
Butyrate Prevents Induction of CXCL10 and Non-Canonical IRF9 Expression by Activated Human Intestinal Epithelial Cells via HDAC Inhibition

 and
and
Abstract
:1. Introduction
2. Results
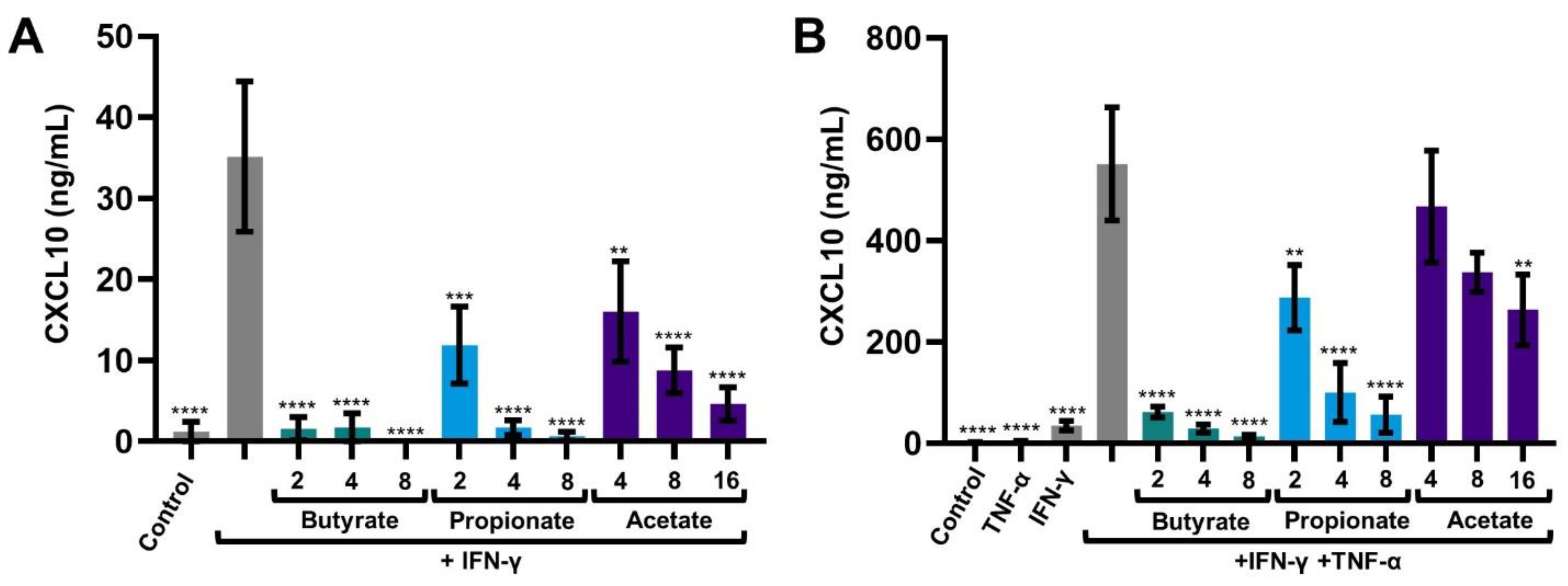
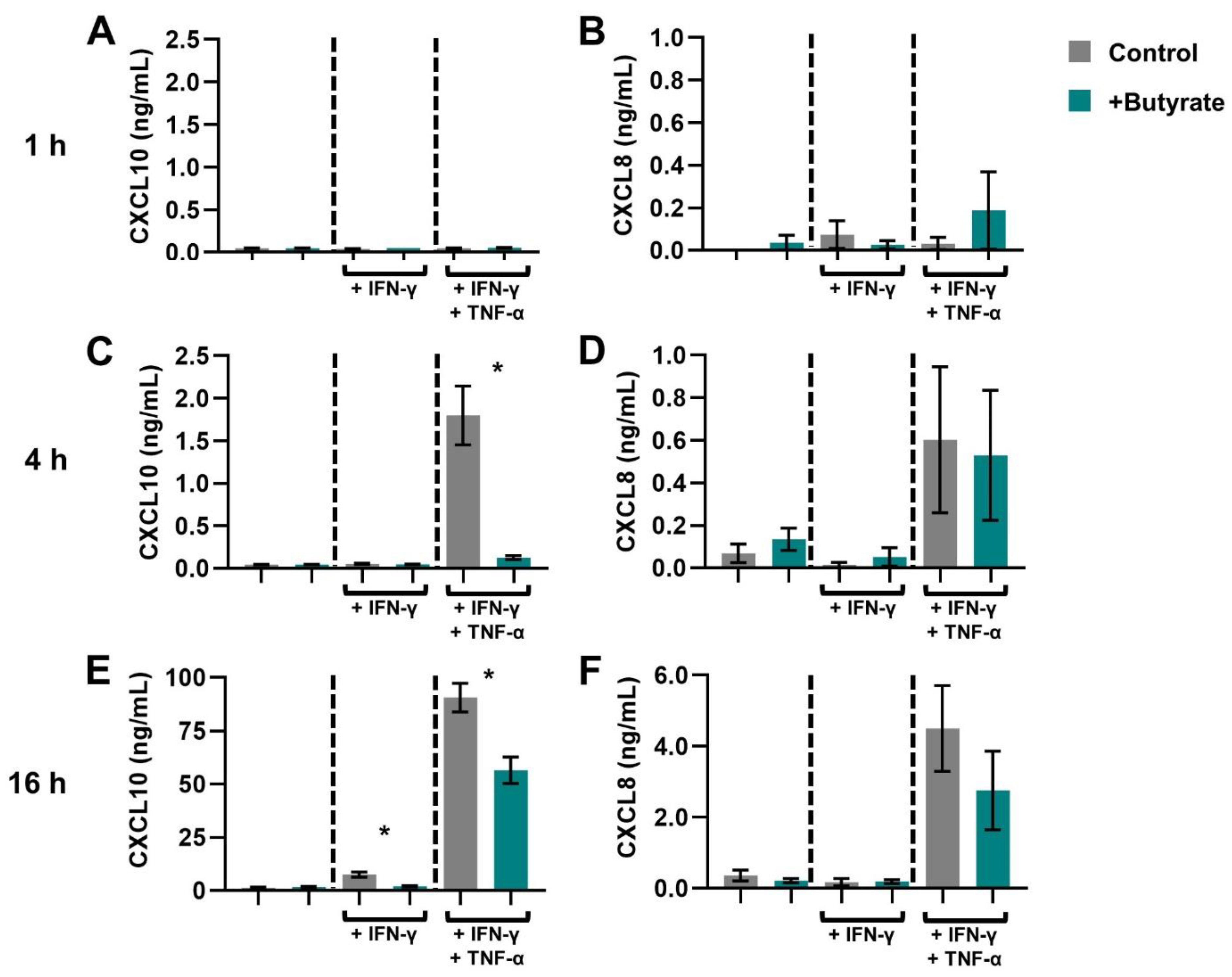
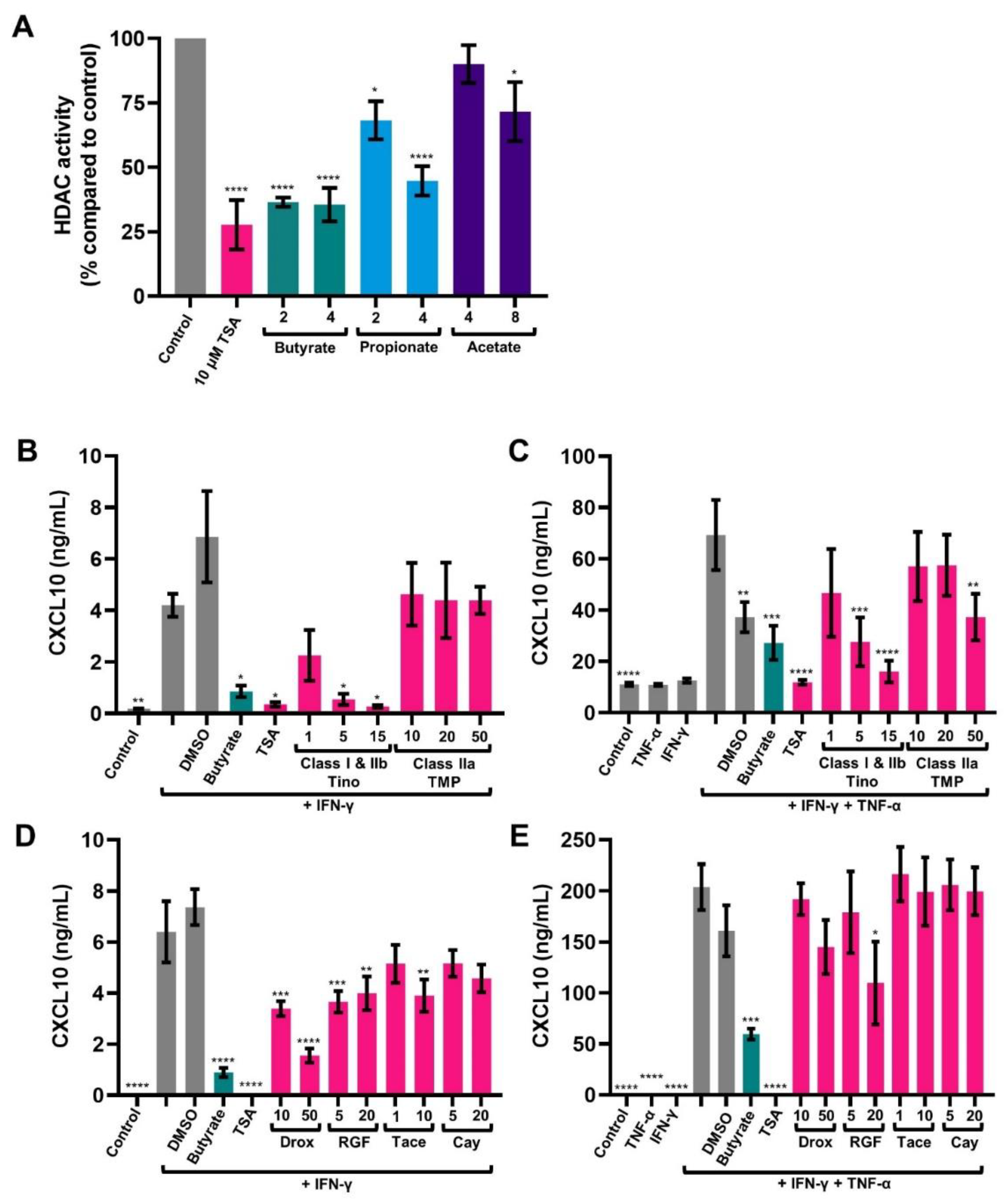
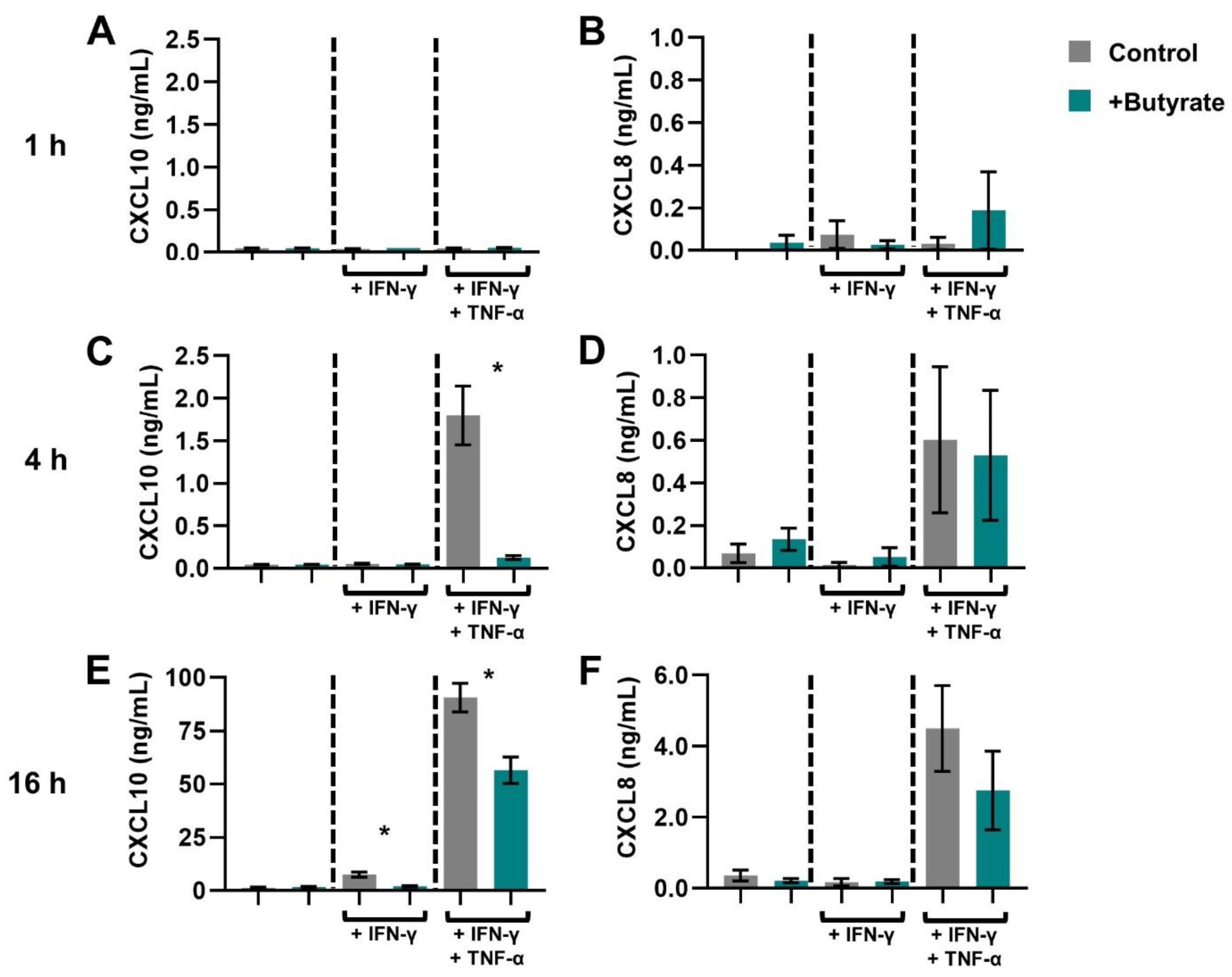
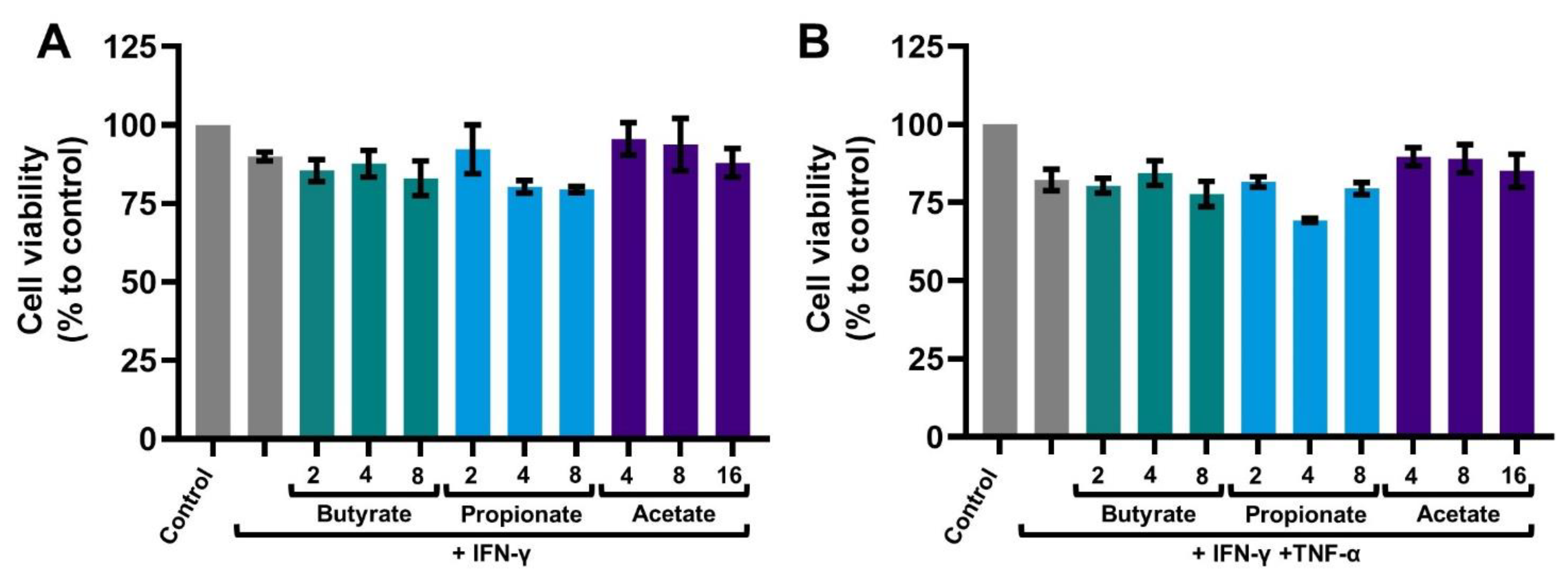
2.1. SCFAs Prevent the Release of CXCL10 by Activated IECs
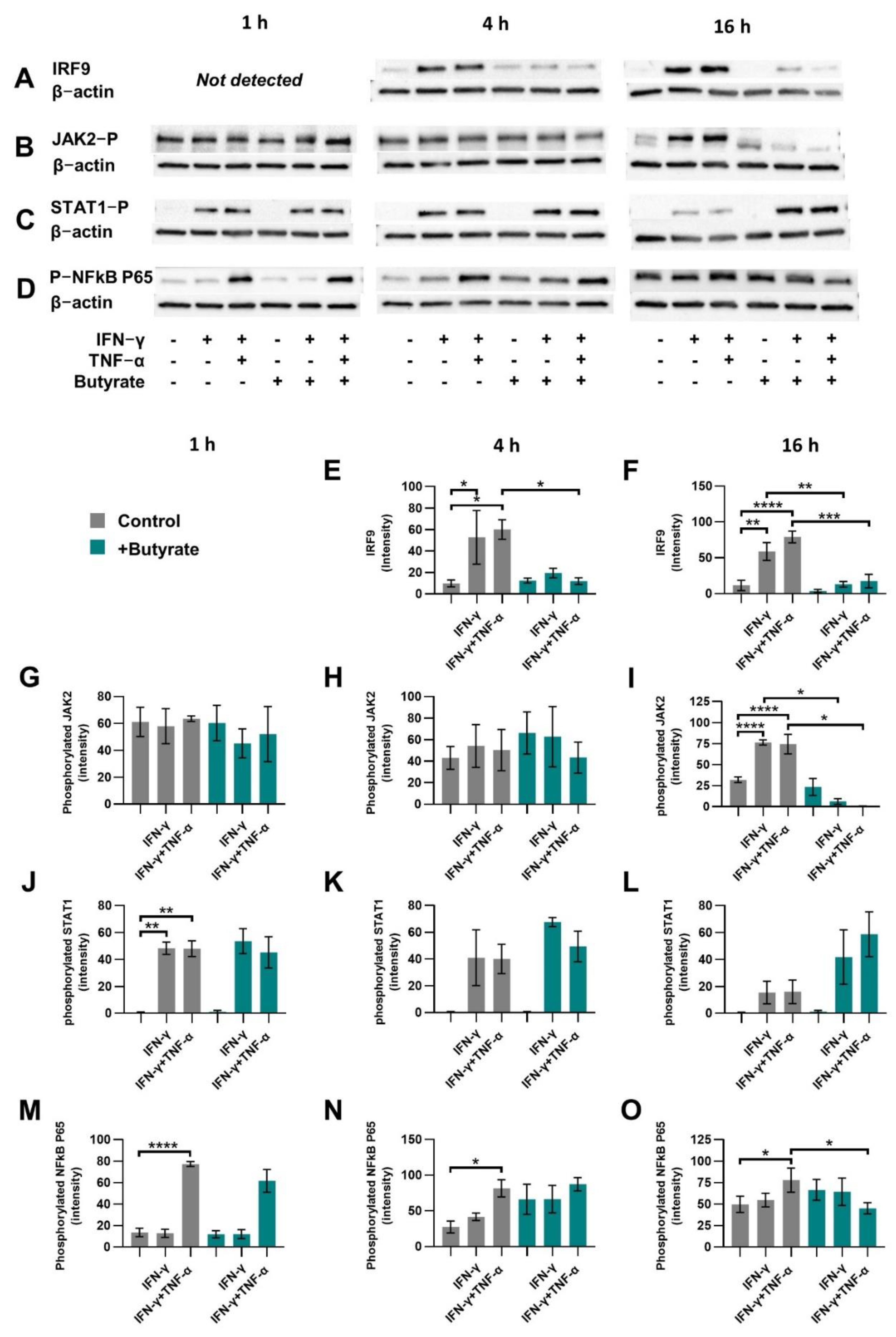
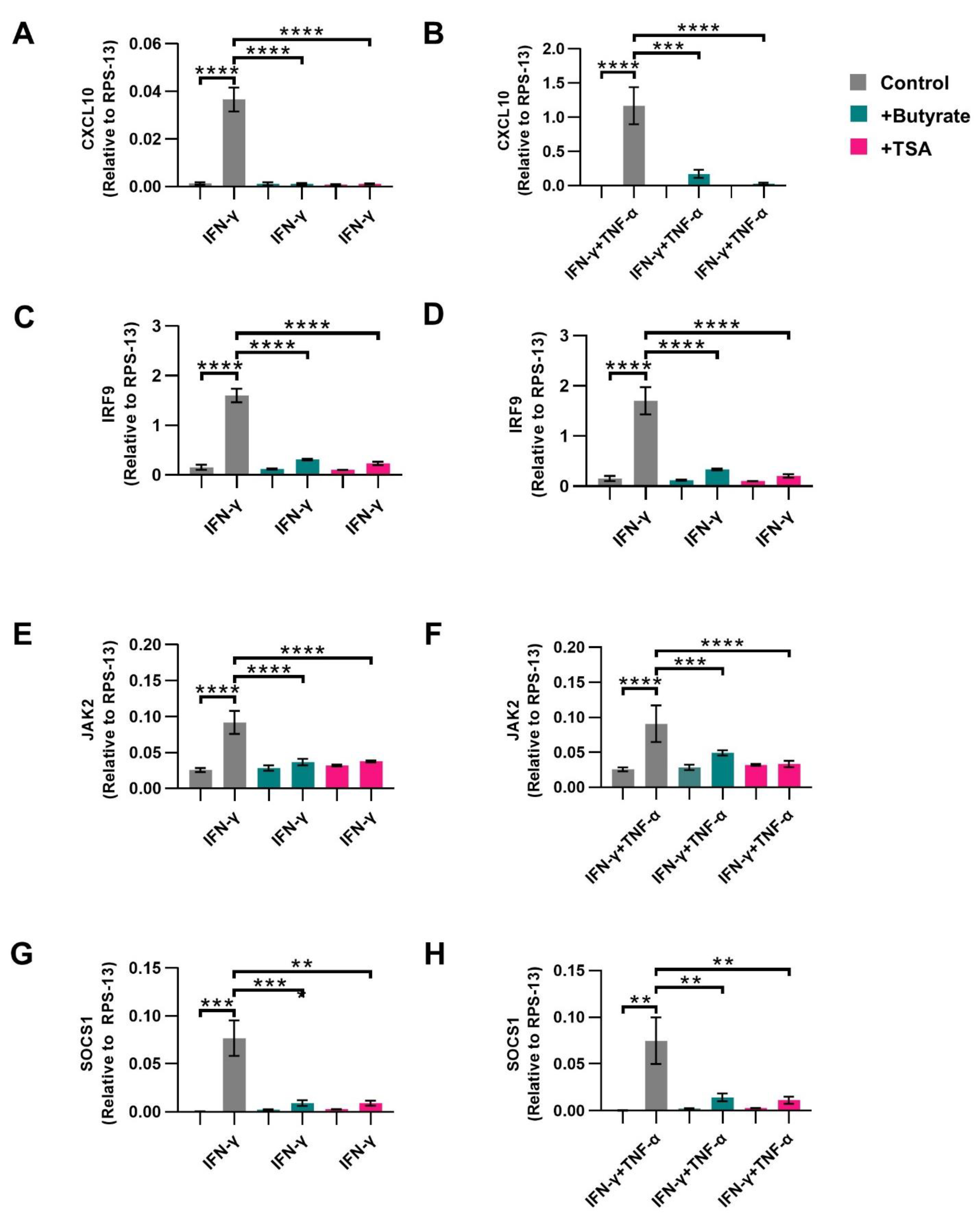
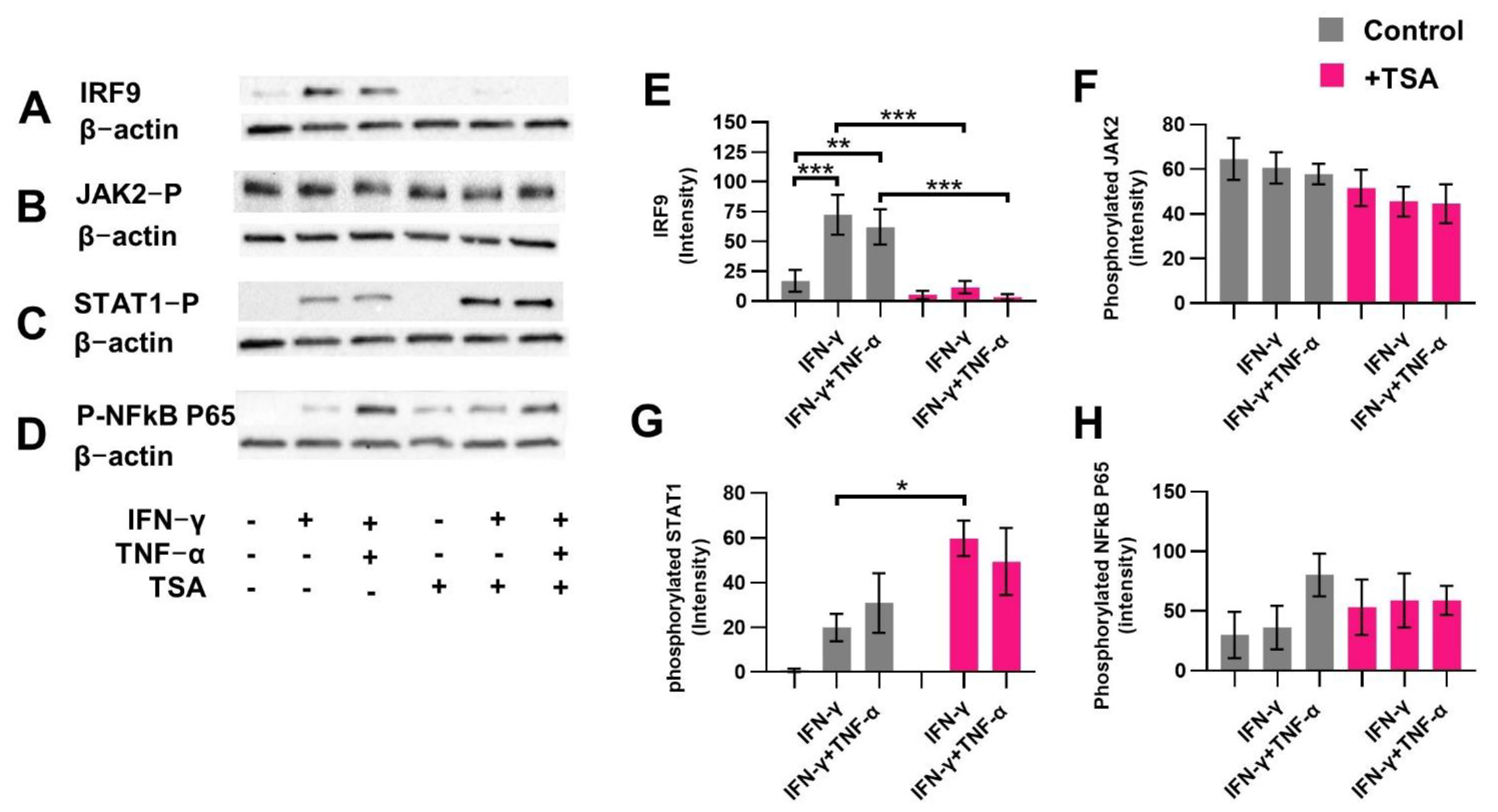
2.2. Butyrate Reduces Proteins Related to the Non-Canonical STAT1 Signaling Cascade
2.3. SCFAs Inhibit HDAC Activity in HT-29
2.4. HDAC Inhibitors Mimic the Effect of Butyrate in Activated IECs
2.5. Butyrate Inhibits CXCL10 Transcription in Activated IECs
2.6. HDAC Inhibitor TSA Prevents Induced IRF9 Expression Similar to Butyrate
3. Discussion
4. Materials and Methods
4.1. Intestinal Epithelial Cell Culture
4.2. Epithelial Activation and SCFAs and HDAC Inhibitor Treatment
4.3. HDAC Activity Assay
4.4. ELISA
4.5. Western Blot
4.6. qPCR
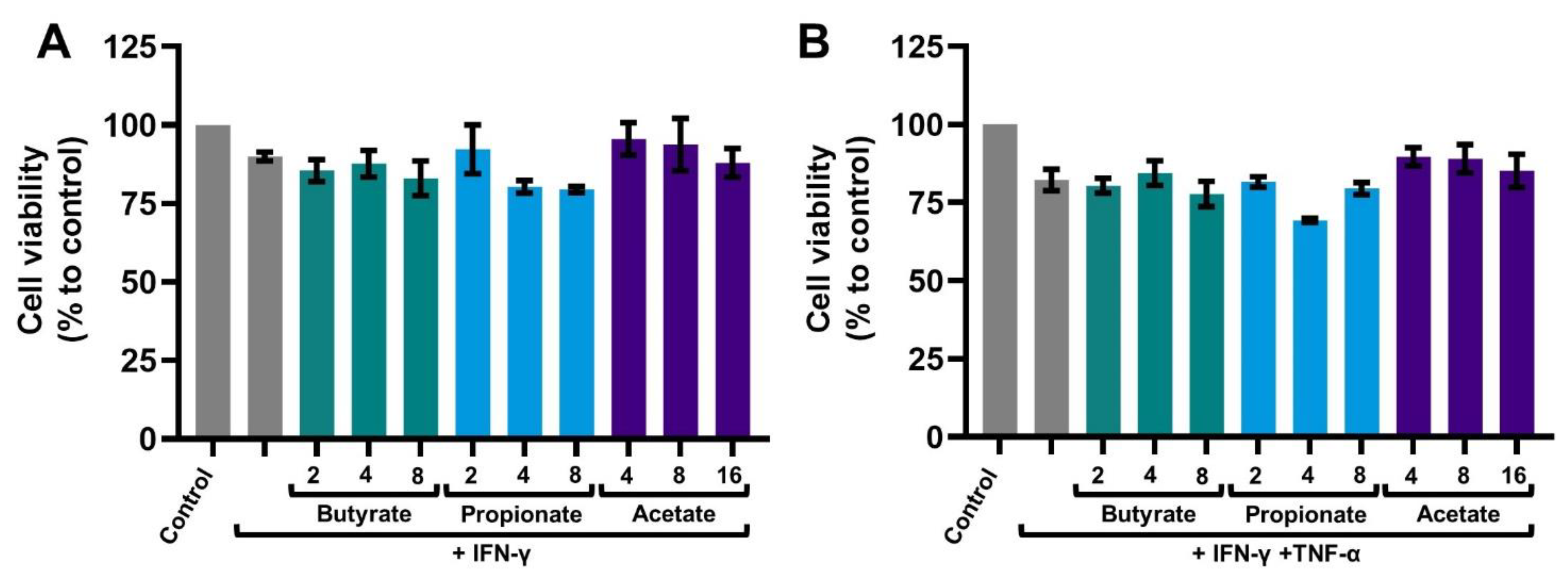
4.7. Viability Assay
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SCFAs | Short Chain Fatty Acids |
| IFN-γ | Interferon-gamma |
| TNF-α | tumor necrosis factor-alpha |
| IECs | Intestinal epithelial cells |
| STAT1 | transducer and activator of transcription 1 |
| HDAC | histone deacetylase |
| JAK1 | Janus kinase 1 |
| JAK2 | Janus kinase 2 |
| IRF9 | Interferon Regulatory Factor 9 |
| CXCL10 | C-X-C motif chemokine ligand 10 |
| qPCR | quantitative polymerase chain reaction |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| SOCS1 | suppressor of cytokine signaling 1 |
| RPS13 | ribosomal protein S13 |
| CXCL8 | C-X-C motif chemokine ligand 8 |
| NFκB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| TSA | Trichostatin A |
Appendix A
References
- Agus, A.; Denizot, J.; Thévenot, J.; Martinez-Medina, M.; Massier, S.; Sauvanet, P.; Bernalier-Donadille, A.; Denis, S.; Hofman, P.; Bonnet, R.; et al. Western Diet Induces a Shift in Microbiota Composition Enhancing Susceptibility to Adherent-Invasive E. Coli Infection and Intestinal Inflammation. Sci. Rep. 2016, 6, 19032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statovci, D.; Aguilera, M.; MacSharry, J.; Melgar, S. The Impact of Western Diet and Nutrients on the Microbiota and Immune Response at Mucosal Interfaces. Front. Immunol. 2017, 8, 838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, C.E.; Renz, H.; Jenmalm, M.C.; Kozyrskyj, A.L.; Allen, K.J.; Vuillermin, P.; Prescott, S.L. The Gut Microbiota and Inflammatory Noncommunicable Diseases: Associations and Potentials for Gut Microbiota Therapies. J. Allergy Clin. Immunol. 2015, 135, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prescott, S.L. Early-Life Environmental Determinants of Allergic Diseases and the Wider Pandemic of Inflammatory Noncommunicable Diseases. J. Allergy Clin. Immunol. 2013, 131, 23–30. [Google Scholar] [CrossRef]
- Kriss, M.; Hazleton, K.Z.; Nusbacher, N.M.; Martin, C.G.; Lozupone, C.A. Low Diversity Gut Microbiota Dysbiosis: Drivers, Functional Implications and Recovery. Curr. Opin. Microbiol. 2018, 44, 34–40. [Google Scholar] [CrossRef]
- Thorburn, A.N.; Macia, L.; Mackay, C.R. Diet, Metabolites, and “Western-Lifestyle” Inflammatory Diseases. Immunity 2014, 40, 833–842. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- Willemsen, L.E.M.; Koetsier, M.A.; van Deventer, S.J.H.; van Tol, E.A.F. Short Chain Fatty Acids Stimulate Epithelial Mucin 2 Expression through Differential Effects on Prostaglandin E1 and E2 Production by Intestinal Myofibroblasts. Gut 2003, 52, 1442–1447. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; He, Z.; Chen, W.; Holzman, I.R.; Lin, J. Effects of Butyrate on Intestinal Barrier Function in a Caco-2 Cell Monolayer Model of Intestinal Barrier. Pediatric Res. 2007, 61, 37–41. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, W.N.; Douangpanya, J.; Mu, S.; Jaeckel, P.; Zhang, M.; Maxwell, J.R.; Rottman, J.B.; Labitzke, K.; Willee, A.; Beckmann, H.; et al. Differing Roles for Short Chain Fatty Acids and GPR43 Agonism in the Regulation of Intestinal Barrier Function and Immune Responses. PLoS ONE 2017, 12, e0180190. [Google Scholar] [CrossRef] [Green Version]
- Miao, W.; Wu, X.; Wang, K.; Wang, W.; Wang, Y.; Li, Z.; Liu, J.; Li, L.; Peng, L. Sodium Butyrate Promotes Reassembly of Tight Junctions in Caco-2 Monolayers Involving Inhibition of MLCK/MLC2 Pathway and Phosphorylation of PKCβ2. Int. J. Mol. Sci. 2016, 17, 1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Kelly, C.J.; Battista, K.D.; Schaefer, R.; Lanis, J.M.; Alexeev, E.E.; Wang, R.X.; Onyiah, J.C.; Kominsky, D.J.; Colgan, S.P. Microbial-Derived Butyrate Promotes Epithelial Barrier Function through IL-10 Receptor–Dependent Repression of Claudin-2. J. Immunol. 2017, 199, 2976–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Ajuwon, K.M. Butyrate Modifies Intestinal Barrier Function in IPEC-J2 Cells through a Selective Upregulation of Tight Junction Proteins and Activation of the Akt Signaling Pathway. PLoS ONE 2017, 12, e0179586. [Google Scholar] [CrossRef] [PubMed]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of Inflammatory Responses by Gut Microbiota and Chemoattractant Receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. The Role of Short-Chain Fatty Acids in Health and Disease, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2014; Volume 121, ISBN 9780128001004. [Google Scholar]
- Lührs, H.; Gerke, T.; Schauber, J.; Dusel, G.; Melcher, R.; Scheppach, W.; Menzel, T. Cytokine-Activated Degradation of Inhibitory ΚB Protein α Is Inhibited by the Short-Chain Fatty Acid Butyrate. Int. J. Colorectal Dis. 2001, 16, 195–201. [Google Scholar] [CrossRef]
- Asarat, M.; Apostolopoulos, V.; Vasiljevic, T.; Donkor, O. Short-Chain Fatty Acids Regulate Cytokines and Th17/Treg Cells in Human Peripheral Blood Mononuclear Cells In Vitro. Immunol. Investig. 2016, 45, 205–222. [Google Scholar] [CrossRef] [Green Version]
- Tedelind, S.; Westberg, F.; Kjerrulf, M.; Vidal, A. Anti-Inflammatory Properties of the Short-Chain Fatty Acids Acetate and Propionate: A Study with Relevance to Inflammatory Bowel Disease. World J. Gastroenterol. 2007, 13, 2826–2832. [Google Scholar] [CrossRef]
- Liu, T.; Li, J.; Liu, Y.; Xiao, N.; Suo, H.; Xie, K.; Yang, C.; Wu, C. Short-Chain Fatty Acids Suppress Lipopolysaccharide-Induced Production of Nitric Oxide and Proinflammatory Cytokines through Inhibition of NF-KB Pathway in RAW264.7 Cells. Inflammation 2012, 35, 1676–1684. [Google Scholar] [CrossRef]
- Cox, M.A.; Jackson, J.; Stanton, M.; Rojas-Triana, A.; Bober, L.; Laverty, M.; Yang, X.; Zhu, F.; Liu, J.; Wang, S.; et al. Short-Chain Fatty Acids Act as Antiinflammatory Mediators by Regulating Prostaglandin E2 and Cytokines. World J. Gastroenterol. 2009, 15, 5549–5557. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Neyrinck, A.M.; Delzenne, N.M. Changes in Gut Microbiota Control Metabolic Diet–Induced Obesity and Diabetes in Mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Jakobsdottir, G.; Xu, J.; Molin, G.; Ahrné, S.; Nyman, M. High-Fat Diet Reduces the Formation of Butyrate, but Increases Succinate, Inflammation, Liver Fat and Cholesterol in Rats, While Dietary Fibre Counteracts These Effects. PLoS ONE 2013, 8, e80476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, M.D.; Singh, P.K. Superantigen Activation of Immune Cells Evokes Epithelial (T84) Transport and Barrier Abnormalities via IFN-y and TNF-a. J. Immunol. 1997, 159, 2382–2390. [Google Scholar] [PubMed]
- Madsen, K.L.; Lewis, S.A.; Tavernini, M.M.; Hibbard, J.; Fedorak, R.N. Interleukin 10 Prevents Cytokine-Induced Disruption of T84 Monolayer Barrier Integrity and Limits Chloride Secretion. Gastroenterology 1997, 113, 151–159. [Google Scholar] [CrossRef]
- Groschwitz, K.R.; Hogan, S.P. Intestinal Barrier Function: Molecular Regulation and Disease Pathogenesis. J. Allergy Clin. Immunol. 2009, 124, 3–20. [Google Scholar] [CrossRef] [Green Version]
- Adams, R.B.; Planchon, S.M.; Roche, J.K. IFN-Gamma Modulation of Epithelial Barrier Function. Time Course, Reversibility, and Site of Cytokine Binding. J. Immunol. 1993, 150, 2356–2363. [Google Scholar]
- Valente, G.; Ozmen, L.; Novelli, F.; Geuna, M.; Palestro, G.; Forni, G.; Garotta, G. Distribution of Interferon-γ Receptor in Human Tissues. Eur. J. Immunol. 1992, 22, 2403–2412. [Google Scholar] [CrossRef]
- Majoros, A.; Platanitis, E.; Kernbauer-Hölzl, E.; Rosebrock, F.; Müller, M.; Decker, T. Canonical and Non-Canonical Aspects of JAK-STAT Signaling: Lessons from Interferons for Cytokine Responses. Front. Immunol. 2017, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Liau, N.P.D.; Laktyushin, A.; Lucet, I.S.; Murphy, J.M.; Yao, S.; Whitlock, E.; Callaghan, K.; Nicola, N.A.; Kershaw, N.J.; Babon, J.J. The Molecular Basis of JAK/STAT Inhibition by SOCS1. Nat. Commun. 2018, 9, 1558. [Google Scholar] [CrossRef]
- Liu, M.; Guo, S.; Hibbert, J.M.; Jain, V.; Singh, N.; Wilson, N.O.; Stiles, J.K. CXCL10/IP-10 in Infectious Diseases Pathogenesis and Potential Therapeutic Implications. Cytokine Growth Factor Rev. 2011, 22, 121–130. [Google Scholar] [CrossRef]
- Yeruva, S.; Ramadori, G.; Raddatz, D. NF-ΚB-Dependent Synergistic Regulation of CXCL10 Gene Expression by IL-1β and IFN-γ in Human Intestinal Epithelial Cell Lines. Int. J. Colorectal Dis. 2008, 23, 305–317. [Google Scholar] [CrossRef] [Green Version]
- Krakauer, T.; Oppenheim, J.J. IL-1 and Tumor Necrosis Factor-Alpha Each up-Regulate Both the Expression of IFN-Gamma Receptors and Enhance IFN-Gamma-Induced HLA-DR Expression on Human Monocytes and a Human Monocytic Cell Line (THP-1). J. Immunol. 1993, 150, 1205–1211. [Google Scholar] [PubMed]
- Robinson, C.M.; Shirey, K.A.; Carlin, J.M. Synergistic Transcriptional Activation of Indoleamine Dioxygenase by IFN-γ and Tumor Necrosis Factor-α. J. Interferon Cytokine Res. 2003, 23, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Dunbar, J.D.; Yang, C.H.; Pfeffer, L.M.; Donner, D.B. Induction of Jak/STAT Signaling by Activation of the Type 1 TNF Receptor. J. Immunol. 1998, 160, 2742–2750. [Google Scholar] [PubMed]
- Ohmori, Y.; Schreiber, R.D.; Hamilton, T.A. Synergy between Interferon-γ and Tumor Necrosis Factor-α in Transcriptional Activation Is Mediated by Cooperation between Signal Transducer and Activator of Transcription 1 and Nuclear Factor ΚB. J. Biol. Chem. 1997, 272, 14899–14907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klampfer, L.; Huang, J.; Swaby, L.A.; Augenlicht, L. Requirement of Histone Deacetylase Activity for Signaling by STAT1. J. Biol. Chem. 2004, 279, 30358–30368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klampfer, L.; Huang, J.; Sasazuki, T.; Shirasawa, S.; Augenlicht, L. Inhibition of Interferon γ Signaling by the Short Chain Fatty Acid Butyrate. Mol. Cancer Res. 2003, 1, 855–862. [Google Scholar] [PubMed]
- Inatomi, O.; Andoh, A.; Kitamura, K.I.; Yasui, H.; Zhang, Z.; Fujiyama, Y. Butyrate Blocks Interferon-γ-Inducible Protein-10 Release in Human Intestinal Subepithelial Myofibroblasts. J. Gastroenterol. 2005, 40, 483–489. [Google Scholar] [CrossRef]
- Chemudupati, M.; Kenney, A.D.; Smith, A.C.; Fillinger, R.J.; Zhang, L.; Zani, A.; Liu, S.-L.; Anderson, M.Z.; Sharma, A.; Yount, J.S. Butyrate Reprograms Expression of Specific Interferon-Stimulated Genes. J. Virol. 2020, 94, e00326-20. [Google Scholar] [CrossRef]
- Nastasi, C.; Candela, M.; Bonefeld, C.M.; Geisler, C.; Hansen, M.; Krejsgaard, T.; Biagi, E.; Andersen, M.H.; Brigidi, P.; Ødum, N.; et al. The Effect of Short-Chain Fatty Acids on Human Monocyte-Derived Dendritic Cells. Sci. Rep. 2015, 5, 16148. [Google Scholar] [CrossRef]
- Ciarlo, E.; Heinonen, T.; Herderschee, J.; Fenwick, C.; Mombelli, M.; le Roy, D.; Roger, T. Impact of the Microbial Derived Short Chain Fatty Acid Propionate on Host Susceptibility to Bacterial and Fungal Infections In Vivo. Sci. Rep. 2016, 6, 37944. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, J.S. A Short Guide to Histone Deacetylases Including Recent Progress on Class II Enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Davie, J.R. Inhibition of Histone Deacetylase Activity by Butyrate. J. Nutr. 2003, 133, 2485–2493. [Google Scholar] [CrossRef] [PubMed]
- Cousens, L.S.; Gallwitz, D.; Alberts, B.M. Different Accessibilities in Chromatin to Histone Acetylase. J. Biol. Chem. 1979, 254, 1716–1723. [Google Scholar] [CrossRef]
- Vinolo, M.A.R.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of Inflammation by Short Chain Fatty Acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef] [Green Version]
- Sunkara, L.T.; Achanta, M.; Schreiber, N.B.; Bommineni, Y.R.; Dai, G.; Jiang, W.; Lamont, S.; Lillehoj, H.S.; Beker, A.; Teeter, R.G.; et al. Butyrate Enhances Disease Resistance of Chickens by Inducing Antimicrobial Host Defense Peptide Gene Expression. PLoS ONE 2011, 6, e27225. [Google Scholar] [CrossRef]
- Waldecker, M.; Kautenburger, T.; Daumann, H.; Busch, C.; Schrenk, D. Inhibition of Histone-Deacetylase Activity by Short-Chain Fatty Acids and Some Polyphenol Metabolites Formed in the Colon. J. Nutr. Biochem. 2008, 19, 587–593. [Google Scholar] [CrossRef]
- Nusinzon, I.; Horvath, C.M. Interferon-Stimulated Transcription and Innate Antiviral Immunity Require Deacetylase Activity and Histone Deacetylase 1. Proc. Natl. Acad. Sci. USA 2003, 100, 14742–14747. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, S.; Potla, R.; Larner, A.C. Histone Deacetylase Activity Is Required to Recruit RNA Polymerase II to the Promoters of Selected Interferon-Stimulated Early Response Genes. J. Biol. Chem. 2004, 279, 40362–40367. [Google Scholar] [CrossRef] [Green Version]
- Krämer, O.H.; Heinzel, T. Phosphorylation-Acetylation Switch in the Regulation of STAT1 Signaling. Mol. Cell. Endocrinol. 2010, 315, 40–48. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.M.; Paulson, M.; Holko, M.; Rice, C.M.; Williams, B.R.G.; Marié, I.; Levy, D.E. Induction of Interferon-Stimulated Gene Expression and Antiviral Responses Require Protein Deacetylase Activity. Proc. Natl. Acad. Sci. USA 2004, 101, 9578–9583. [Google Scholar] [CrossRef] [Green Version]
- Turgeon, N.; Gagné, J.M.; Blais, M.; Gendron, F.P.; Boudreau, F.; Asselin, C. The Acetylome Regulators Hdac1 and Hdac2 Differently Modulate Intestinal Epithelial Cell Dependent Homeostatic Responses in Experimental Colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.J.; Li, Q.L.; Zhang, J.; Huang, A.L. Histone Deacetylation Is Involved in Activation of CXCL 10 upon IFNγ Stimulation. Mol. Cells 2006, 22, 163–167. [Google Scholar] [PubMed]
- Canani, R.B.; Costanzo, M.d.; Leone, L.; Pedata, M.; Meli, R.; Calignano, A. Potential Beneficial Effects of Butyrate in Intestinal and Extraintestinal Diseases. World J. Gastroenterol. 2011, 17, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Segain, J.P.; Raingeard de la Blétière, D.; Bourreille, A.; Leray, V.; Gervois, N.; Rosales, C.; Ferrier, L.; Bonnet, C.; Blottière, H.M.; Galmiche, J.P. Butyrate Inhibits Inflammatory Responses through NFkappaB Inhibition: Implications for Crohn’s Disease. Gut 2000, 47, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Inan, M.S.; Rasoulpour, R.J.; Yin, L.; Hubbard, A.K.; Rosenberg, D.W.; Giardina, C. The Luminal Short-Chain Fatty Acid Butyrate Modulates NF-ΚB Activity in a Human Colonic Epithelial Cell Line. Gastroenterology 2000, 118, 724–734. [Google Scholar] [CrossRef]
- Asarat, M.; Vasiljevic, T.; Apostolopoulos, V.; Donkor, O. Short-Chain Fatty Acids Regulate Secretion of IL-8 from Human Intestinal Epithelial Cell Lines in Vitro. Immunol. Investig. 2015, 44, 678–693. [Google Scholar] [CrossRef]
- Li, M.; van Esch, B.C.A.M.; Henricks, P.A.J.; Folkerts, G.; Garssen, J. The Anti-Inflammatory Effects of Short Chain Fatty Acids on Lipopolysaccharide- or Tumor Necrosis Factor α-Stimulated Endothelial Cells via Activation of GPR41/43 and Inhibition of HDACs. Front. Pharmacol. 2018, 9, 533. [Google Scholar] [CrossRef] [Green Version]
- Rauch, I.; Rosebrock, F.; Hainzl, E.; Heider, S.; Majoros, A.; Wienerroither, S.; Strobl, B.; Stockinger, S.; Kenner, L.; Müller, M.; et al. Noncanonical Effects of IRF9 in Intestinal Inflammation: More than Type I and Type III Interferons. Mol. Cell. Biol. 2015, 35, 2332–2343. [Google Scholar] [CrossRef] [Green Version]
- Chou, D.H.; Holson, E.B.; Wagner, F.F.; Tang, A.J.; Maglathlin, R.L.; Lewis, T.A.; Schreiber, S.L.; Wagner, B.K. Inhibition of Histone Deacetylase 3 Protects Beta Cells from Cytokine-Induced Apoptosis. Chem. Biol. 2012, 19, 669–673. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.Z.; Yu, S.; Yan, P.A.; Gong, D.Y.; Wu, F.L.; He, Z.; Yuan, Y.Y.; Zhao, A.Y.; Tang, X.; Zhang, R.Q.; et al. Crotonoside Exhibits Selective Post-Inhibition Effect in AML Cells via Inhibition of FLT3 and HDAC3/6. Oncotarget 2017, 8, 103087–103099. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Yang, W.; Zeng, H.; Hu, C.; Zhang, Y.; Ding, N.; Fan, G.; Shao, L.; Kuang, B. Droxinostat Sensitizes Human Colon Cancer Cells to Apoptotic Cell Death via Induction of Oxidative Stress. Cell. Mol. Biol. Lett. 2018, 23, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leus, N.G.J.; van der Wouden, P.E.; van den Bosch, T.; Hooghiemstra, W.T.R.; Ourailidou, M.E.; Kistemaker, L.E.M.; Bischoff, R.; Gosens, R.; Haisma, H.J.; Dekker, F.J. HDAC 3-Selective Inhibitor RGFP966 Demonstrates Anti-Inflammatory Properties in RAW 264.7 Macrophages and Mouse Precision-Cut Lung Slices by Attenuating NF-ΚB P65 Transcriptional Activity. Biochem. Pharmacol. 2016, 108, 58–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.Y.; Kee, H.J.; Jin, L.; Ryu, Y.; Sun, S.; Kim, G.R.; Jeong, M.H. Inhibition of Class IIa Histone Deacetylase Activity by Gallic Acid, Sulforaphane, TMP269, and Panobinostat. Biomed. Pharmacother. 2018, 101, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Clarhaut, J.; Tranoy-Opalinski, I.; Gesson, J.P.; Roche, J.; Papot, S. Synthesis and Biological Evaluation of Glucuronide Prodrugs of the Histone Deacetylase Inhibitor CI-994 for Application in Selective Cancer Chemotherapy. Bioorganic Med. Chem. 2008, 16, 8109–8116. [Google Scholar] [CrossRef] [PubMed]
- Mehrling, T.; Chen, Y. The Alkylating-HDAC Inhibition Fusion Principle: Taking Chemotherapy to the Next Level with the First in Class Molecule EDO-S101. Anti-Cancer Agents Med. Chem. 2015, 16, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Goverse, G.; Molenaar, R.; Macia, L.; Tan, J.; Erkelens, M.N.; Konijn, T.; Knippenberg, M.; Cook, E.C.L.; Hanekamp, D.; Veldhoen, M.; et al. Diet-Derived Short Chain Fatty Acids Stimulate Intestinal Epithelial Cells To Induce Mucosal Tolerogenic Dendritic Cells. J. Immunol. 2017, 198, 2172–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Iglesias, A.A.; Herrero, A.B.; Chesi, M.; San-Segundo, L.; González-Méndez, L.; Hernández-García, S.; Misiewicz-Krzeminska, I.; Quwaider, D.; Martín-Sánchez, M.; Primo, D.; et al. Preclinical Anti-Myeloma Activity of EDO-S101, a New Bendamustine-Derived Molecule with Added HDACi Activity, through Potent DNA Damage Induction and Impairment of DNA Repair. J. Hematol. Oncol. 2017, 10, 127. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, B.; Chen, D.; Setroikromo, R.; Haisma, H.J.; Quax, W.J. Histone Deacetylase Inhibitors Sensitize TRAIL-Induced Apoptosis in Colon Cancer Cells. Cancers 2019, 11, 645. [Google Scholar] [CrossRef] [Green Version]
- Jiao, F.Z.; Wang, Y.; Zhang, H.Y.; bin Zhang, W.; Wang, L.W.; Gong, Z.J. Histone Deacetylase 2 Inhibitor CAY10683 Alleviates Lipopolysaccharide Induced Neuroinflammation through Attenuating TLR4/NF-ΚB Signaling Pathway. Neurochem. Res. 2018, 43, 1161–1170. [Google Scholar] [CrossRef]
- Kikuchi, S.; Suzuki, R.; Ohguchi, H.; Yoshida, Y.; Lu, D.; Cottini, F.; Jakubikova, J.; Bianchi, G.; Harada, T.; Gorgun, G.; et al. Class IIa HDAC Inhibition Enhances ER Stress-Mediated Cell Death in Multiple Myeloma. Leukemia 2015, 29, 1918–1927. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, H.; Chen, Q.; Jiao, F.Z.; bin Zhang, W.; Gong, Z.J. The Protective Mechanism of CAY10683 on Intestinal Mucosal Barrier in Acute Liver Failure through LPS/TLR4/MyD88 Pathway. Mediat. Inflamm. 2018, 2018, 7859601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerbeth, L.; Glauben, R. Histone Deacetylases in the Inflamed Intestinal Epithelium—Promises of New Therapeutic Strategies. Front. Med. 2021, 8, 351. [Google Scholar] [CrossRef] [PubMed]
- Paparo, L.; Nocerino, R.; Ciaglia, E.; Di Scala, C.; De Caro, C.; Russo, R.; Trinchese, G.; Aitoro, R.; Amoroso, A.; Bruno, C.; et al. Butyrate as a bioactive human milk protective component against food allergy. Allergy 2021, 76, 1398–1415. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.C.; Piper, C.J.; Matei, D.E.; Blair, P.A.; Rendeiro, A.F.; Orford, M.; Alber, D.G.; Krausgruber, T.; Catalan, D.; Klein, N.; et al. Microbiota-Derived Metabolites Suppress Arthritis by Amplifying Aryl-Hydrocarbon Receptor Activation in Regulatory B Cells. Cell Metab. 2020, 31, 837–851. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450, Erratum in Nature 2014, 506, 254. [Google Scholar] [CrossRef]
- Mariño, E.; Richards, J.L.; McLeod, K.H.; Stanley, D.; Yap, Y.-A.; Knight, J.; McKenzie, C.; Kranich, J.; Oliveira, A.C.; Rossello, F.J.; et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat. Immunol. 2017, 18, 552–562. [Google Scholar] [CrossRef] [PubMed]
- García-Vallejo, J.J.; van het Hof, B.; Robben, J.; van Wijk, J.A.E.; van Die, I.; Joziasse, D.H.; van Dijk, W. Approach for Defining Endogenous Reference Genes in Gene Expression Experiments. Anal. Biochem. 2004, 329, 293–299. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class I | Class IIb | Class IIa | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HDAC | 1 | 2 | 3 | 8 | 6 | 10 | 4 | 5 | 7 | 9 | |||
| TSA | |||||||||||||
| Butyrate | |||||||||||||
| TMP269 | |||||||||||||
| Tinostamustine | |||||||||||||
| Tacedinaline | |||||||||||||
| Droxinostat | |||||||||||||
| RGFP966 | |||||||||||||
| Cay10683 | |||||||||||||
| mM | μM | nM | pM | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korsten, S.G.P.J.; Peracic, L.; van Groeningen, L.M.B.; Diks, M.A.P.; Vromans, H.; Garssen, J.; Willemsen, L.E.M. Butyrate Prevents Induction of CXCL10 and Non-Canonical IRF9 Expression by Activated Human Intestinal Epithelial Cells via HDAC Inhibition. Int. J. Mol. Sci. 2022, 23, 3980. https://doi.org/10.3390/ijms23073980
Korsten SGPJ, Peracic L, van Groeningen LMB, Diks MAP, Vromans H, Garssen J, Willemsen LEM. Butyrate Prevents Induction of CXCL10 and Non-Canonical IRF9 Expression by Activated Human Intestinal Epithelial Cells via HDAC Inhibition. International Journal of Molecular Sciences. 2022; 23(7):3980. https://doi.org/10.3390/ijms23073980
Chicago/Turabian StyleKorsten, Sandra G. P. J., Laura Peracic, Luka M. B. van Groeningen, Mara A. P. Diks, Herman Vromans, Johan Garssen, and Linette E. M. Willemsen. 2022. "Butyrate Prevents Induction of CXCL10 and Non-Canonical IRF9 Expression by Activated Human Intestinal Epithelial Cells via HDAC Inhibition" International Journal of Molecular Sciences 23, no. 7: 3980. https://doi.org/10.3390/ijms23073980
APA StyleKorsten, S. G. P. J., Peracic, L., van Groeningen, L. M. B., Diks, M. A. P., Vromans, H., Garssen, J., & Willemsen, L. E. M. (2022). Butyrate Prevents Induction of CXCL10 and Non-Canonical IRF9 Expression by Activated Human Intestinal Epithelial Cells via HDAC Inhibition. International Journal of Molecular Sciences, 23(7), 3980. https://doi.org/10.3390/ijms23073980