
Changing Landscape of Cancer Vaccines—Novel Proteomics Platform for New Antigen Compositions




Abstract
1. Introduction
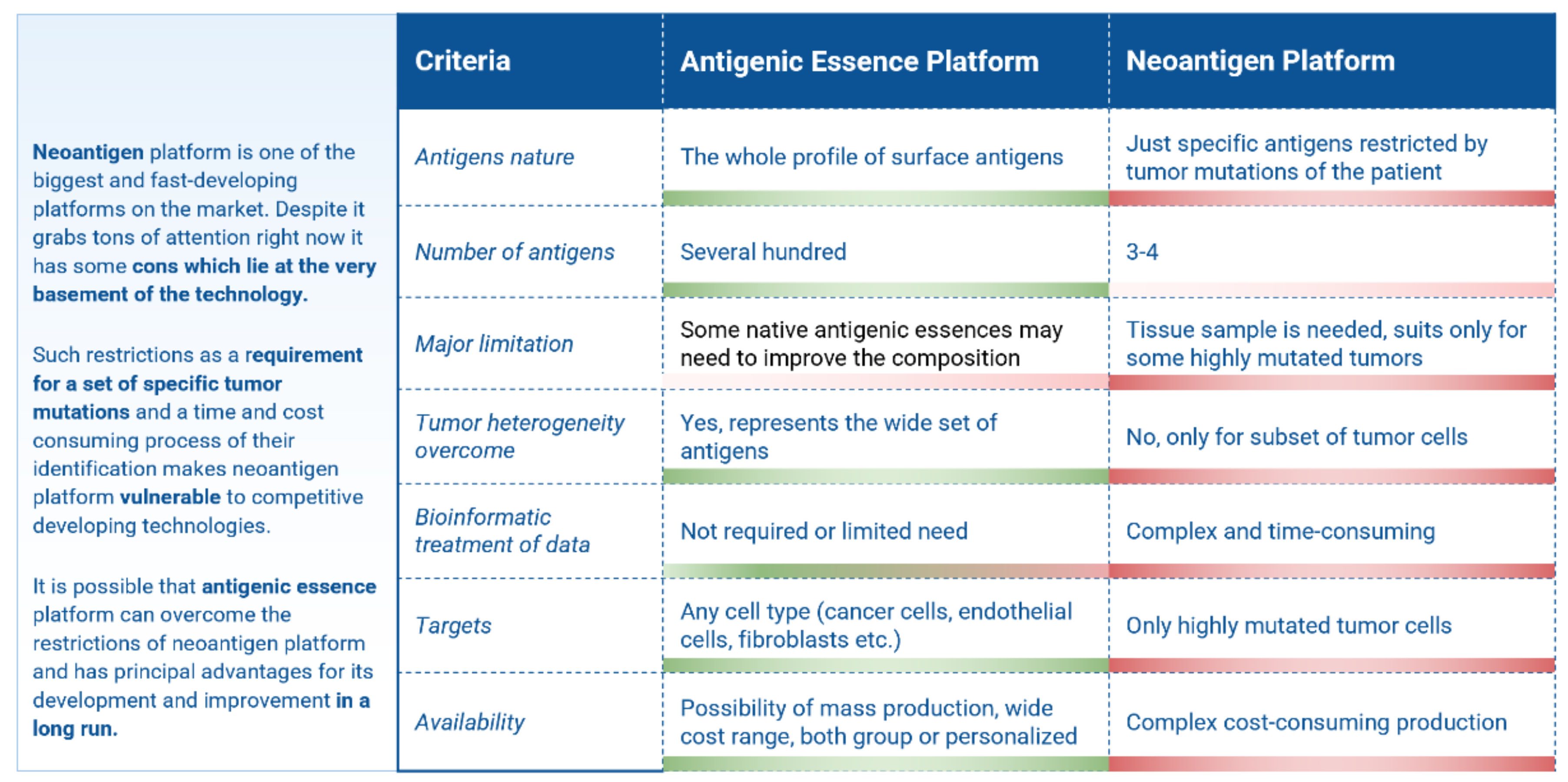
- A clear immunological basis, mechanism of action, and full-fledged source of the entire variety of native antigens are persistent drivers behind the creation of cellular cancer vaccines.
- Despite the development of many cell-based cancer vaccines, they do not pass clinical trials. The high activity of this direction is explained by the fact that cell-based vaccines are promising in their clinical trials but fail to meet the stringent requirements of regulators in terms of efficiency.
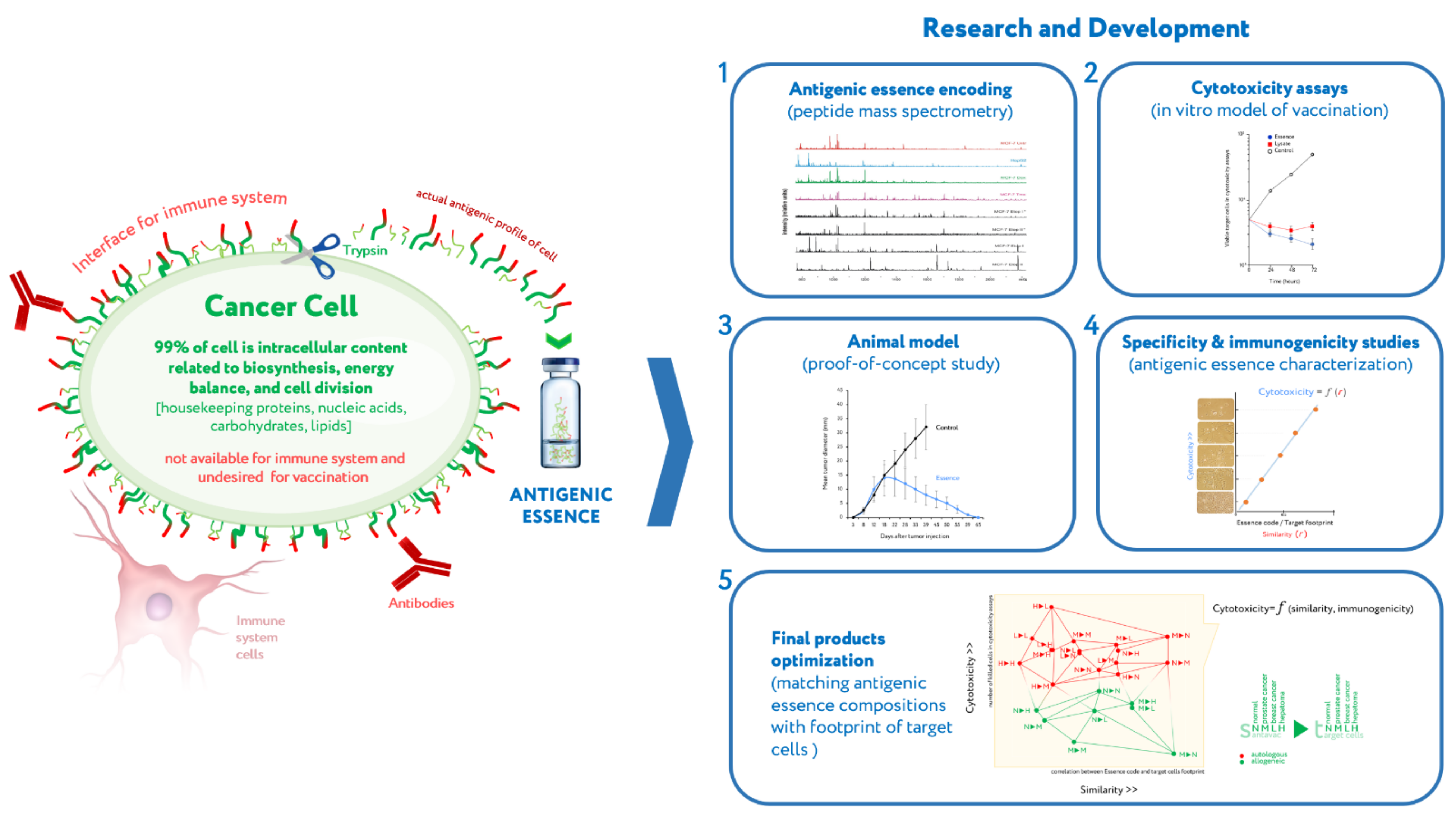
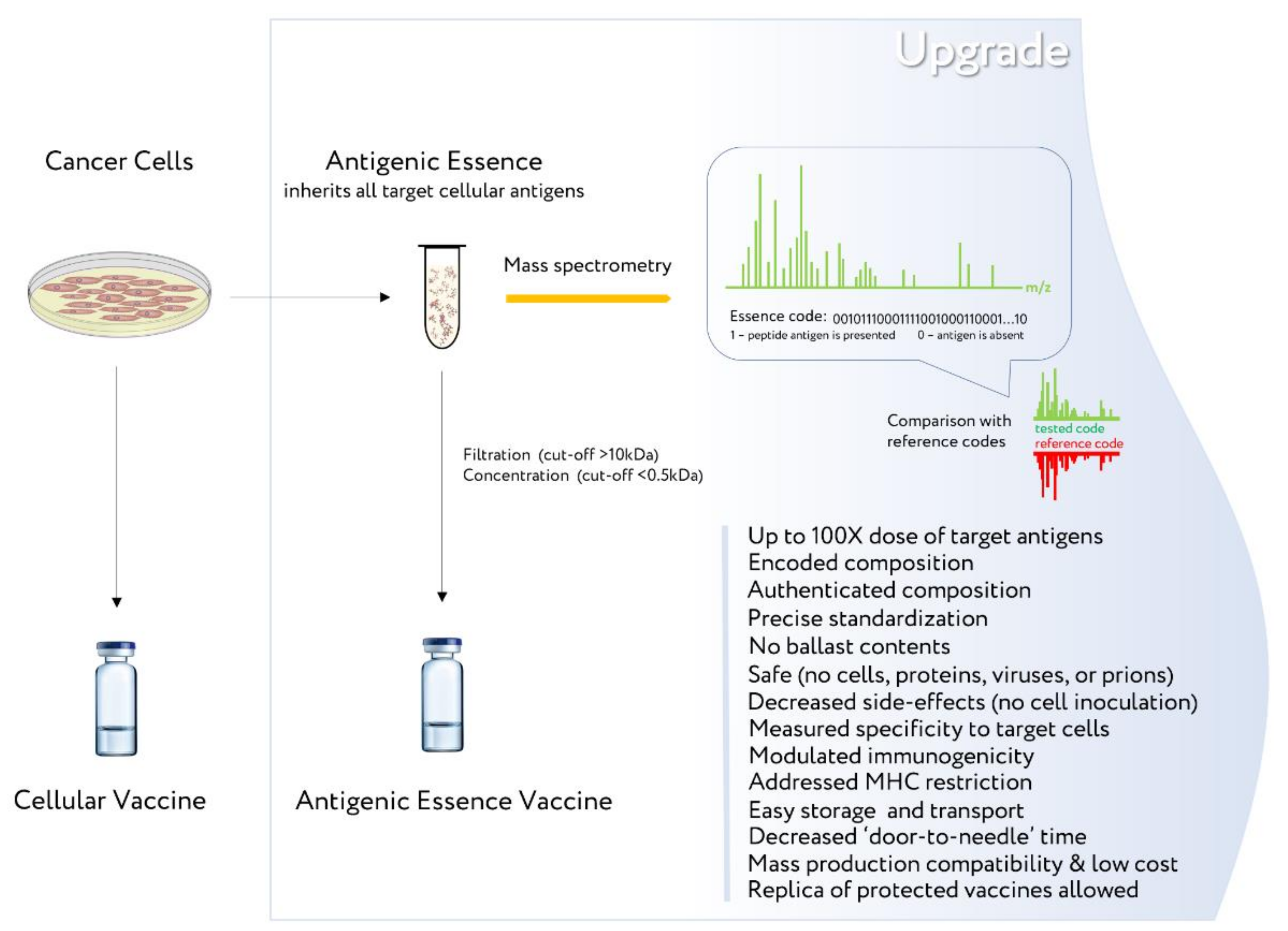
- Upgrading the existing cellular vaccines to address their shortcomings may allow developers to overcome common stumbling points and revitalize the field of cancer vaccines.
2. List of Vaccines Suitable for the Upgrade
3. Limitations
4. Legal and Ethical Aspects of Cellular Vaccines Upgrade
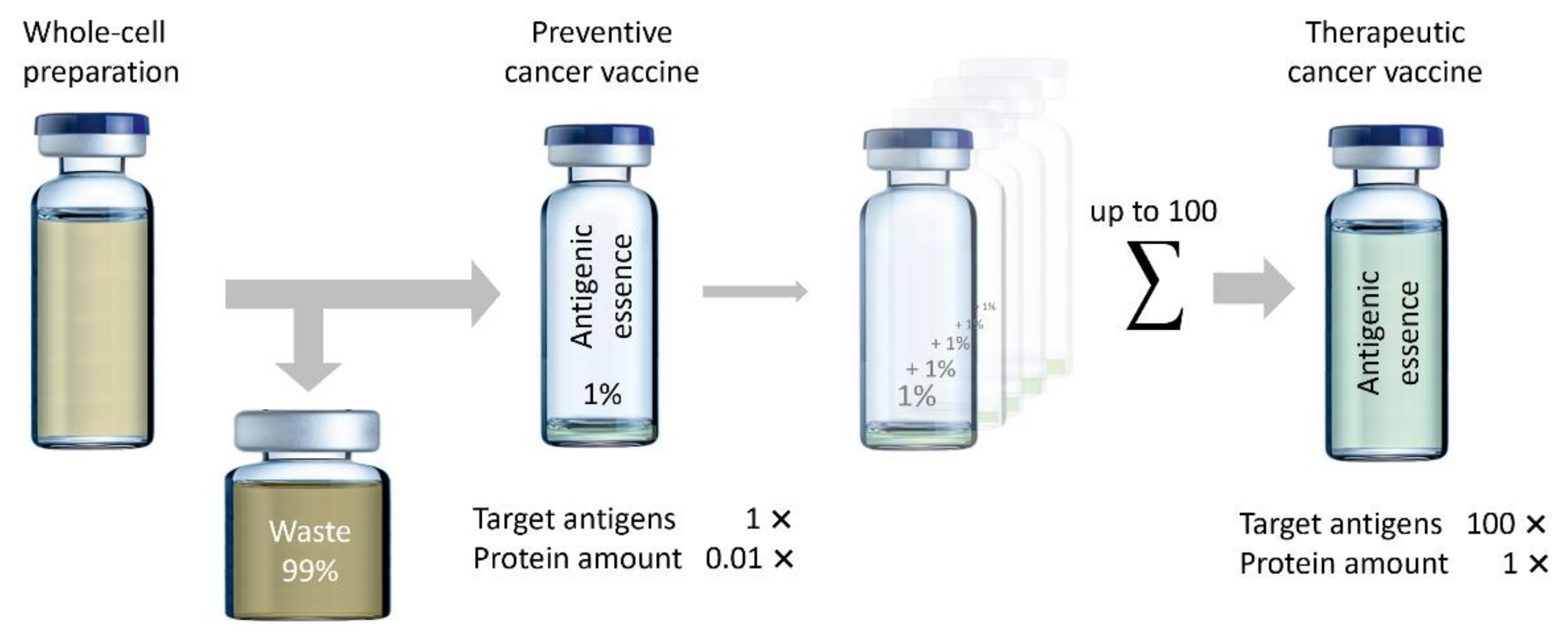
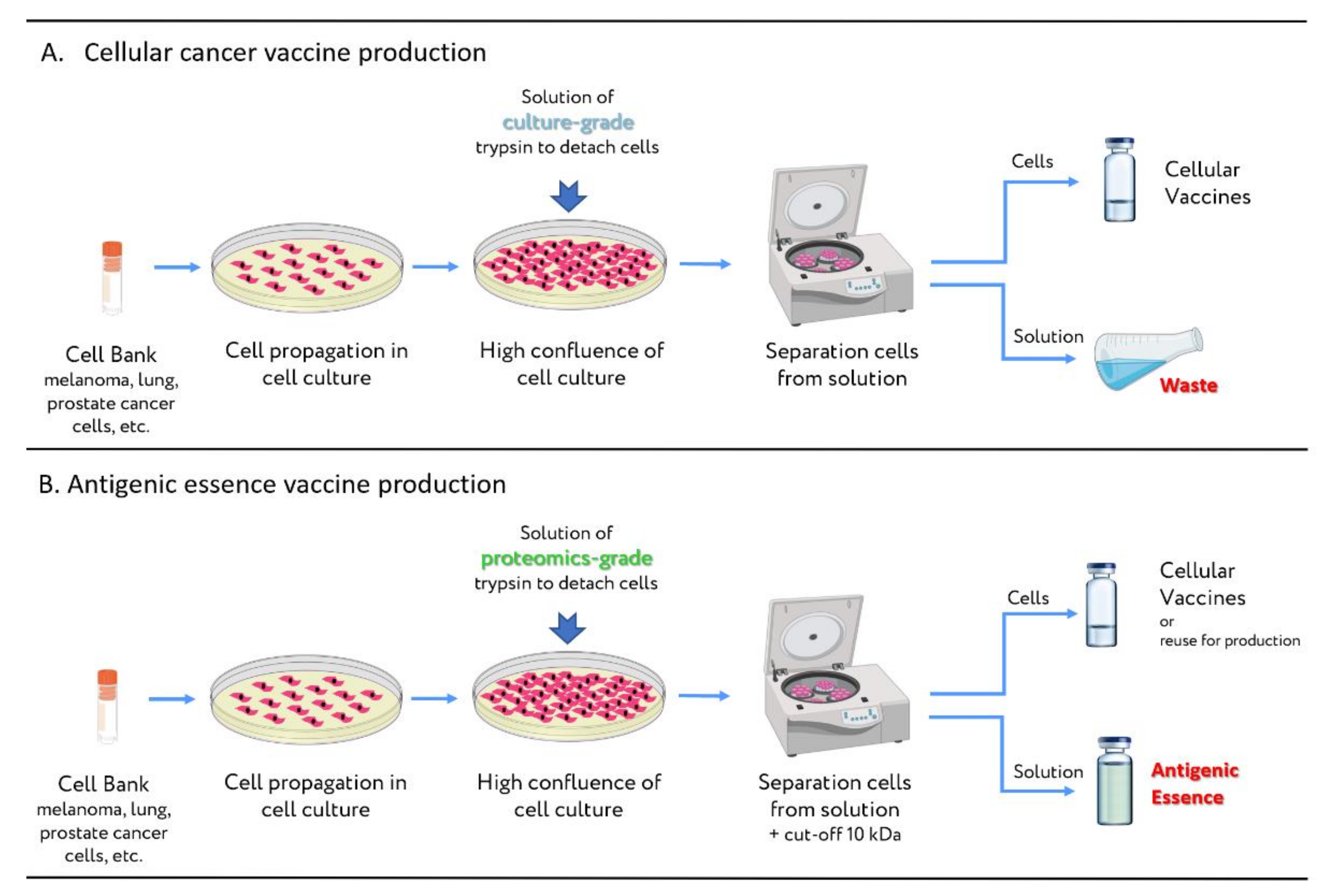
5. Manufacturing
6. Consortium
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berger, M.; Kreutz, F.T.; Horst, J.L.; Baldi, A.C.; Koff, W.J. Phase I study with an autologous tumor cell vaccine for locally advanced or metastatic prostate cancer. J. Pharm. Pharm. Sci. 2007, 10, 144–152. [Google Scholar] [PubMed]
- Nemunaitis, J.; Sterman, D.; Jablons, D.; Smith, J.W.; Fox, B.; Maples, P.; Hamilton, S.; Borellini, F.; Lin, A.; Morali, S.; et al. Granulocyte-macrophage colony-stimulating factor gene-modified autologous tumor vaccines in non-small-cell lung cancer. J. Natl. Cancer Inst. 2004, 96, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Rüttinger, D.; van den Engel, N.K.; Winter, H.; Schlemmer, M.; Pohla, H.; Grützner, S.; Wagner, B.; Schendel, D.J.; Fox, B.A.; Jauch, K.W.; et al. Adjuvant therapeutic vaccination in patients with non-small cell lung cancer made lymphopenic and reconstituted with autologous PBMC: First clinical experience and evidence of an immune response. J. Transl. Med. 2007, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Schulof, R.S.; Mai, D.; Nelson, M.A.; Paxton, H.M.; Cox, J.W., Jr.; Turner, M.L.; Mills, M.; Hix, W.R.; Nochomovitz, L.E.; Peters, L.C. Active specific immunotherapy with an autologous tumor cell vaccine in patients with resected non-small cell lung cancer. Mol. Biother. 1988, 1, 30–36. [Google Scholar] [PubMed]
- De Weger, V.A.; Turksma, A.W.; Voorham, Q.J.M.; Euler, Z.; Bril, H.; Van Den Eertwegh, A.J.; Bloemena, E.; Pinedo, H.M.; Vermorken, J.B.; Van Tinteren, H.; et al. Clinical effects of adjuvant active specific immunotherapy differ between patients with microsatellite-stable and microsatellite-instable colon cancer. Clin. Cancer Res. 2012, 18, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Hanna, M.G.; Hoover, H.C.; Vermorken, J.B.; Harris, J.E.; Pinedo, H.M. Adjuvant active specific immunotherapy of stage II and stage III colon cancer with an autologous tumor cell vaccine: First randomized phase III trials show promise. Vaccine 2001, 19, 2576–2582. [Google Scholar] [CrossRef]
- Harris, J.E.; Ryan, L.; Hoover, H.C.; Stuart, R.K.; Oken, M.M.; Benson, A.B.; Mansour, E.; Haller, D.G.; Manola, J.; Hanna, M.G. Adjuvant active specific immunotherapy for stage II and III colon cancer with an autologous tumor cell vaccine: Eastern Cooperative Oncology Group study E5283. J. Clin. Oncol. 2000, 18, 148–157. [Google Scholar] [CrossRef]
- Ockert, D.; Schirrmacher, V.; Beck, N.; Stoelben, E.; Ahlert, T.; Flechtenmacher, J.; Hagmüller, E.; Buchcik, R.; Nagel, M.; Saeger, H.D. Newcastle disease virus-infected intact autologous tumor cell vaccine for adjuvant active specific immunotherapy of resected colorectal carcinoma. Clin. Cancer Res. 1996, 2, 21–28. [Google Scholar] [CrossRef]
- Baars, A.; Van Riel, J.M.G.H.; Cuesta, M.A.; Jaspars, E.H.; Pinedo, H.M.; Van den Eertwegh, A.J.M. Metastasectomy and active specific immunotherapy for a large single melanoma metastasis. Hepatogastroenterology 2002, 49, 691–693. [Google Scholar]
- Berd, D.; Maguire, H.C.; McCue, P.; Mastrangelo, M.J. Treatment of metastatic melanoma with an autologous tumor-cell vaccine: Clinical and immunologic results in 64 patients. J. Clin. Oncol. 1990, 8, 1858–1867. [Google Scholar] [CrossRef]
- Méndez, R.; Ruiz-Cabello, F.; Rodríguez, T.; Del Campo, A.; Paschen, A.; Schadendorf, D.; Garrido, F. Identification of different tumor escape mechanisms in several metastases from a melanoma patient undergoing immunotherapy. Cancer Immunol. Immunother. 2007, 56, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Seigne, J.; Diaz, J.; Muro-Cacho, C.; Extermann, M.; Farmelo, M.J.; Friberg, M.; Alsarraj, M.; Mahany, J.J.; Pow-Sang, J.; et al. Phase I trial of a B7-1 (CD80) gene modified autologous tumor cell vaccine in combination with systemic interleukin-2 in patients with metastatic renal cell carcinoma. J. Urol. 2002, 167, 1995–2000. [Google Scholar] [CrossRef]
- Fishman, M.; Hunter, T.B.; Soliman, H.; Thompson, P.; Dunn, M.; Smilee, R.; Farmelo, M.J.; Noyes, D.R.; Mahany, J.J.; Lee, J.H.; et al. Phase II trial of B7-1 (CD-86) transduced, cultured autologous tumor cell vaccine plus subcutaneous interleukin-2 for treatment of stage IV renal cell carcinoma. J. Immunother. 2008, 31, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, Y.; Kono, T.; Yasumoto, R.; Kishimoto, T.; Wang, C.Y.; Haas, G.P.; Nishisaka, N. Antitumor effect on murine renal cell carcinoma by autologous tumor vaccines genetically modified with granulocyte-macrophage colony-stimulating factor and interleukin-6 cells. J. Immunother. 2001, 24, 205–211. [Google Scholar] [CrossRef]
- Copier, J.; Dalgleish, A. Whole-cell vaccines: A failure or a success waiting to happen? Curr. Opin Mol. Ther. 2010, 12, 14–20. [Google Scholar] [PubMed]
- Bodey, B.; Bodey, B.J.; Siegel, S.E.; Kaiser, H.E. Failure of cancer vaccines: The significant limitations of this approach to immunotherapy. Anticancer Res. 2000, 20, 2665–2676. [Google Scholar] [PubMed]
- Tan, A.C.L.; Goubier, A.; Kohrt, H.E. A quantitative analysis of therapeutic cancer vaccines in phase 2 or phase 3 trial. J. Immunother. Cancer 2015, 3, 48. [Google Scholar] [CrossRef]
- Lokhov, P.G.; Balashova, E.E. Antigenic essence: Upgrade of cellular cancer vaccines. Cancers 2021, 13, 774. [Google Scholar] [CrossRef]
- Lokhov, P.G. Method for Producing an Antitumoral Vaccine Based on Surface Endothelial Cell Antigens. U.S. Patent No. 9844586, 19 December 2007. [Google Scholar]
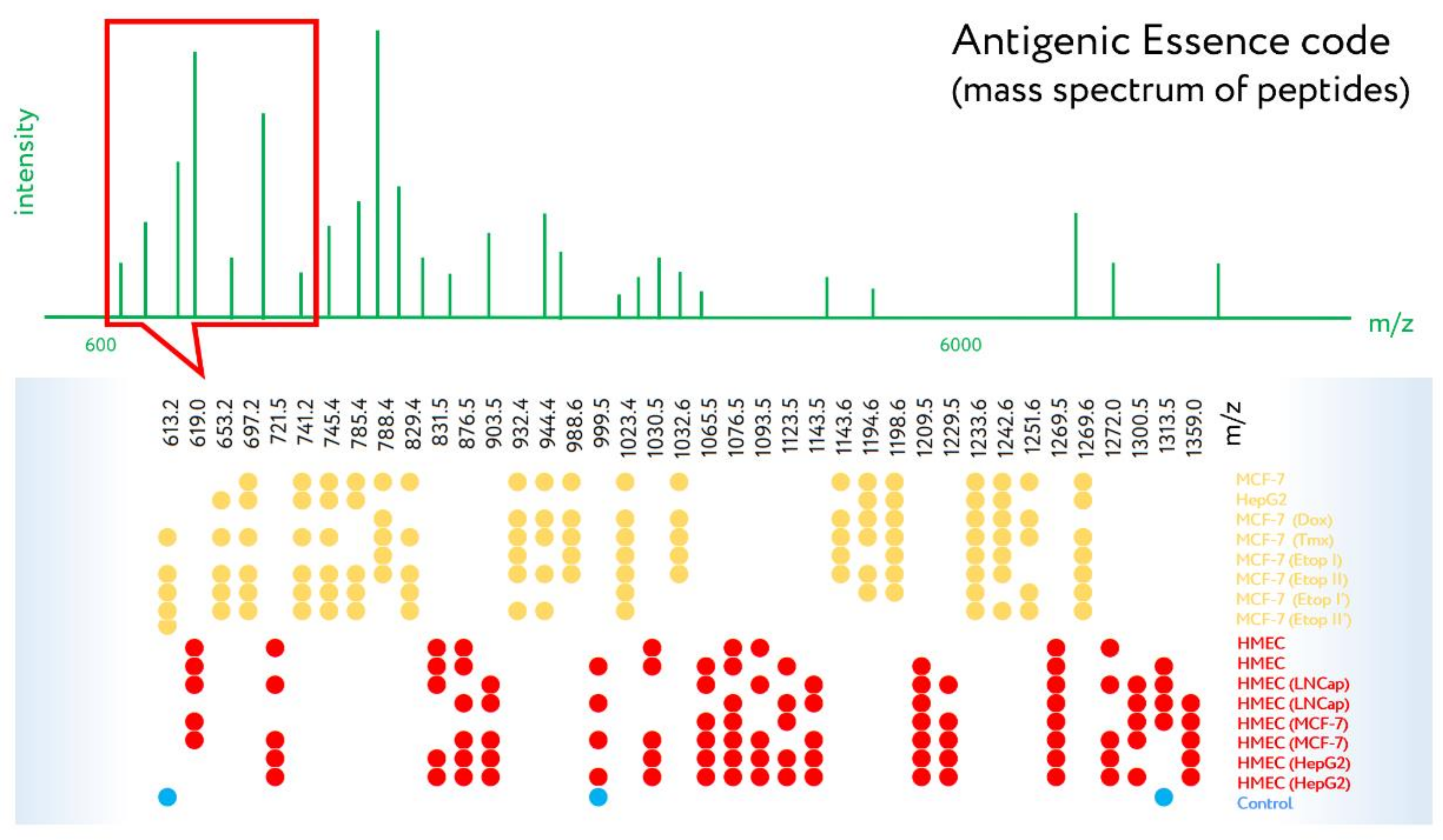
- Lokhov, P.; Balashova, E.; Dashtiev, M. Cell proteomic footprint. Rapid Commun. Mass Spectrom. 2009, 23, 680–682. [Google Scholar] [CrossRef]
- Lokhov, P.G.; Balashova, E.E. Cellular cancer vaccines: An update on the development of vaccines generated from cell surface antigens. J. Cancer 2010, 1, 230–241. [Google Scholar] [CrossRef]
- Balashova, E.E.; Lokhov, P.G. Proteolytically-cleaved fragments of cell-surface proteins from live tumor cells stimulate anti-tumor immune response in vitro. J. Carcinog. Mutagen. 2010, 1, 1–3. [Google Scholar] [CrossRef]
- Balashova, E.E.; Lokhov, P.G. Proteolytically-cleaved fragments of cell surface proteins stimulate a cytotoxic immune response against tumor-activated endothelial cells in vitro. J. Cancer Sci. Ther. 2010, 2, 126–131. [Google Scholar] [CrossRef]
- Lokhov, P.G.; Balashova, E.E. Universal cancer vaccine: An update on the design of cancer vaccines generated from endothelial cells. Hum. Vaccin. Immunother. 2013, 9, 1549–1552. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lokhov, P.G.; Balashova, E.E. Tumor-induced endothelial cell surface heterogeneity directly affects endothelial cell escape from a cell-mediated immune response in vitro. Hum. Vaccin. Immunother. 2013, 9, 198–209. [Google Scholar] [CrossRef][Green Version]
- Lokhov, P.G.; Balashova, E.E. Design of universal cancer vaccines using natural tumor vessel-specific antigens (SANTAVAC). Hum. Vaccin. Immunother. 2015, 11, 689–698. [Google Scholar] [CrossRef]
- Lokhov, P.G.; Balashova, E.E. Allogeneic antigen composition for preparing universal cancer vaccines. J. Immunol. Res. 2016, 2016, 5031529. [Google Scholar] [CrossRef]
- Lokhov, P.G.; Mkrtichyan, M.; Mamikonyan, G.; Balashova, E.E. SANTAVAC: Summary of research and development. Vaccines 2019, 7, 186. [Google Scholar] [CrossRef]
- Balashova, E.E.; Dashtiev, M.I.; Lokhov, P.G. Proteomic footprinting of drug-treated cancer cells as a measure of cellular vaccine efficacy for the prevention of cancer recurrence. Mol. Cell. Proteom. 2012, 11, M111.01448. [Google Scholar] [CrossRef]
- Habal, N.; Gupta, R.K.; Bilchik, A.J.; Yee, R.; Leopoldo, Z.; Ye, W.; Elashoff, R.M.; Morton, D.L. CancerVax, an allogeneic tumor cell vaccine, induces specific humoral and cellular immune responses in advanced colon cancer. Ann. Surg. Oncol. 2001, 8, 389–401. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Claessen, A.M.E.; Van Tinteren, H.; Gall, H.E.; Ezinga, R.; Meijer, S.; Scheper, R.J.; Meijer, C.J.L.M.; Bloemena, E.; Ransom, J.H.; et al. Active specific immunotherapy for stage II and stage III human colon cancer: A randomised trial. Lancet 1999, 353, 345–350. [Google Scholar] [CrossRef]
- Emens, L.A.; Armstrong, D.; Biedrzycki, B.; Davidson, N.; Davis-Sproul, J.; Fetting, J.; Jaffee, E.; Onners, B.; Piantadosi, S.; Reilly, R.T.; et al. A phase I vaccine safety and chemotherapy dose-finding trial of an allogeneic GM-CSF-secreting breast cancer vaccine given in a specifically timed sequence with immunomodulatory doses of cyclophosphamide and doxorubicin. Hum. Gene Ther. 2004, 15, 313–337. [Google Scholar] [CrossRef] [PubMed]
- Dols, A.; Smith, J.W.; Meijer, S.L.; Fox, B.A.; Hu, H.M.; Walker, E.; Rosenheim, S.; Moudgil, T.; Doran, T.; Wood, W.; et al. Vaccination of women with metastatic breast cancer, using a costimulatory gene (CD80)-modified, HLA-A2-matched, allogeneic, breast cancer cell line: Clinical and immunological results. Hum. Gene Ther. 2003, 14, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Asquith, J.M.; Leatherman, J.M.; Kobrin, B.J.; Petrik, S.; Laiko, M.; Levi, J.; Daphtary, M.M.; Biedrzycki, B.; Wolff, A.C.; et al. Timed sequential treatment with cyclophosphamide, doxorubicin, and an allogeneic granulocyte-macrophage colony-stimulating factor—Secreting breast tumor vaccine: A chemotherapy dose-ranging factorial study of safety and immune activation. J. Clin. Oncol. 2009, 27, 5911–5918. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Gupta, R.; Petrik, S.; Laiko, M.; Leatherman, J.M.; Asquith, J.M.; Daphtary, M.M.; Garrett-Mayer, E.; Davidson, N.E.; Hirt, K.; et al. A feasibility study of cyclophosphamide, trastuzumab, and an allogeneic GM-CSF-secreting breast tumor vaccine for HER2+ metastatic breast cancer. Cancer Immunol. Res. 2014, 2, 949–961. [Google Scholar] [CrossRef]
- Gückel, B.; Stumm, S.; Rentzsch, C.; Marmé, A.; Mannhardt, G.; Wallwiener, D. A CD80-transfected human breast cancer cell variant induces HER-2/neu-specific T cells in HLA-A*02-matched situations in vitro as well as in vivo. Cancer Immunol. Immunother. 2005, 54, 129–140. [Google Scholar] [CrossRef]
- Berd, D.; Sato, T.; Maguire, H.C.; Kairys, J.; Mastrangelo, M.J. Immunopharmacologic analysis of an autologous, hapten-modified human melanoma vaccine. J. Clin. Oncol. 2004, 22, 403–415. [Google Scholar] [CrossRef]
- Hirschowitz, E.A.; Foody, T.; Hidalgo, G.E.; Yannelli, J.R. Immunization of NSCLC patients with antigen-pulsed immature autologous dendritic cells. Lung Cancer 2007, 57, 365–372. [Google Scholar] [CrossRef]
- Pruitt, S.K.; Kirk, A.D.; Bollinger, R.R.; Marsh, H.C.; Collins, B.H.; Levin, J.L.; Mault, J.R.; Heinle, J.S.; Ibrahim, S.; Rudolph, A.R.; et al. The effect of soluble complement receptor type 1 on hyperacute rejection of porcine xenografts. Transplantation 1994, 57, 363–370. [Google Scholar] [CrossRef]
- Lai, L.; Kolber-Simonds, D.; Park, K.W.; Cheong, H.T.; Greenstein, J.L.; Im, G.S.; Samuel, M.; Bonk, A.; Rieke, A.; Day, B.N.; et al. Production of α-1,3-galactosyltransferase knockout pigs by nuclear transfer cloning. Science 2002, 295, 1089–1092. [Google Scholar] [CrossRef]
- Hirschowitz, E.A.; Mullins, A.; Prajapati, D.; Baeker, T.; Kloecker, G.; Foody, T.; Damron, K.; Love, C.; Yannelli, J.R. Pilot study of 1650-G: A simplified cellular vaccine for lung cancer. J. Thorac. Oncol. 2011, 6, 169–173. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Jahan, T.; Ross, H.; Sterman, D.; Richards, D.; Fox, B.; Jablons, D.; Aimi, J.; Lin, A.; Hege, K. Phase 1/2 trial of autologous tumor mixed with an allogeneic GVAX® vaccine in advanced-stage non-small-cell lung cancer. Cancer Gene Ther. 2006, 13, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Nemunaitis, J.; Dillman, R.O.; Schwarzenberger, P.O.; Senzer, N.; Cunningham, C.; Cutler, J.; Tong, A.; Kumar, P.; Pappen, B.; Hamilton, C.; et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J. Clin. Oncol. 2006, 24, 4721–4730. [Google Scholar] [CrossRef] [PubMed]
- Nemunaitis, J.; Nemunaitis, M.; Senzer, N.; Snitz, P.; Bedell, C.; Kumar, P.; Pappen, B.; Maples, P.B.; Shawler, D.; Fakhrai, H. Phase II trial of Belagenpumatucel-L, a TGF-Β2 antisense gene modified allogeneic tumor vaccine in advanced non small cell lung cancer (NSCLC) patients. Cancer Gene Ther. 2009, 16, 620–624. [Google Scholar] [CrossRef]
- Giaccone, G.; Bazhenova, L.A.; Nemunaitis, J.; Tan, M.; Juhász, E.; Ramlau, R.; Van Den Heuvel, M.M.; Lal, R.; Kloecker, G.H.; Eaton, K.D.; et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur. J. Cancer 2015, 51, 2321–2329. [Google Scholar] [CrossRef]
- Raez, L.E.; Cassileth, P.A.; Schlesselman, J.J.; Sridhar, K.; Padmanabhan, S.; Fisher, E.Z.; Baldie, P.A.; Podack, E.R. Allogeneic vaccination with a B7.1 HLA-A gene-modified adenocarcinoma cell line in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2004, 22, 2800–2807. [Google Scholar] [CrossRef] [PubMed]
- Raez, L.E.; Walker, G.R.; Baldie, P.; Fisher, E.; Gomez, J.E.; Tolba, K.; Santos, E.S.; Podack, E.R. CD8 T cell response in a phase I study of therapeutic vaccination of advanced NSCLC with allogeneic tumor cells secreting endoplasmic reticulum-chaperone gp96-Ig-peptide complexes. Adv. Lung Cancer 2013, 2, 9–18. [Google Scholar] [CrossRef][Green Version]
- Mitchell, M.S.; Kan-Mitchell, J.; Kempf, R.A.; Harel, W.; Shau, H.; Lind, S. Active Specific Immunotherapy for Melanoma: Phase I Trial of Allogeneic Lysates and a Novel Adjuvant. Cancer Res. 1988, 48, 5883–5893. [Google Scholar]
- Vaishampayan, U.; Abrams, J.; Darrah, D.; Jones, V.; Mitchell, M.S. Active immunotherapy of metastatic melanoma with allogeneic melanoma lysates and interferon αlpha. Clin. Cancer Res. 2002, 8, 3696–3701. [Google Scholar]
- Sosman, J.A.; Unger, J.M.; Liu, P.Y.; Flaherty, L.E.; Park, M.S.; Kempf, R.A.; Thompson, J.A.; Terasaki, P.I.; Sondak, V.K. Adjuvant immunotherapy of resected, intermediate-thickness, node-negative melanoma with an allogeneic tumor vaccine: Impact of HLA class I antigen expression on outcome. J. Clin. Oncol. 2002, 20, 2067–2075. [Google Scholar] [CrossRef]
- von Euw, E.M.; Barrio, M.M.; Furman, D.; Levy, E.M.; Bianchini, M.; Peguillet, I.; Lantz, O.; Vellice, A.; Kohan, A.; Chacón, M.; et al. A phase I clinical study of vaccination of melanoma patients with dendritic cells loaded with allogeneic apoptotic/necrotic melanoma cells. Analysis of toxicity and immune response to the vaccine and of IL-10-1082 promoter genotype as predictor of disease. J. Transl. Med. 2008, 6, 6. [Google Scholar] [CrossRef]
- Mordoh, J.; Kairiyama, C.; Bover, L.; Solarolo, E. Allogeneic cells vaccine increases disease-free survival in stage III melanoma patients: A non randomized phase II study. Medicina 1997, 57, 421–427. [Google Scholar] [PubMed]
- Barrio, M.M.; De Motta, P.T.; Kaplan, J.; Von Euw, E.M.; Bravo, A.I.; Chacón, R.D.; Mordoh, J. A phase I study of an allogeneic cell vaccine (VACCIMEL) with GM-CSF in melanoma patients. J. Immunother. 2006, 29, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Palucka, A.K.; Ueno, H.; Connolly, J.; Kerneis-Norvell, F.; Blanck, J.P.; Johnston, D.A.; Fay, J.; Banchereau, J. Dendritic cells loaded with killed allogeneic melanoma cells can induce objective clinical responses and MART-1 specific CD8+ T-cell immunity. J. Immunother. 2006, 29, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Berd, D. M-Vax: An autologous, hapten-modified for human cancer. Expert Rev. Vaccines 2004, 2, 335–342. [Google Scholar] [CrossRef]
- Ribas, A.; Camacho, L.H.; Lee, S.M.; Hersh, E.M.; Brown, C.K.; Richards, J.M.; Rodriguez, M.J.; Prieto, V.G.; Glaspy, J.A.; Oseguera, D.K.; et al. Multicenter phase II study of matured dendritic cells pulsed with melanoma cell line lysates in patients with advanced melanoma. J. Transl. Med. 2010, 8, 89. [Google Scholar] [CrossRef]
- Aris, M.; Bravo, A.I.; Pampena, M.B.; Blanco, P.A.; Carri, I.; Koile, D.; Yankilevich, P.; Levy, E.M.; Barrio, M.M.; Mordoh, J. Changes in the TCRβ repertoire and tumor immune signature from a cutaneous melanoma patient immunized with the CSF-470 vaccine: A case report. Front. Immunol. 2018, 9, 955. [Google Scholar] [CrossRef]
- Mordoh, J.; Pampena, M.B.; Aris, M.; Blanco, P.A.; Lombardo, M.; von Euw, E.M.; Mac Keon, S.; Crow, M.Y.; Bravo, A.I.; O’Connor, J.M.; et al. Phase II study of adjuvant immunotherapy with the CSF-470 vaccine plus Bacillus Calmette-Guerin plus recombinant human granulocyte macrophage-colony stimulating factor vs medium-dose Interferon alpha 2B in Stages IIB, IIC, and III cutaneous melanoma patie. Front. Immunol. 2017, 8, 625. [Google Scholar] [CrossRef]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef]
- Hopkins, A.C.; Yarchoan, M.; Durham, J.N.; Yusko, E.C.; Rytlewski, J.A.; Robins, H.S.; Laheru, D.A.; Le, D.T.; Lutz, E.R.; Jaffee, E.M. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight 2018, 3, 13. [Google Scholar] [CrossRef]
- Eric, L.; Yeo, C.J.; Lillemoe, K.D.; Biedrzycki, B.; Kobrin, B.; Herman, J.; Sugar, E.; Piantadosi, S.; Cameron, J.L.; Solt, S.; et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma: A phase II trial of safety, efficacy, and immune activation. Ann. Surg. 2011, 253, 328–335. [Google Scholar] [CrossRef]
- Simons, J.W.; Carducci, M.A.; Mikhak, B.; Lim, M.; Biedrzycki, B.; Borellini, F.; Clift, S.M.; Hege, K.M.; Ando, D.G.; Piantadosi, S.; et al. Phase I/II trial of an allogeneic cellular immunotherapy in hormone-naïve prostate cancer. Clin. Cancer Res. 2006, 12, 3394–3401. [Google Scholar] [CrossRef] [PubMed]
- Small, E.J.; Sacks, N.; Nemunaitis, J.; Urba, W.J.; Dula, E.; Centeno, A.S.; Nelson, W.G.; Ando, D.; Howard, C.; Borellini, F.; et al. Granulocyte macrophage colony-stimulating factor-secreting allogeneic cellular immunotherapy for hormone-refractory prostate cancer. Clin. Cancer Res. 2007, 13, 3883–3891. [Google Scholar] [CrossRef] [PubMed]
- Van den Eertwegh, A.J.M.; Versluis, J.; Van den Berg, H.P.; Santegoets, S.J.A.M.; Van Moorselaar, R.J.A.; Van der Sluis, T.M.; Gall, H.E.; Harding, T.C.; Jooss, K.; Lowy, I.; et al. Combined immunotherapy with granulocyte-macrophage colony-stimulating factor-transduced allogeneic prostate cancer cells and ipilimumab in patients with metastatic castration-resistant prostate cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 509–517. [Google Scholar] [CrossRef]
- Higano, C.S.; Corman, J.M.; Smith, D.C.; Centeno, A.S.; Steidle, C.P.; Gittleman, M.; Simons, J.W.; Sacks, N.; Aimi, J.; Small, E.J. Phase 1/2 dose-escalation study of a GM-CSF-secreting, allogeneic, cellular immunotherapy for metastatic hormone-refractory prostate cancer. Cancer 2008, 113, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Doehn, C.; Böhmer, T.; Jocham, D. Technology evaluation: Onyvax-P, Onyvax. Curr. Opin. Mol. Ther. 2005, 7, 511–519. [Google Scholar] [PubMed]
- Brill, T.H.; Kübler, H.R.; Randenborgh, H.V.; Fend, F.; Pohla, H.; Breul, J.; Hartung, R.; Paul, R.; Schendel, D.J.; Gansbacher, B. Allogeneic retrovirally transduced, IL-2- and IFN-γ-secreting cancer cell vaccine in patients with hormone refractory prostate cancer—A phase I clinical trial. J. Gene Med. 2007, 9, 547–560. [Google Scholar] [CrossRef]
- Brill, T.H.; Kübler, H.R.; Pohla, H.; Buchner, A.; Fend, F.; Schuster, T.; Van Randenborgh, H.; Paul, R.; Kummer, T.; Plank, C.; et al. Therapeutic vaccination with an interleukin-2-interferon-γ-secreting allogeneic tumor vaccine in patients with progressive castration-resistant prostate cancer: A phase I/II trial. Hum. Gene Ther. 2009, 20, 1641–1651. [Google Scholar] [CrossRef]
- Eaton, J.D.; Perry, M.J.A.; Nicholson, S.; Guckian, M.; Russell, N.; Whelan, M.; Kirby, R.S. Allogeneic whole-cell vaccine: A phase I/II study in men with hormone-refractory prostate cancer. BJU Int. 2002, 89, 19–26. [Google Scholar] [CrossRef]
- Schijns, V.E.J.C.; Pretto, C.; Devillers, L.; Pierre, D.; Hofman, F.M.; Chen, T.C.; Mespouille, P.; Hantos, P.; Glorieux, P.; Bota, D.A.; et al. First clinical results of a personalized immunotherapeutic vaccine against recurrent, incompletely resected, treatment-resistant glioblastoma multiforme (GBM) tumors, based on combined allo- and auto-immune tumor reactivity. Vaccine 2015, 33, 2690–2696. [Google Scholar] [CrossRef]
- Westermann, J.; Flörcken, A.; Willimsky, G.; Van Lessen, A.; Kopp, J.; Takvorian, A.; Jöhrens, K.; Lukowsky, A.; Schönemann, C.; Sawitzki, B.; et al. Allogeneic gene-modified tumor cells (RCC-26/IL-7/CD80) as a vaccine in patients with metastatic renal cell cancer: A clinical phase-I study. Gene Ther. 2011, 18, 354–363. [Google Scholar] [CrossRef]
- Smith, B.D.; Kasamon, Y.L.; Kowalski, J.; Gocke, C.; Murphy, K.; Miller, C.B.; Garrett-Mayer, E.; Tsai, H.L.; Qin, L.; Chia, C.; et al. K562/GM-CSF immunotherapy reduces tumor burden in chronic myeloid leukemia patients with residual disease on imatinib mesylate. Clin. Cancer Res. 2010, 16, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Okaji, Y.; Tsuno, N.H.; Tanaka, M.; Yoneyama, S.; Matsuhashi, M.; Kitayama, J.; Saito, S.; Nagura, Y.; Tsuchiya, T.; Yamada, J.; et al. Pilot study of anti-angiogenic vaccine using fixed whole endothelium in patients with progressive malignancy after failure of conventional therapy. Eur. J. Cancer 2008, 44, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Moasser, M.M. The oncogene HER2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469–6487. [Google Scholar] [CrossRef] [PubMed]
- Andersson, B. Studies on the regulation of avidity at the level of the single antibody-forming cell. The effect of antigen dose and time after immunization. J. Exp. Med. 1970, 132, 77–88. [Google Scholar] [CrossRef]
- Billeskov, R.; Beikzadeh, B.; Berzofskya, J.A. The effect of antigen dose on T cell-targeting vaccine outcome. Hum. Vaccines Immunother. 2019, 15, 407–411. [Google Scholar] [CrossRef]
- Billeskov, R.; Wang, Y.; Solaymani-Mohammadi, S.; Frey, B.; Kulkarni, S.; Andersen, P.; Agger, E.; Sui, Y.; Berzofsky, J. Low antigen dose in adjuvant-based vaccination selectively induces CD4 T cells with enhanced functional avidity and protective efficacy. J. Immunol. 2017, 198, 3494–3506. [Google Scholar] [CrossRef]
- Alexander-Miller, M.; Leggatt, G.; Berzofsky, J. Selective expansion of high- or low-avidity cytotoxic T lymphocytes and efficacy for adoptive immunotherapy. Proc. Natl. Acad. Sci. USA 1996, 93, 4102–4107. [Google Scholar] [CrossRef]
- Alexander-Miller, M.; Leggatt, G.; Sarin, A.; Berzofsky, J. Role of antigen, CD8, and cytotoxic T lymphocyte (CTL) avidity in high dose antigen induction of apoptosis of effector CTL. J. Exp. Med. 1996, 184, 485–492. [Google Scholar] [CrossRef]
- Alexander-Miller, M.A.; Derby, M.; Sarin, A.; Henkart, P.; Berzofsky, J. Supraoptimal peptide-major histocompatibility complex causes a decrease in bc1–2 levels and allows tumor necrosis factor alpha receptor II-mediated apoptosis of cytotoxic T lymphocytes. J. Exp. Med. 1998, 188, 1391–1399. [Google Scholar] [CrossRef]
- Derby, M.; Alexander-Miller, M.; Tse, R.; Berzofsky, J. High-avidity CTL exploit two complementary mechanisms to provide better protection against viral infection than low-avidity CTL. J. Immunol. 2001, 166, 1690–1697. [Google Scholar] [CrossRef]
- Zeh, H., 3rd; Perry-Lalley, D.; Dudley, M.; Rosenberg, S.; Yang, J. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J. Immunol. 1999, 162, 989–994. [Google Scholar] [PubMed]
- Yee, C.; Savage, P.; Lee, P.; Davis, M.; Greenberg, P. Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J. Immunol. 1999, 162, 2227–2234. [Google Scholar] [PubMed]
- Billeskov, R.; Lindenstrom, T.; Woodworth, J.; Vilaplana, C.; Cardona, P.; Cassidy, J.; Mortensen, R.; Agger, E.; Andersen, P. High antigen dose is detrimental to post-exposure vaccine protection against tuberculosis. Front. Immunol. 2018, 8, 1973. [Google Scholar] [CrossRef] [PubMed]
- Si, C.; Xu, M.; Lu, M.; Yu, Y.; Yang, M.; Yan, M.; Zhou, L.; Yang, X. In vivo antitumor activity evaluation of cancer vaccines prepared by various antigen forms in a murine hepatocellular carcinoma model. Oncol. Lett. 2017, 14, 7391–7397. [Google Scholar] [CrossRef][Green Version]
- Buckwalter, M.; Srivastava, P. Form of antigen dictates immunity: Irradiated cell vs. whole cell lysate vaccination (48.16). J. Immunol. 2007, 178, S77 LP-S77. [Google Scholar]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018. JAMA J. Am. Med. Assoc. 2020, 323, 844–853. [Google Scholar] [CrossRef]
- Srivastava, S.; Riddell, S.R. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015, 36, 494–502. [Google Scholar] [CrossRef]
- Jaffee, E.M.; Van Dang, C.; Agus, D.B.; Alexander, B.M.; Anderson, K.C.; Ashworth, A.; Barker, A.D.; Bastani, R.; Bhatia, S.; Bluestone, J.A.; et al. Future cancer research priorities in the USA: A Lancet Oncology Commission. Lancet Oncol. 2017, 18, e653–e706. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellular Vaccine (Sponsor, Developer) | Description | Cancer Origin | Phase | Reference/Clinical Trial ID | |
|---|---|---|---|---|---|
| 1 | CanVaxin (CancerVax Corp.; John Wayne Cancer Institute) | Administration of 3 allogeneic melanoma cell lines (M10-V, M24-V, and M101-V pooled in equal amounts) with BCG 1. Median OS 2: 12.9 months. | Colon | I | Habal, Gupta, et al. [30] |
| 2 | OncoVax (Vaccinogen Inc) | Vaccination with autologous colon cancer cells mixed with live BCG. Significant improvement in overall and disease-free survival in the IIIa study. | Colon | III | Vermorken, Claessenet, et al. [31]; NCT02448173 |
| 3 | ONYCR1-3 (ONYvax) | Allogeneic adenocarcinoma cell-based vaccines mixed with BCG or alum adjuvant. | Colon | I/II | NCT00007826 (Arm II and III) |
| 4 | HyperAcute-Breast cancer (NewLink Genetics Corporation) | Genetically modified allogeneic tumor cells expressing the xenoantigen αGal. | Breast | I/II | NCT00090480 |
| 5 | Allogeneic GM-CSF-secreting whole-cell breast cancer vaccine (Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins) | The GM-CSF 3-producing 3SKBR3-7 and 2T47D-V cells combined into a single vaccine formulation. | Breast | I | Emens, Armstrong, et al. [32] |
| 6 | Allogeneic cellular cancer vaccine (Robert W. Franz Cancer Research Center, Earle A. Chiles Research Institute) | Vaccination with MDA-MB-231, an HLA-A2(+), HER2/neu(+) allogeneic breast cancer cell line genetically modified to express the costimulatory molecule CD80 (B7-1). | Breast | I | Dols, Smith, et al. [33]. |
| 7 | Allogeneic cellular cancer vaccine (Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins; NCI) | Allogeneic GM-CSF-secreting breast cancer cell lines (SKBR3 and T47D) alone or with CY and DOX. | Breast | I | Emens, Asquith, et al. [34]; NCT00093834. |
| 8 | Allo GM-CSF-secreting vaccine (Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins) | Allogeneic GM-CSF-secreting breast cancer vaccine (two parts 2T47D-V and one part 3SKBR3-7) with trastuzumab and cyclophosphamide. | Breast | II | Chen, Gupta, et al. [35]; NCT00399529 |
| 9 | KS2422-vacc (University Hospital Tuebingen; Paul Ehrlich Institute, Langen) | Allogeneic breast cancer cell line, KS24.22, genetically modified to express CD80 and Her-2/neu. | Breast | I | Gückel, Stumm, et al. [36]; NCT01127074 |
| 10 | BriaVax (BriaCell Therapeutics Corporation) | BriaVax is derived from a human breast cancer cell line (SV-BR-1-GM) that expresses the protein Her2/neu, which is overexpressed in some epithelial cancers like breast and ovarian cancers. It was designed to produce and secrete GM-CSF. | Breast Ovary | I/II | NCT03066947 |
| 11 | O-Vax (AVAX Technologies) | DNP-modified autologous ovarian tumor cell vaccine. Median OS: 22.7 months. | Ovary | I/ II | Berd, Sato, et al. [37]; NCT00660101 |
| 12 | Autologous DC vaccine (Edward Hirschowitz; University of Kentucky; NCI) | Autologous dendritic cells loaded with allogeneic non-small cell lung cancer cells (NSCLC 4 cell lines that over-expresses Her2/neu, CEA, WT1, Mage2, and survivin). | Lung | II | Hirschowitz, Foody, et al. [38]; NCT0010311 |
| 13 | MelCancerVac (Herlev Hospital; University of Copenhagen) | Autologous dendritic cells pulsed with allogeneic melanoma cell lysate (MelCancerVac) in combination with the Cox-2 inhibitor of celecoxib. | Lung | II | NCT00442754 |
| 14 | HyperAcute-Lung Cancer (tergenpumatucel-L) (NewLink Genetics Corporation; NCI) | The vaccine consists of genetically modified allogeneic NSCLC tumor cells with the αGal moiety on the cell surfaces. | Lung | I/II | Pruitt, Kirk, et al. [39]; Lai, Kolber-Simonds, et al. [40]; NCT00073398 |
| 15 | 1650-G vaccine (University of Kentucky) | Allogeneic NSCLC cell line 1650 mixed with GM-CSF. | Lung | I/II | Hirschowitz, Mullins, et al. [41]; NCT00654030 |
| 16 | Viagenpumatucel-L (HS-110) (Heat Biologics) | Allogeneic vaccine derived from irradiated human lung cancer cells genetically engineered to continually secrete gp96-Ig. | Lung | I/II | NCT02117024 |
| 17 | GVAX lung cancer vaccine (Southwest Oncology Group; NCI) | K562 cells genetically modified to secrete GM-CSF combined with autologous lung tumor cells. Median OS: 5.4 months. | Lung | I/II | Nemunaitis, Jahan, et al. [42]; NCT00074295 |
| 18 | Belagenpumatucel-L (Lucanix) (NovaRx Corporation) | Administration of Belagenpumatucel-L (a cocktail of 4 irradiated allogeneic NSCLC cell lines transfected with TGF-β2 antisense transgene). Median OS in II trial: 14.5 months. | Lung | II/III | Nemunaitis, Dillman et al. [43]; Nemunaitis, Nemunaitis, et al. [44]; Giaccone, Bazhenova, et al. [45] NCT01058785; NCT00676507 |
| 19 | Allogeneic tumor cell-based vaccine (H. Lee Moffitt Cancer Center and Research Institute; NCI; NIH) | Allogeneic lung adenocarcinoma cells are combined with a bystander K562 cell line transfected with hCD40L and hGM-CSF. | Lung | II | NCT00601796 |
| 20 | Allogeneic B7.1/HLA-A1 (University of Miami) | Administration of irradiated whole-cell (AD100) allogeneic vaccine transfected to express B7.1 along with either HLA-A1 or HLA-A2. Median OS: 18 months. | Lung | I/II | Raez, Cassileth, et al. [46]; NCT00534209 |
| 21 | Allogeneic vaccine (University of Miami) | Allogeneic tumor cells secreting endoplasmic reticulum-chaperone gp96-Ig-peptide complexes. Median OS: 16.5 months. | Lung | I | Raez, Walker, et al. [47]. |
| 22 | CanVaxin (CancerVax Corporation) | Allogeneic whole-cell vaccine consisting of three melanoma lines combined with BCG as an adjuvant. In phase II median OS and 5-year rate of survival were significantly higher in stage III melanoma. | Melanoma | III | NCT00052156; NCT00052130 |
| 23 | Melacine (Corixa Corporation) | Administration of Mel-D and Mel-S cell lysates (Melacine) with DETOX. | Melanoma | I | Mitchell, Kanmitchell, et al. [48] |
| 24 | Melacine (Corixa Corporation) | Administration of Melacine with CY and IFN-α i.v. after 4 doses of Melacine. Median OS: 12.5 months. | Melanoma | II/III | Vaishampayan, Abrams, et al. [49]; NCT00002767 |
| 25 | Melacine (Corixa Corporation) | Melacine administration. Investigation of the impact of class I antigen expression on relapse-free survival after adjuvant therapy with the vaccine (5 years relapse-free survival). | Melanoma | III | Sosman, Unger, et al. [50] |
| 26 | Autologous DC vaccine (Centro de Investigaciones Oncológicas FUCA et al.) | Ex vivo loading of autologous DCs with antigens from apoptotic/necrotic allogeneic melanoma cells and subsequent adoptive transfer. Apoptotic-necrotic (Apo-Nec) tumor cells were prepared as a batch of four cell lines (MEL-XY1; MEL-XY2; MEL-XY3 and MEL-XX4). | Melanoma | I | Von Euw, Barrio, et al. [51]. |
| 27 | A2/4-1BBL melanoma vaccine (Hadassah Medical Organization) | Vaccination with irradiated M20/A2B cells. | Melanoma | II/III | NCT01898039; NCT01861938 |
| 28 | VACCIMEL (Laboratorio Pablo Cassará S.R.L. et al.) | Administration of Cyp followed by VACCIMEL (a mixture of 3 allogeneic cell lines IIB-MEL-J, IIB-MEL-LES, and IIB-MEL-IAN). | Melanoma | II/III | Mordoh, Kairiyama, et al. [52]; NCT01729663 |
| 29 | VACCIMEL (Laboratorio Pablo Cassará S.R.L. et al.) | Administration of VACCIMEL with rhGM-CSF. | Melanoma | I | Barrio, De Motta, et al. [53] |
| 30 | Dendritic cell vaccine (Baylor Institute for Immunology Research) | Administration of autologous monocyte-derived DCs loaded ex vivo with killed allogeneic Colo829 melanoma cells and activated with GM-CSF, IL-4, TNF-α, and CD40 ligand. Median OS: 22.5 months. | Melanoma | I | Palucka, Ueno, et al. [54] |
| 31 | BIBW2 component A and B (Boehringer Ingelheim) | Allogeneic tumor vaccine BIWB 2 containing melanoma cells transfected with the human IL-2 gene. | Melanoma | I | NCT02203864 |
| 32 | M-Vax (DNP-VACC) (AVAX Tech.) | DNP-modified autologous tumor cells. 5-year OS rate was 46%. | Melanoma | I/ II | David Berd [55]. |
| 33 | Autologous dendritic cell-allogeneic melanoma tumor cell lysate vaccine (Jonsson Comprehensive Cancer Center; NCI) | Matured dendritic cells pulsed ex vivo with 3 melanoma cell line lysates (IDD-3). | Melanoma | II | Ribas, Camacho, et al. [56]; NCT00107159 |
| 34 | CSF470 Vaccine (Laboratorio Pablo Cassará S.R.L.) | Allogeneic 4 lethally irradiated cutaneous melanoma cell lines (MEL-XY1, MEL-XY2, MEL-XY3, and MEL-XX4). | Melanoma | II/III | Aris, Bravo, et al. [57]; Mordoh, Pampena, et al. [58] |
| 35 | GVAX (Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins) | Allogeneic pancreatic tumor cells transfected with a GM-CSF gene administered in combination with Ipilimumab (an antibody that blocks negative signals to T cells). | Pancreas | I | Le, Lutz, et al. [59]; Hopkins, Yarchoan, et al. [60] |
| 36 | PANC 10.05 and PANC 6.03 vaccines (Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins; Viragh Foundation) | A pancreatic vaccine secreting a GM-CSF and consists of equal numbers of pancreatic cancer cells (Panc 6.03) and (Panc 10.05) into a single vaccine. | Pancreas | II | NCT01088789 |
| 37 | GVAX (Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins; NCI) | Allogenic pancreatic tumor cell vaccine transfected with GM-CSF used with cyclophosphamide. | Pancreas | II | NCT00727441 |
| 38 | GVAX | Irradiated GM-CSF transfected allogeneic whole-cell tumor lines. Two pancreas cancer cell lines (PANC 10.05 and PANC 6.03) were combined. The median disease-free survival is 17.3 months with a median OS of 24.8 months. | Pancreas | I/II | Lutz, Yeo, et al. [61]; NCT00084383 |
| 39 | GVAX | The first administration of an allogeneic prostate cancer cell lines (PC3 and LNCap) modified to secrete GM-CSF. | Prostate | I/II | Simons, Carducci, et al. [62] |
| 40 | GVAX | Administration of allogeneic prostate cancer cell lines (PC3 and LNCap) modified to secrete GM-CSF. Median OS: 34.9 months (high dose); 24.0 months (low dose). | Prostate | I/II | Small, Sacks, et al. [63] |
| 41 | GVAX | Administration of GVAX plus ipilimumab (fully human IgG CTLA-4 blocking Ab). Median OS: 29.2 months. | Prostate | I | Van den Eertwegh, Versluis, et al. [64] |
| 42 | GVAX | Administration of 2 allogeneic prostate-carcinoma cell lines (PC3 and LNCap) modified to secrete GM-CSF. Median OS: 35.0 months (high-dose); 20.0 months (mid-dose); 23.1 months (low-dose). | Prostate | I/II | Higano, Corman, Smith, et al. [65] |
| 43 | ONY-P1 (ONYVAX-P) (GemVax & Kael Co., Ltd.; NCI) | The vaccine is derived from three irradiated allogeneic prostate cancer cell lines that represent different stages of prostate cancer. | Prostate | II | Doehn, Torsten, et al. [66] NCT00514072 |
| 44 | Allogeneic vaccine (Institut für Experimentelle Onkologie und Therapieforschung) | Administration of LNCaP modified using retroviral vector to secrete IL-2 and IFN-γ. Median OS: 32 months. | Prostate | I/II | Brill, Kuebler, et al. [67] Brill, Kuebler Pohla, et al. [68] |
| 45 | Allogeneic whole-cell vaccine (OnyVax Ltd.) | Administration of whole-cell vaccine consisting of a mixture of 3 prostate cancer cell lines (Pr1-4) along with Mycobacterium vaccine (SRL172). | Prostate | I/II | Eaton, Perry, et al. [69] |
| 46 | DC-APCC (Mayo Clinic; NCI) | Allogenic whole prostate carcinoma cell (APCC) vaccine co-administered with ex vivo generated dendritic cells. | Prostate | II | NCT00814892 |
| 47 | Neuroblastoma vaccine (Baylor College of Medicine; Center for Cell and Gene Therapy, Baylor College of Medicine) | Vaccination with unmodified SKNLP, with gene-modified SJNB-JF-IL2 and SJNB-JF-LTN neuroblastoma cells. | Brain | NCT01192555 | |
| 48 | Gliovac (ERC1671) (Daniela A. Bota; University of California; Epitopoietic Research Corporation) | Autologous and allogeneic tumor cell vaccines against glioblastoma based on irradiated DNFB-modified tumor cells. | Brain | II | Schijns, Pretto, et al. [70]; NCT01903330 |
| 49 | Autologous DC vaccine (Mayo Clinic; NCI) | Allogeneic glioma tumor lysate-pulsed autologous dendritic cell vaccine. | Brain | I | NCT03360708 |
| 50 | RCC26/IL-7/CD80 vaccine (Charite’-University Medicine et al.) | Administration of renal cancer cell line (RCC26) genetically modified to express IL-7 and CD80 (B7-1). Median OS: 40 months. | Kidney | I | Westermen, Flörcken, et al. [71] |
| 51 | MGN1601 (Mologen AG) | Genetically modified allogeneic tumor cells for the Expression of IL-7, GM-CSF, CD80, and CD154. | Kidney | I/II | NCT01265368 |
| 52 | Allogeneic myeloma GM-CSF Vaccine (Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins; CellGen Corporation) | Allogeneic GM-CSF secreting myeloma vaccine in combination with lenalidomide. | Blood | II | NCT01349569 |
| 53 | K562/GM-CSF vaccine (Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins; Cell Genesis Inc) | The vaccine produced from chronic myeloid leukemia (CML) cell line modified to secrete GM-CSF administered with imatinib mesylate. | Blood | I | Smith, Kasamon, et al. [72] |
| 54 | Allogeneic tumor cell vaccine (K562) (NCI) | Allogeneic tumor cell vaccine produced from cell line K562. | Lung Esophagus Pleura Thymus | I | NCT01143545 |
| 55 | ADKV (N.N. Petrov National Medical Research Center of Oncology) | Autologous dendritic cell vaccine (ADKV) loaded with allogeneic tumor lysate expression of cancer-testis antigens (CTA). | Soft tissue sarcomas | I/II | NCT01883518 |
| 56 | Allogenic tumor cell vaccine (K562) (NCI; NIH Clinical Center) | The vaccine produced from irradiated K562 erythroleukemia cells expressing GM-CSF (K562-GM cells). | Sarcoma Melanoma Epithelium Pleura | I | NCT01313429 |
| 57 | Tumor cell vaccine (Hadassah Medical Organization; International Center for Cell Therapy & Cancer Immunotherapy) | Vaccination with allogeneic tumor cell lines that share MHC determinants with the patient aiming to overcome the possible restriction of antigen presentation. | Solid tumors | II | NCT00148993 |
| 58 | Antiangiogenic cancer vaccine (University of Tokyo) | Vaccine using glutaraldehyde-fixed human umbilical vein endothelial cells (HUVECs). | Brain Colon | I | Okaji et al. [73] |
| Criteria | Antigenic Essence Vaccines | Whole Cancer Cell Vaccines (Irradiated Cells, Whole Cell Lysate or Fixed Cells) |
|---|---|---|
| Antigens | Full diversity of native antigens desirable for vaccination. | Full diversity of native antigens, a vast majority of which are intracellular antigens undesirable for vaccination. |
| Immunogenicity | Low (‘as is’), or decreased (compromised compositions 1), or increased [18,28]. | Low (‘as is’), or increased (overexpression of some antigens and release of cytokines by irradiated cells); fixed cells have lower immunogenicity than whole cell lysate [85], irradiated cells have higher immunogenicity than whole cell lysate [86]. |
| Bioinformatic processing of data | No, moderate, or enhanced processing of mass spectrometry data. 2 | No |
| Target cell killing rate (in vitro) | Directly connected with antigenic essence composition [28,29]. | There is no method to connect the antigen composition of whole cells with immune response. |
| Limitations | The lifetime of tryptic peptides is limited in the body; some epitopes may be fragmented, and the secondary structure of some peptide epitopes may be broken. | The vaccination dose includes all antigens of the cell, so less than 1% of the maximum allowed dose would include targeted antigens. |
| MHC-restriction | Not addressed or addressed 2 [18]. | Not addressed. |
| Immunopeptidome | Enriched in comparison with whole cells [18]. | As a trace amount in most other antigens. |
| Vaccine type | Preventive and therapeutic. | Only therapeutic. |
| Availability for mass production | Can be adapted for mass production (the same cancer cells can be used to produce antigens many times). | Cells for vaccination are used only once. |
| Stability | Stable (as peptide composition). | Unstable (as live or fixed cells, or as protein mixture). |
| Safety | High (no supramolecular structures, prions, or viruses present). | Low (tight control is required to exclude the presence of dangerous agents). |
| Quality Control | Improved (included control of antigen composition). | Moderate (no test related to control of antigen profiles of cells). |
| Clinical trials | Not yet conducted. | Failed in all clinical trials. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lokhov, P.G.; Lichtenberg, S.; Balashova, E.E. Changing Landscape of Cancer Vaccines—Novel Proteomics Platform for New Antigen Compositions. Int. J. Mol. Sci. 2022, 23, 4401. https://doi.org/10.3390/ijms23084401
Lokhov PG, Lichtenberg S, Balashova EE. Changing Landscape of Cancer Vaccines—Novel Proteomics Platform for New Antigen Compositions. International Journal of Molecular Sciences. 2022; 23(8):4401. https://doi.org/10.3390/ijms23084401
Chicago/Turabian StyleLokhov, Petr G., Steven Lichtenberg, and Elena E. Balashova. 2022. "Changing Landscape of Cancer Vaccines—Novel Proteomics Platform for New Antigen Compositions" International Journal of Molecular Sciences 23, no. 8: 4401. https://doi.org/10.3390/ijms23084401
APA StyleLokhov, P. G., Lichtenberg, S., & Balashova, E. E. (2022). Changing Landscape of Cancer Vaccines—Novel Proteomics Platform for New Antigen Compositions. International Journal of Molecular Sciences, 23(8), 4401. https://doi.org/10.3390/ijms23084401