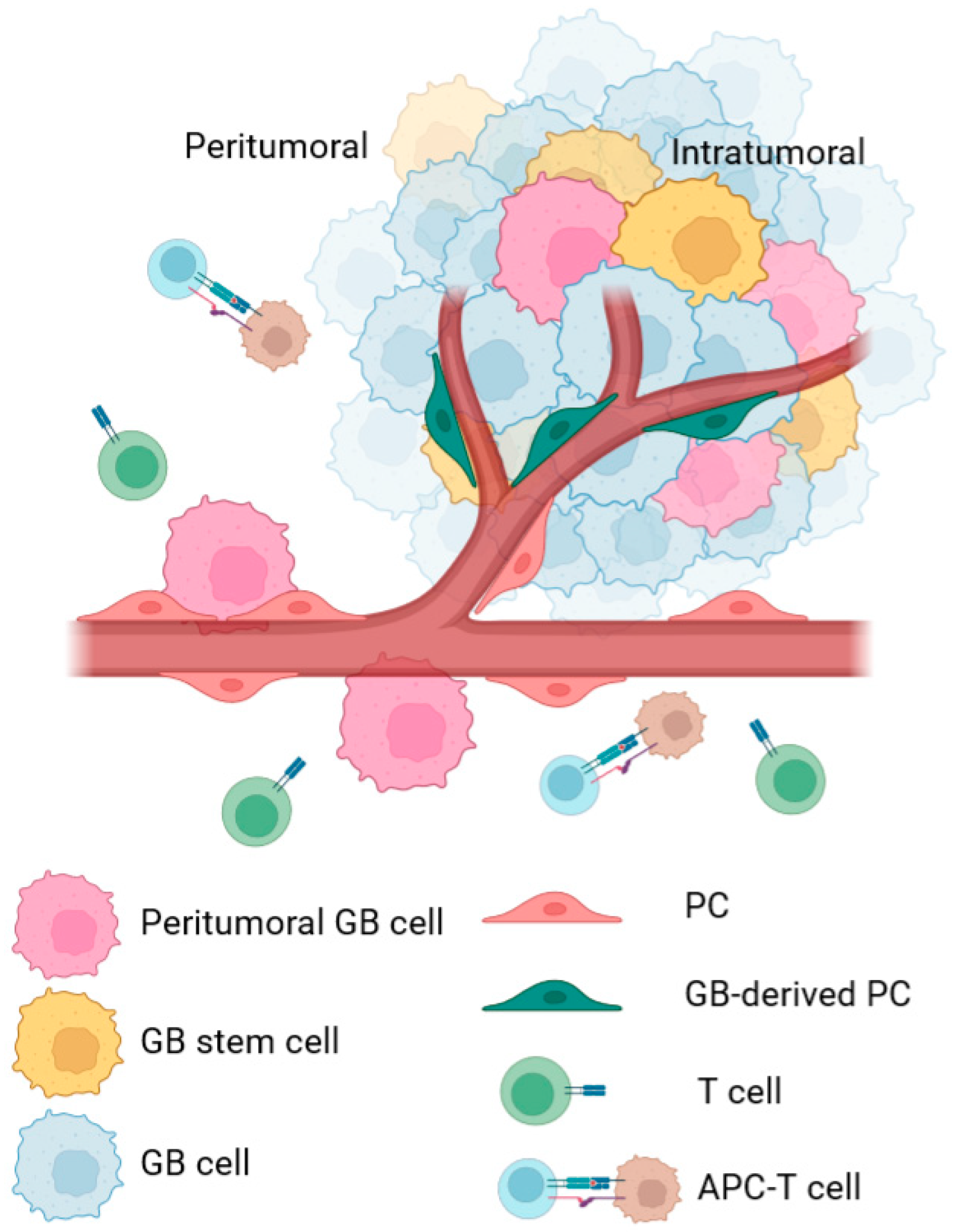
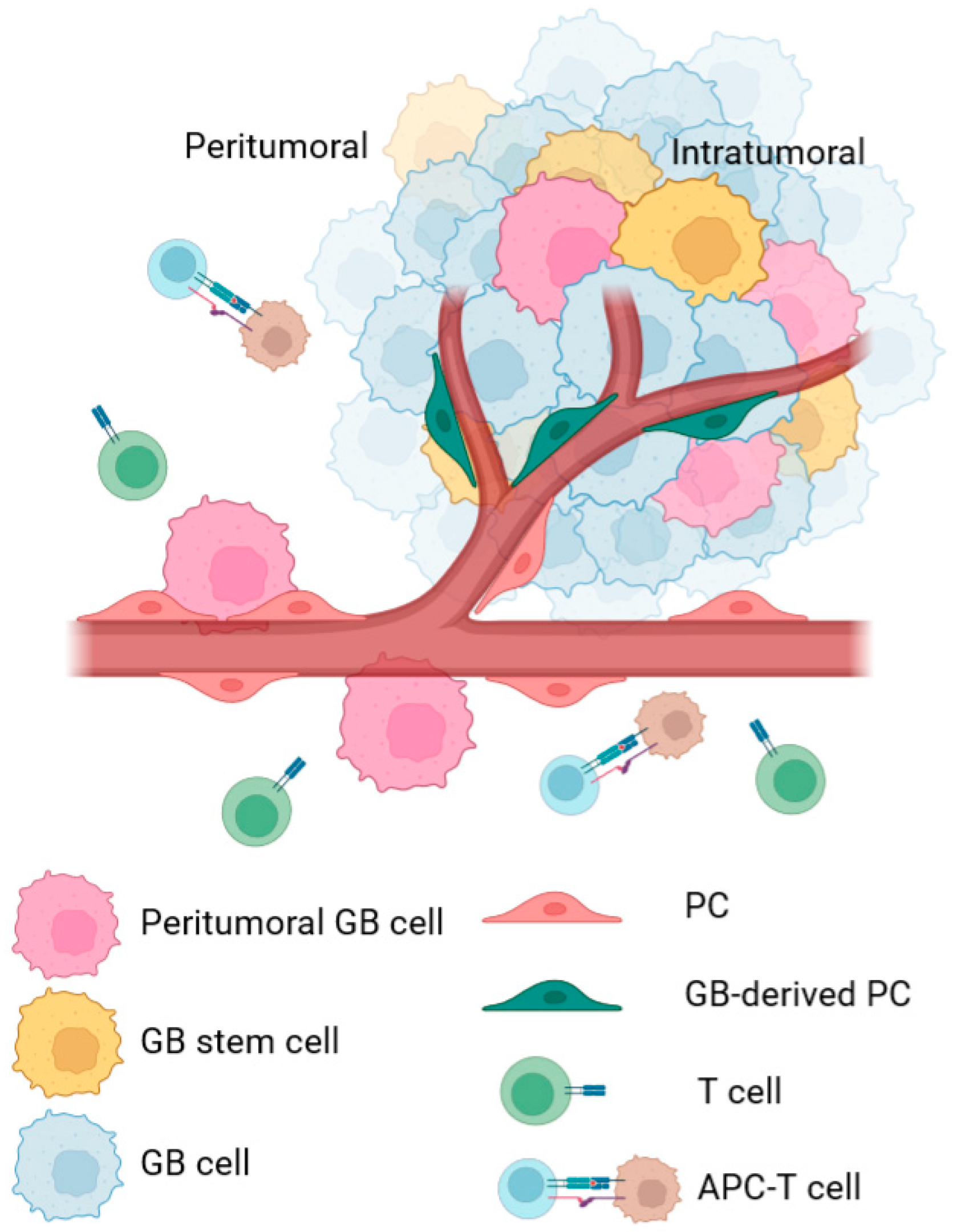
2. CMA in PC during GB Progression
2.1. Intratumoral PC
2.2. Peritumoral PC
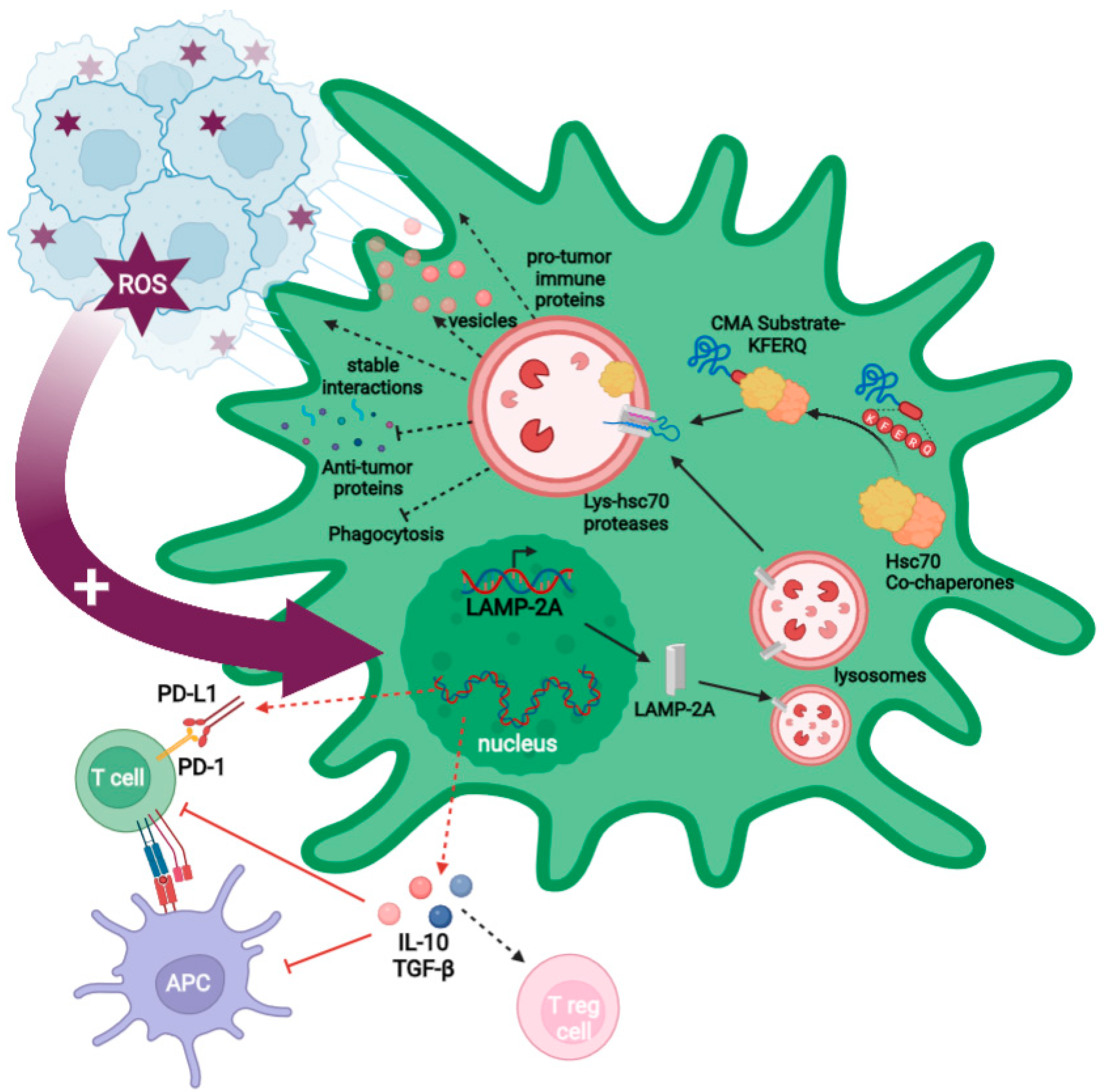
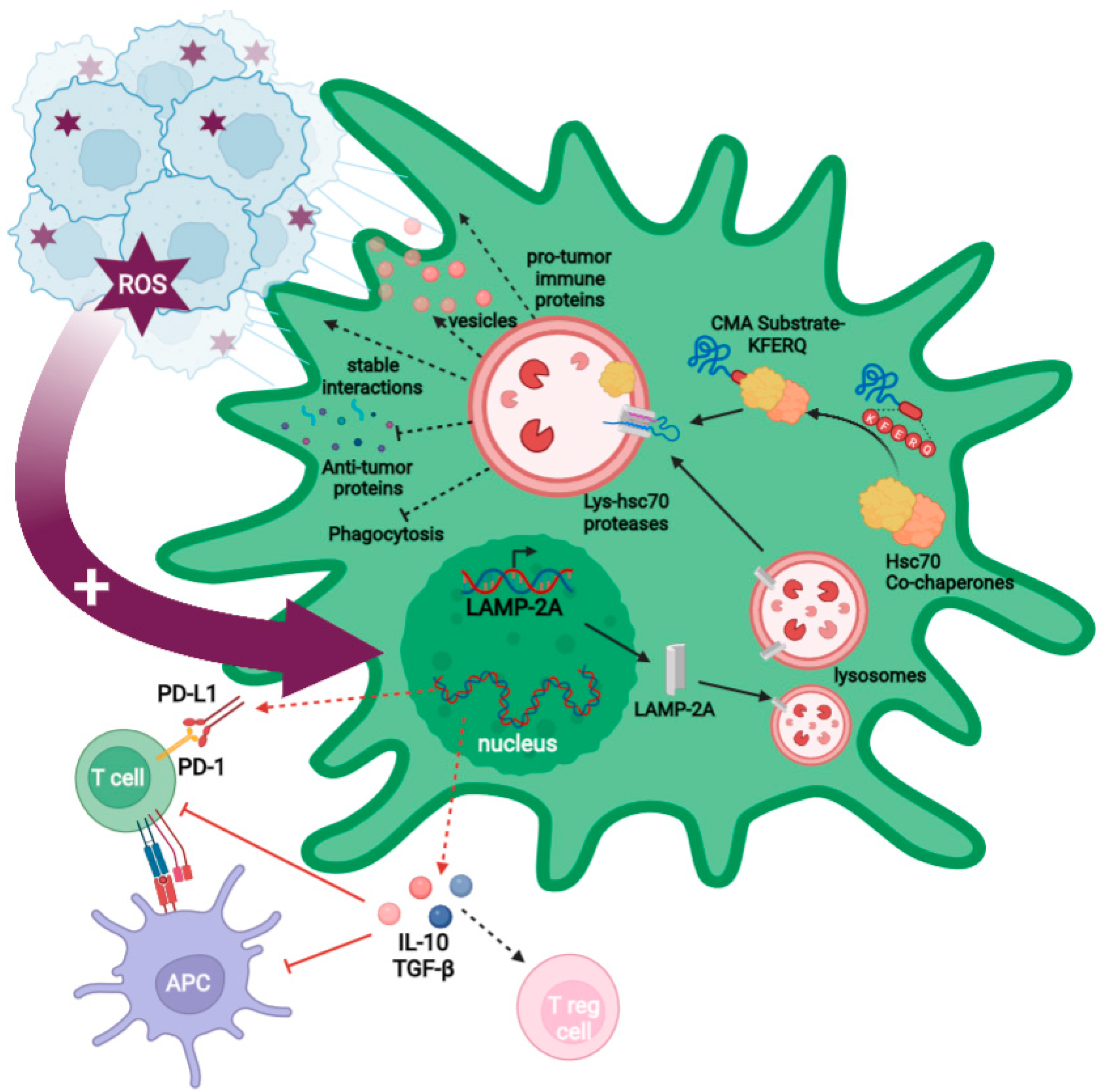
2.3. GB-Induced CMA in PC
2.3.1. GB-Induced CMA in PC Is Required for GB Immune Evasion
2.3.2. GB-Induced CMA Upregulation in PC Is Required for Stable PC–GB Interactions
2.3.3. GB-Induced CMA in PC Is Required to Modulate PC Secretome and PC MSC-like Properties That Favor Tumor Progression
3. Possible Therapeutic Approaches Based on CMA Inhibition in PC to Counter GB Progression
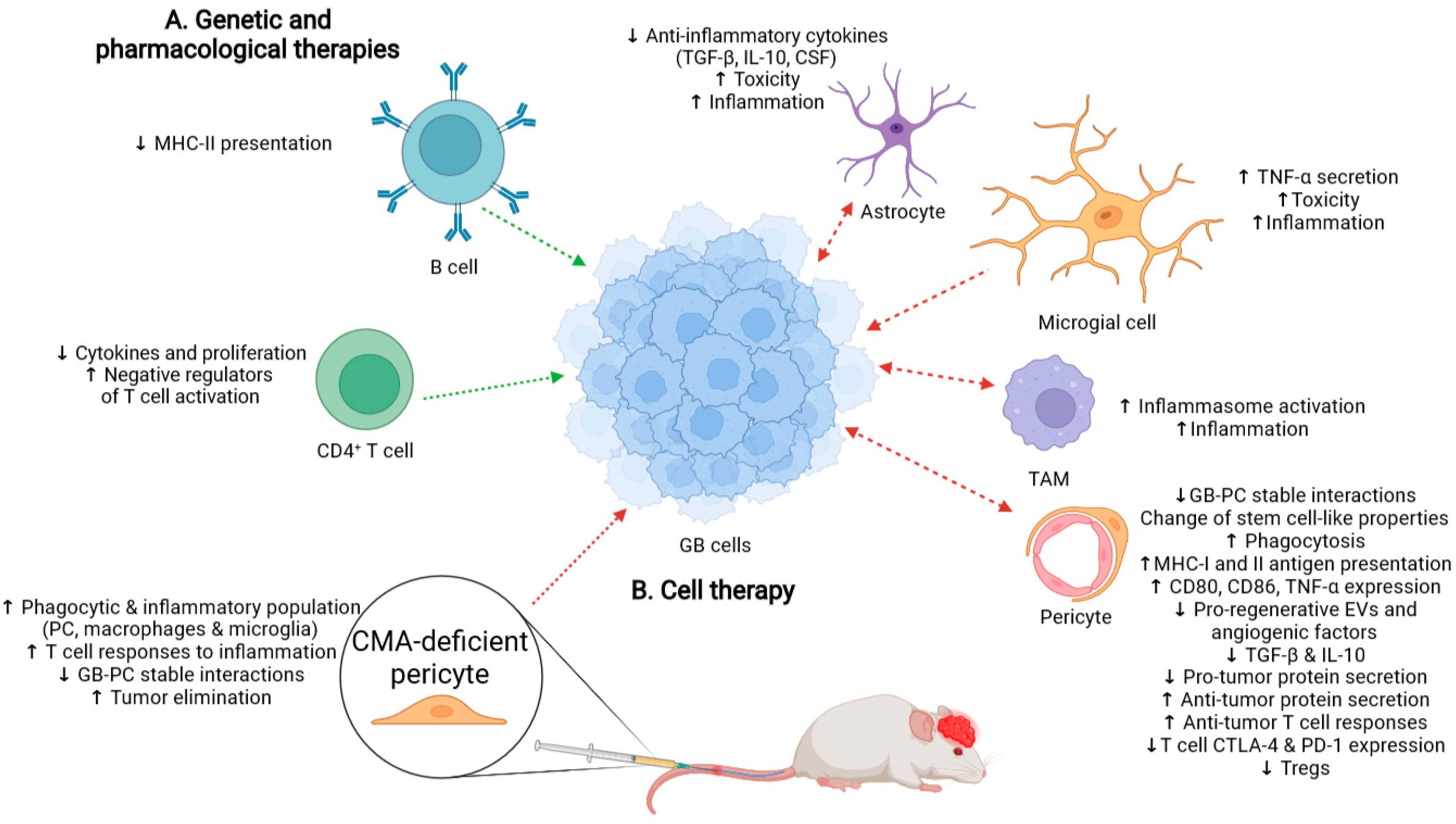
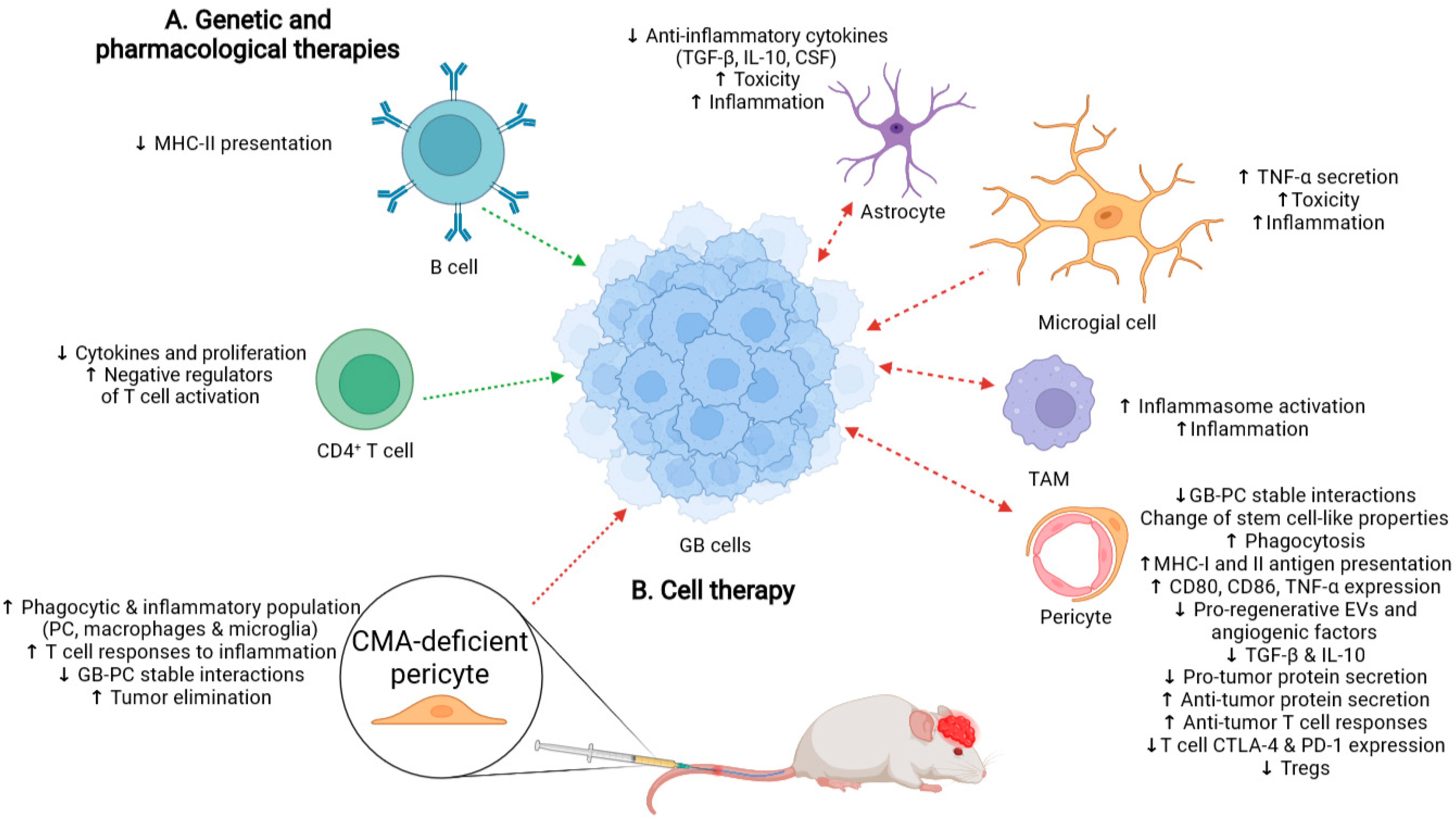
3.1. Possible Side-Effects Following Therapy That Promotes a Generalized CMA Inhibition
3.2. Cell Therapy with CMA-Deficient PC as an Alternative to Treat GB
4. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Molina, M.L.; Valdor, R. The effect of glioblastoma on pericytes. Curr. Tissue Microenviron. Rep. 2020, 1, 171–181. [Google Scholar] [CrossRef]
- Llaguno, S.R.A.; Parada, L.F. Cell of origin of glioma: Biological and clinical implications. Br. J. Cancer 2016, 115, 1445–1450. [Google Scholar] [CrossRef] [PubMed]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma multiforme: A review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef]
- Mousavi, S.M.; Derakhshan, M.; Baharloii, F.; Dashti, F.; Mirazimi, S.M.A.; Mahjoubin-Tehran, M.; Hosseindoost, S.; Goleij, P.; Rahimian, N.; Hamblin, M.R.; et al. Non-coding RNAs and glioblastoma: Insight into their roles in metastasis. Mol. Ther. Oncolytics 2022, 24, 262–287. [Google Scholar] [CrossRef] [PubMed]
- Lah, T.T.; Novak, M.; Breznik, B. Brain malignancies: Glioblastoma and brain metastases. Sem. Cancer Biol. 2020, 60, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Verhaak, R.G.; Canoll, P. The cellular origin for malignant glioma and prospects for clinical advancements. Expert Rev. Mol. Diagn. 2012, 12, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbańska, K.; Sokołowska, J.; Szmidt, M.; Sysa, P. Glioblastoma multiforme—An overview. Contemp. Oncol. 2014, 18, 307–312. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining tumor niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Xie, T.; Fang, J.; Xue, W.; Kang, H.; Tong, H.; Guo, Y.; Zhang, B.; Wang, S.; Yang, Y.; et al. Dynamic MR imaging for functional vascularization depends on tissue factor signaling in glioblastoma. Cancer Biol. Ther. 2018, 19, 416–426. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell–like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [Green Version]
- Lemée, J.-M.; Clavreul, A.; Menei, P. Intratumoral heterogeneity in glioblastoma: Don’t forget the peritumoral brain zone. Neuro-Oncology 2015, 17, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive mechanisms in glioblastoma. Neuro-Oncology 2015, 17, vii9–vii14. [Google Scholar] [CrossRef]
- Hernández, A.; Domènech, M.; Muñoz-Mármol, A.M.; Carrato, C.; Balana, C. Glioblastoma: Relationship between metabolism and immunosuppressive microenvironment. Cells 2021, 10, 3529. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee-Chang, C.; Rashidi, A.; Miska, J.; Zhang, P.; Pituch, K.C.; Hou, D.; Xiao, T.; Fischietti, M.; Kang, S.J.; Appin, C.L.; et al. Myeloid-derived suppressive cells promote b cell–mediated immunosuppression via transfer of PD-L1 in glioblastoma. Cancer Immunol. Res. 2019, 7, 1928–1943. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Valdor, R.; García-Bernal, D.; Riquelme, D.; Martinez, C.M.; Moraleda, J.M.; Cuervo, A.M.; Macian, F.; Martinez, S. Glioblastoma ablates pericytes antitumor immune function through aberrant up-regulation of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2019, 116, 20655–20665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) 1. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Lescat, L.; Herpin, A.; Mourot, B.; Véron, V.; Guiguen, Y.; Bobe, J.; Seiliez, I. CMA restricted to mammals and birds: Myth or reality? Autophagy 2018, 14, 1267–1270. [Google Scholar] [CrossRef]
- Kon, M.; Cuervo, A.M. Chaperone-mediated autophagy in health and disease. FEBS Lett. 2010, 584, 1399–1404. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Bourdenx, M.; Martín-Segura, A.; Scrivo, A.; Rodriguez-Navarro, J.A.; Kaushik, S.; Tasset, I.; Diaz, A.; Storm, N.J.; Xin, Q.; Juste, Y.R.; et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 2021, 184, 2696–2714.e25. [Google Scholar] [CrossRef] [PubMed]
- Ikami, Y.; Terasawa, K.; Sakamoto, K.; Ohtake, K.; Harada, H.; Watabe, T.; Yokoyama, S.; Hara-Yokoyama, M. The two-domain architecture of LAMP2A regulates its interaction with Hsc70. Exp. Cell Res. 2022, 411, 112986. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Tomaz, M.; de Souza, I.; Rocha, C.R.R.; Gomes, L.R. The role of chaperone-mediated autophagy in cell cycle control and its implications in cancer. Cells 2020, 9, 2140. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Aguirre-Hernandez, C.; Scrivo, A.; Eliscovich, C.; Arias, E.; Bravo-Cordero, J.J.; Cuervo, A.M. Monitoring spatiotemporal changes in chaperone-mediated autophagy in vivo. Nat. Commun. 2020, 11, 645. [Google Scholar] [CrossRef] [PubMed]
- Madrigal-Matute, J.; de Bruijn, J.; van Kuijk, K.; Riascos-Bernal, D.F.; Diaz, A.; Tasset, I.; Martín-Segura, A.; Gijbels, M.J.J.; Sander, B.; Kaushik, S.; et al. Protective role of chaperone-mediated autophagy against atherosclerosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2121133119. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Villarroya, J.; Diaz-Carretero, A.; Patel, B.; Urbanska, A.M.; Thi, M.M.; Villarroya, F.; Santambrogio, L.; Cuervo, A.M. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell 2015, 14, 249–264. [Google Scholar] [CrossRef]
- Dong, S.; Wang, Q.; Kao, Y.-R.; Diaz, A.; Tasset, I.; Kaushik, S.; Thiruthuvanathan, V.; Zintiridou, A.; Nieves, E.; Dzieciatkowska, M.; et al. Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 2021, 591, 117–123. [Google Scholar] [CrossRef]
- Qiao, L.; Ma, J.; Zhang, Z.; Sui, W.; Zhai, C.; Xu, D.; Wang, Z.; Lu, H.; Zhang, M.; Zhang, C.; et al. Deficient chaperone-mediated autophagy promotes inflammation and atherosclerosis. Circ. Res. 2021, 129, 1141–1157. [Google Scholar] [CrossRef]
- Valdor, R.; Mocholi, E.; Botbol, Y.; Guerrero-Ros, I.; Chandra, D.; Koga, H.; Gravekamp, C.; Cuervo, A.M.; Macian, F. Chaperone-mediated autophagy regulates t cell responses through targeted degradation of negative regulators of t cell activation. Nat. Immunol. 2014, 15, 1046–1054. [Google Scholar] [CrossRef] [Green Version]
- Valdor, R.; Macian, F. Autophagy and the regulation of the immune response. Pharmacol. Res. 2012, 66, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Arias, E.; Cuervo, A.M. Pros and cons of chaperone-mediated autophagy in cancer biology. Trends Endocrinol. Metab. 2020, 31, 53–66. [Google Scholar] [CrossRef] [PubMed]
- de Souza, A.S.C.; Gonçalves, L.B.; Lepique, A.P.; de Araujo-Souza, P.S. The role of autophagy in tumor immunology—Complex mechanisms that may be explored therapeutically. Front. Oncol. 2020, 10, 603661. [Google Scholar] [CrossRef]
- Gómez-Sintes, R.; Arias, E. Chaperone-mediated autophagy and disease: Implications for cancer and neurodegeneration. Mol. Asp. Med. 2021, 82, 101025. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.R.; Menck, C.F.M.; Cuervo, A.M. Chaperone-mediated autophagy prevents cellular transformation by regulating myc proteasomal degradation. Autophagy 2017, 13, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Molina, M.L.; García-Bernal, D.; Martinez, S.; Valdor, R. Autophagy in the immunosuppressive perivascular microenvironment of glioblastoma. Cancers 2020, 12, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auzmendi-Iriarte, J.; Matheu, A. Impact of chaperone-mediated autophagy in brain aging: Neurodegenerative diseases and glioblastoma. Front. Aging Neurosci. 2021, 12, 630743. [Google Scholar] [CrossRef] [PubMed]
- Auzmendi-Iriarte, J.; Otaegi-Ugartemendia, M.; Carrasco-Garcia, E.; Azkargorta, M.; Diaz, A.; Saenz-Antoñanzas, A.; Andermatten, J. Andrés.; García-Puga, M.; Garcia, I.; Elua-Pinin, A.; et al. Chaperone-mediated autophagy controls proteomic and transcriptomic pathways to maintain glioma stem cell activity. Cancer Res. 2022, 82, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Molina, M.L.; García-Bernal, D.; Salinas, M.D.; Rubio, G.; Aparicio, P.; Moraleda, J.M.; Martínez, S.; Valdor, R. Chaperone-mediated autophagy ablation in pericytes reveals new glioblastooma prognostic merkers and efficient treatment against tumor progression. Front. Cell Dev. Biol. 2022, 10, 797945. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [Green Version]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-brain barrier dysfunction amplifies the development of neuroinflammation: Understanding of cellular events in brain microvascular endothelial cells for prevention and treatment of BBB dysfunction. Front. Cell. Neurosci. 2021, 15, 344. [Google Scholar] [CrossRef]
- Prazeres, P.H.D.M.; Sena, I.F.G.; Borges, I.D.T.; de Azevedo, P.O.; Andreotti, J.P.; de Paiva, A.E.; de Almeida, V.M.; Guerra, D.A.D.P.; Pinheiro dos Santos, G.S.; Mintz, A.; et al. Pericytes are heterogeneous in their origin within the same tissue. Dev. Biol. 2017, 427, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.E. Diversity within pericytes. Clin. Exp. Pharmacol. Physiol. 2000, 27, 842–846. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picoli, C.C.; Gonçalves, B.Ô.P.; Santos, G.S.P.; Rocha, B.G.S.; Costa, A.C.; Resende, R.R.; Birbrair, A. Pericytes cross-talks within the tumor microenvironment. Biochim. Biophys. Acta-Rev. Cancer 2021, 1876, 188608. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Korte, N.; Nortley, R.; Sethi, H.; Tang, Y.; Attwell, D. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 2018, 136, 507–523. [Google Scholar] [CrossRef] [Green Version]
- Nakagomi, T.; Kubo, S.; Nakano-Doi, A.; Sakuma, R.; Lu, S.; Narita, A.; Kawahara, M.; Taguchi, A.; Matsuyama, T. Brain vascular pericytes following ischemia have multipotential stem cell activity to differentiate into neural and vascular lineage cells. Stem Cells 2015, 33, 1962–1974. [Google Scholar] [CrossRef] [PubMed]
- Ochs, K.; Sahm, F.; Opitz, C.A.; Lanz, T.V.; Oezen, I.; Couraud, P.-O.; von Deimling, A.; Wick, W.; Platten, M. Immature mesenchymal stem cell-like pericytes as mediators of immunosuppression in human malignant glioma. J. Neuroimmunol. 2013, 265, 106–116. [Google Scholar] [CrossRef]
- Gaceb, A.; Barbariga, M.; Özen, I.; Paul, G. The pericyte secretome: Potential impact on regeneration. Biochimie 2018, 155, 16–25. [Google Scholar] [CrossRef]
- Bose, A.; Barik, S.; Banerjee, S.; Ghosh, T.; Mallick, A.; Majumdar, S.B.; Goswami, K.K.; Bhuniya, A.; Banerjee, S.; Baral, R.; et al. Tumor-derived vascular pericytes anergize th cells. J. Immunol. 2013, 191, 971–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, R.; Compte, M.; Álvarez-Vallina, L.; Sanz, L. Immune regulation by pericytes: Modulating innate and adaptive immunity. Front. Immunol. 2016, 7, 480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdor, R.; García-Bernal, D.; Bueno, C.; Ródenas, M.; Moraleda, J.M.; Macian, F.; Martínez, S. Glioblastoma progression is assisted by induction of immunosuppressive function of pericytes through interaction with tumor cells. Oncotarget 2017, 8, 68614–68626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segura-Collar, B.; Garranzo-Asensio, M.; Herranz, B.; Hernández-SanMiguel, E.; Cejalvo, T.; Casas, B.S.; Matheu, A.; Pérez-Núñez, Á.; Sepúlveda-Sánchez, J.M.; Hernández-Laín, A.; et al. Tumor-derived pericytes driven by EGFR mutations govern the vascular and immune microenvironment of gliomas. Cancer Res. 2021, 81, 2142–2156. [Google Scholar] [CrossRef]
- Alieva, M.; Leidgens, V.; Riemenschneider, M.J.; Klein, C.A.; Hau, P.; van Rheenen, J. Intravital imaging of glioma border morphology reveals distinctive cellular dynamics and contribution to tumor cell invasion. Sci. Rep. 2019, 9, 2054. [Google Scholar] [CrossRef]
- Zhang, G.-L.; Wang, C.-F.; Qian, C.; Ji, Y.-X.; Wang, Y.-Z. Role and mechanism of neural stem cells of the subventricular zone in glioblastoma. World J. Stem Cells 2021, 13, 877–893. [Google Scholar] [CrossRef] [PubMed]
- Yabo, Y.A.; Niclou, S.P.; Golebiewska, A. Cancer cell heterogeneity and plasticity: A paradigm shift in glioblastoma. Neuro-Oncology 2021, 24, 669–682. [Google Scholar] [CrossRef]
- Auzmendi-Iriarte, J.; Matheu, A. Intrinsic role of chaperone-mediated autophagy in cancer stem cell maintenance. Autophagy 2022, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Birbrair, A.; Zhang, T.; Wang, Z.-M.; Messi, M.L.; Olson, J.D.; Mintz, A.; Delbono, O. Type-2 pericytes participate in normal and tumoral angiogenesis. Am. J. Physiol.-Cell Physiol. 2014, 307, C25–C38. [Google Scholar] [CrossRef] [Green Version]
- Gaceb, A.; Özen, I.; Padel, T.; Barbariga, M.; Paul, G. Pericytes secrete pro-regenerative molecules in response to platelet-derived growth factor-BB. J. Cereb. Blood Flow Metab. 2018, 38, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Caspani, E.M.; Crossley, P.H.; Redondo-Garcia, C.; Martinez, S. Glioblastoma: A pathogenic crosstalk between tumor cells and pericytes. PLoS ONE 2014, 9, e101402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Y.; Lu, B. The emerging roles of circular RNAs in vessel co-option and vasculogenic mimicry: Clinical insights for anti-angiogenic therapy in cancers. Cancer Metastasis Rev. 2022, 41, 173–191. [Google Scholar] [CrossRef] [PubMed]
- Lah Turnšek, T.; Jiao, X.; Novak, M.; Jammula, S.; Cicero, G.; Ashton, A.W.; Joyce, D.; Pestell, R.G. an update on glioblastoma biology, genetics, and current therapies: Novel inhibitors of the g protein-coupled receptor CCR5. Int. J. Mol. Sci. 2021, 22, 4464. [Google Scholar] [CrossRef]
- Natsumeda, M.; Aoki, H.; Miyahara, H.; Yajima, N.; Uzuka, T.; Toyoshima, Y.; Kakita, A.; Takahashi, H.; Fujii, Y. Induction of autophagy in temozolomide treated malignant gliomas. Neuropathology 2011, 31, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-N.; Yang, K.-D.; Chen, C.; He, Z.-C.; Wang, Q.-H.; Feng, H.; Lv, S.-Q.; Wang, Y.; Mao, M.; Liu, Q.; et al. Pericytes augment glioblastoma cell resistance to temozolomide through CCL5-CCR5 paracrine signaling. Cell Res. 2021, 31, 1072–1087. [Google Scholar] [CrossRef]
- Salinas, R.D.; Durgin, J.S.; O’Rourke, D.M. Potential of glioblastoma-targeted chimeric antigen receptor (CAR) T-cell therapy. CNS Drugs 2020, 34, 127–145. [Google Scholar] [CrossRef]
- Majc, B.; Novak, M.; Kopitar-Jerala, N.; Jewett, A.; Breznik, B. Immunotherapy of glioblastoma: Current strategies and challenges in tumor model development. Cells 2021, 10, 265. [Google Scholar] [CrossRef]
- Macri, C.; Wang, F.; Tasset, I.; Schall, N.; Page, N.; Briand, J.-P.; Cuervo, A.M.; Muller, S. Modulation of deregulated chaperone-mediated autophagy by a phosphopeptide. Autophagy 2015, 11, 472–486. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Chen, R.; Xu, J.; Lin, Y.; Wang, R.; Chi, Z. Vitamin E inhibits activated chaperone-mediated autophagy in rats with status epilepticus. Neuroscience 2009, 161, 73–77. [Google Scholar] [CrossRef]
- Finn, P.F.; Mesires, N.T.; Vine, M.; Dice, J.F. Effects of small molecules on chaperone-mediated autophagy. Autophagy 2005, 1, 141–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Tasset, I.; Cuervo, A.M.; Muller, S. In vivo remodeling of altered autophagy-lysosomal pathway by a phosphopeptide in lupus. Cells 2020, 9, 2328. [Google Scholar] [CrossRef] [PubMed]
- Macian, F. Autophagy in T cell function and aging. Front. Cell Dev. Biol. 2019, 7, 213. [Google Scholar] [CrossRef]
- Kaushik, S.; Tasset, I.; Arias, E.; Pampliega, O.; Wong, E.; Martinez-Vicente, M.; Cuervo, A.M. Autophagy and the Hallmarks of Aging. Ageing Res. Rev. 2021, 72, 101468. [Google Scholar] [CrossRef]
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810. [Google Scholar] [CrossRef] [Green Version]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-mediated autophagy is required for tumor growth. Sci. Transl. Med. 2011, 3, 109ra117. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Li, P.; Lin, Y.; Lott, J.M.; Hislop, A.D.; Canaday, D.H.; Brutkiewicz, R.R.; Blum, J.S. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity 2005, 22, 571–581. [Google Scholar] [CrossRef] [Green Version]
- Page, N.; Gros, F.; Schall, N.; Décossas, M.; Bagnard, D.; Briand, J.-P.; Muller, S. HSC70 blockade by the therapeutic peptide P140 affects autophagic processes and endogenous MHCII presentation in murine lupus. Ann. Rheum. Dis. 2011, 70, 837–843. [Google Scholar] [CrossRef] [Green Version]
- Valdor, R.; Macian, F. Mechanisms of self-inactivation in anergic T cells. Inmunología 2010, 29, 20–33. [Google Scholar] [CrossRef]
- Wang, R.; Liu, Y.; Liu, L.; Chen, M.; Wang, X.; Yang, J.; Gong, Y.; Ding, B.-S.; Wei, Y.; Wei, X. Tumor cells induce LAMP2a expression in tumor-associated macrophage for cancer progression. EBioMedicine 2019, 40, 118–134. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Li, L.; Guo, J.; Liu, A.; Wu, J.; Wang, H.; Shi, J.; Pang, D.; Cao, Q. M2 tumor-associated macrophages produce interleukin-17 to suppress oxaliplatin-induced apoptosis in hepatocellular carcinoma. Oncotarget 2017, 8, 44465–44476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-specific IPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in parkinson’s disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; An, D.; Xu, J.; Shao, B.; Li, X.; Shi, J. Ac2-26 induces IKKβ degradation through chaperone-mediated autophagy via HSPB1 in NCM-treated microglia. Front. Mol. Neurosci. 2018, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.-M.; Yang, Q.; Xie, X.-Q.; Liao, C.-Y.; Lin, H.; Liu, T.-T.; Yin, L.; Shu, H.-B. Sumoylation promotes the stability of the DNA sensor CGAS and the adaptor STING to regulate the kinetics of response to DNA virus. Immunity 2016, 45, 555–569. [Google Scholar] [CrossRef] [Green Version]
- Linares, J.F.; Zhang, X.; Martinez-Ordoñez, A.; Duran, A.; Kinoshita, H.; Kasashima, H.; Nakanishi, N.; Nakanishi, Y.; Carelli, R.; Cappelli, L.; et al. PKCλ/ι inhibition activates an ULK2-mediated interferon response to repress tumorigenesis. Mol. Cell 2021, 81, 4509–4526.e10. [Google Scholar] [CrossRef]
- Sackstein, R.; Merzaban, J.S.; Cain, D.W.; Dagia, N.M.; Spencer, J.A.; Lin, C.P.; Wohlgemuth, R. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat. Med. 2008, 14, 181–187. [Google Scholar] [CrossRef]
- Abdi, R.; Moore, R.; Sakai, S.; Donnelly, C.B.; Mounayar, M.; Sackstein, R. HCELL expression on murine MSC licenses pancreatotropism and confers durable reversal of autoimmune diabetes in NOD mice. Stem Cells 2015, 33, 1523–1531. [Google Scholar] [CrossRef] [Green Version]
- Tabatabai, G.; Herrmann, C.; von Kürthy, G.; Mittelbronn, M.; Grau, S.; Frank, B.; Möhle, R.; Weller, M.; Wick, W. VEGF-dependent induction of CD62E on endothelial cells mediates glioma tropism of adult haematopoietic progenitor cells. Brain 2008, 131, 2579–2595. [Google Scholar] [CrossRef] [Green Version]
- García-Bernal, D.; García-Arranz, M.; García-Guillén, A.I.; García-Hernández, A.M.; Blanquer, M.; García-Olmo, D.; Sackstein, R.; Moraleda, J.M.; Zapata, A.G. Exofucosylation of adipose mesenchymal stromal cells alters their secretome profile. Front. Cell Dev. Biol. 2020, 8, 584074. [Google Scholar] [CrossRef]
- Shibahara, T.; Ago, T.; Tachibana, M.; Nakamura, K.; Yamanaka, K.; Kuroda, J.; Wakisaka, Y.; Kitazono, T. Reciprocal interaction between pericytes and macrophage in poststroke tissue repair and functional recovery. Stroke 2020, 51, 3095–3106. [Google Scholar] [CrossRef]
- Sun, R.; Kong, X.; Qiu, X.; Huang, C.; Wong, P.-P. The emerging roles of pericytes in modulating tumor microenvironment. Front. Cell Dev. Biol. 2021, 9, 1037. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salinas, M.D.; Valdor, R. Chaperone-Mediated Autophagy in Pericytes: A Key Target for the Development of New Treatments against Glioblastoma Progression. Int. J. Mol. Sci. 2022, 23, 8886. https://doi.org/10.3390/ijms23168886
Salinas MD, Valdor R. Chaperone-Mediated Autophagy in Pericytes: A Key Target for the Development of New Treatments against Glioblastoma Progression. International Journal of Molecular Sciences. 2022; 23(16):8886. https://doi.org/10.3390/ijms23168886
Chicago/Turabian StyleSalinas, María Dolores, and Rut Valdor. 2022. "Chaperone-Mediated Autophagy in Pericytes: A Key Target for the Development of New Treatments against Glioblastoma Progression" International Journal of Molecular Sciences 23, no. 16: 8886. https://doi.org/10.3390/ijms23168886
APA StyleSalinas, M. D., & Valdor, R. (2022). Chaperone-Mediated Autophagy in Pericytes: A Key Target for the Development of New Treatments against Glioblastoma Progression. International Journal of Molecular Sciences, 23(16), 8886. https://doi.org/10.3390/ijms23168886