
Gut Mucosal Microbiome Is Perturbed in Rheumatoid Arthritis Mice and Partly Restored after TDAG8 Deficiency or Suppression by Salicylanilide Derivative

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
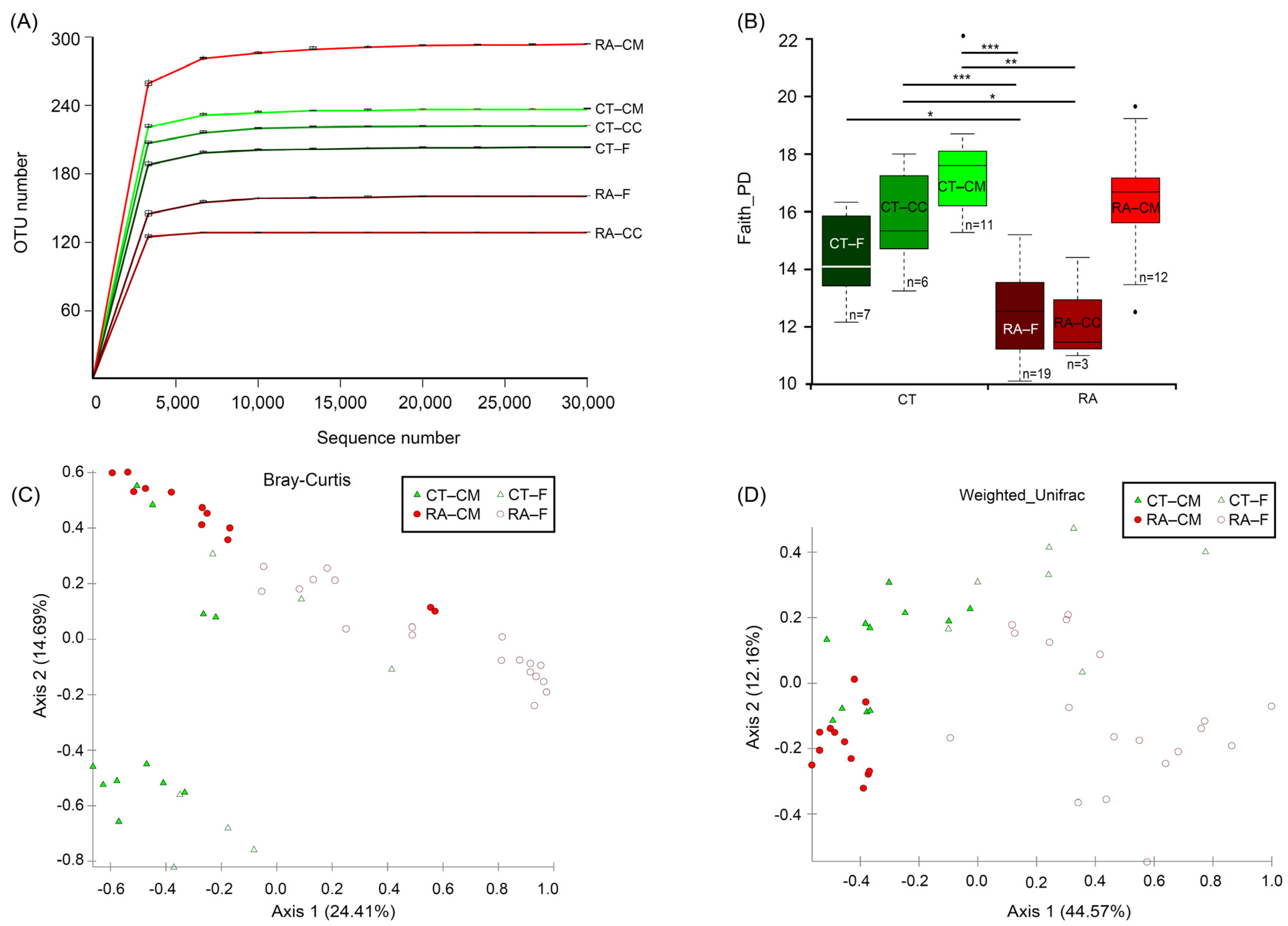
2.1. Differences in Microbiomes of RA Mice
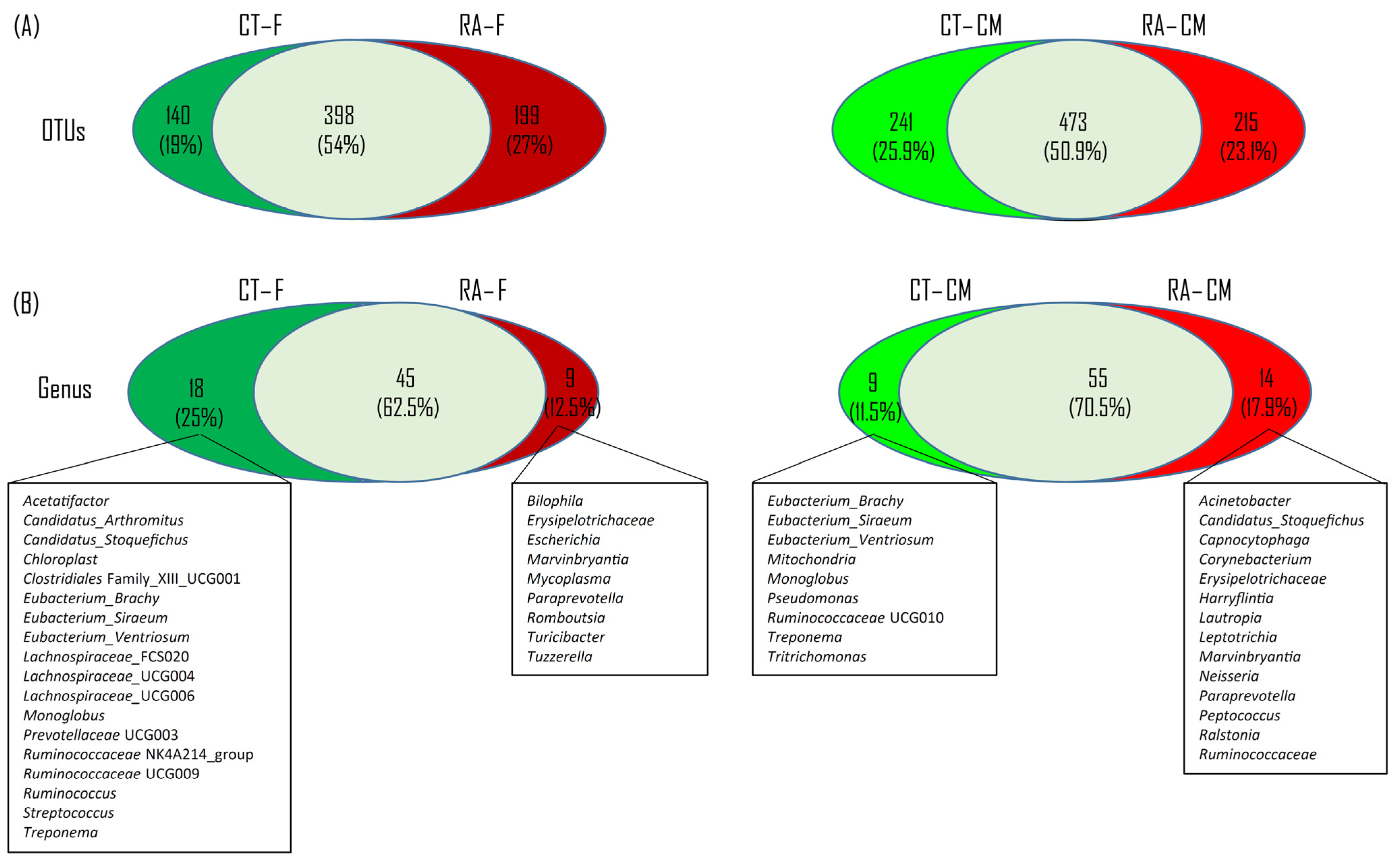
2.2. Core Microbiome
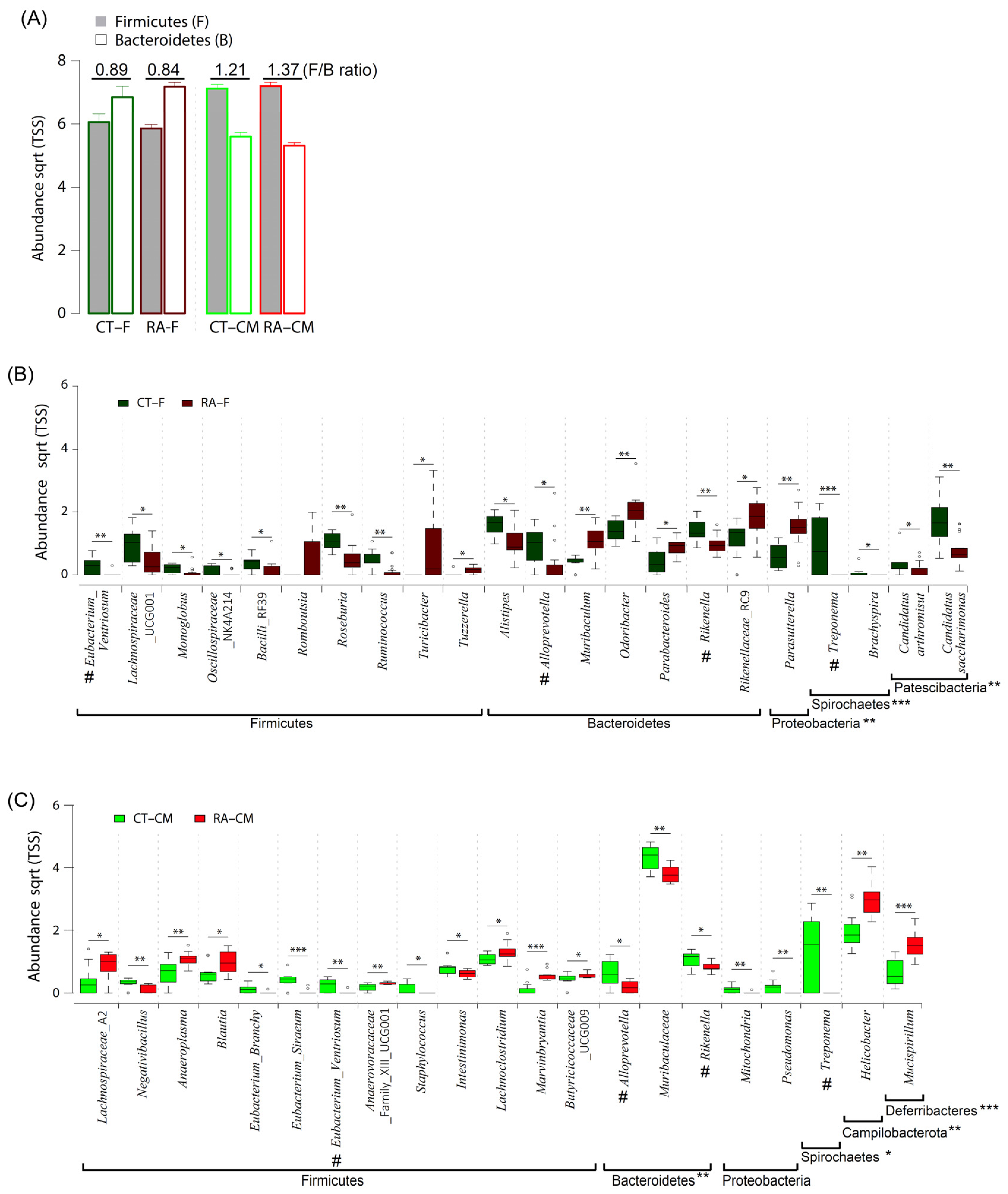
2.3. Featured Microbial Taxa by Using LEfSe
2.4. Featured Microbial Taxa Using Wilcoxon Rank-Sum Tests
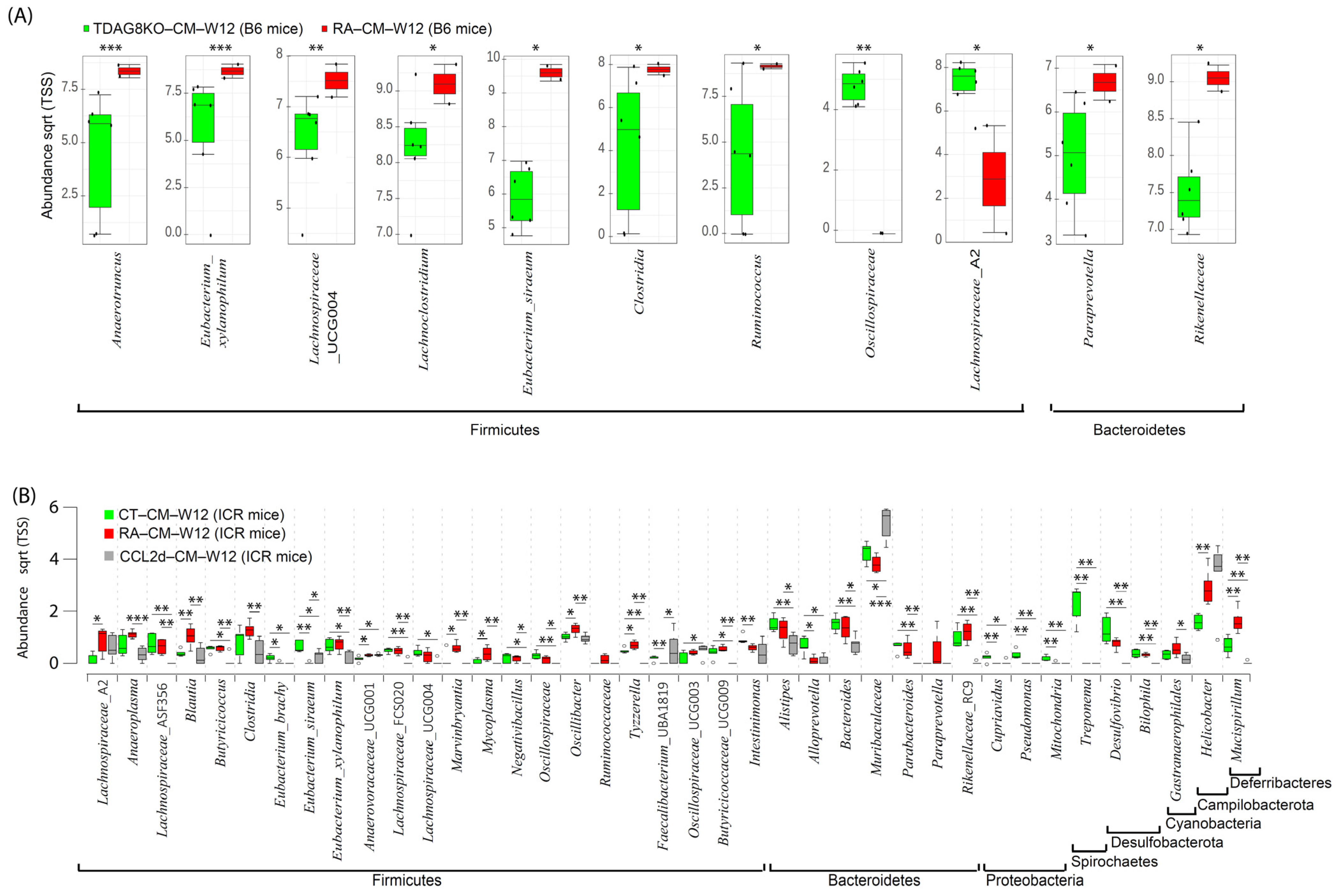
2.5. Restoration of Microbiome in Cecal Mucosa in TDAG8−/− and CCL-2d-Treated Mice
3. Discussion
4. Materials and Methods
4.1. Agents
4.2. Animals
4.3. Arthritis Induction and Drug Treatment
4.4. Sample Collection
4.5. DNA Extraction and Sequencing
4.6. Bioinformatics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wells, P.M.; Williams, F.M.K.; Matey-Hernandez, M.L.; Menni, C.; Steves, C.J. RA and the microbiome: Do host genetic factors provide the link? J. Autoimmun. 2019, 99, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Abramson, S.B. The microbiome and rheumatoid arthritis. Nat. Rev. Rheumatol. 2011, 7, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Vandana, U.K.; Barlaskar, N.H.; Gulzar, A.B.M.; Laskar, I.H.; Kumar, D.; Paul, P.; Pandey, P.; Mazumder, P.B. Linking gut microbiota with the human diseases. Bioinformation 2020, 16, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Scherer, H.U.; Haupl, T.; Burmester, G.R. The etiology of rheumatoid arthritis. J. Autoimmun. 2020, 110, 102400. [Google Scholar] [CrossRef] [PubMed]
- Konig, M.F. The microbiome in autoimmune rheumatic disease. Best Pract. Res. Clin. Rheumatol. 2020, 34, 101473. [Google Scholar] [CrossRef]
- Vaahtovuo, J.; Munukka, E.; Korkeamaki, M.; Luukkainen, R.; Toivanen, P. Fecal microbiota in early rheumatoid arthritis. J. Rheumatol. 2008, 35, 1500–1505. [Google Scholar]
- Scher, J.U.; Ubeda, C.; Equinda, M.; Khanin, R.; Buischi, Y.; Viale, A.; Lipuma, L.; Attur, M.; Pillinger, M.H.; Weissmann, G.; et al. Periodontal disease and the oral microbiota in new-onset rheumatoid arthritis. Arthritis Rheum. 2012, 64, 3083–3094. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef]
- Jeong, Y.; Kim, J.W.; You, H.J.; Park, S.J.; Lee, J.; Ju, J.H.; Park, M.S.; Jin, H.; Cho, M.L.; Kwon, B.; et al. Gut Microbial Composition and Function Are Altered in Patients with Early Rheumatoid Arthritis. J. Clin. Med. 2019, 8, 693. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chen, Q.; Lin, P.; Xu, R.; He, D.; Ji, W.; Bian, Y.; Shen, Y.; Li, Q.; Liu, C.; et al. Characteristics of Gut Microbiota in Patients With Rheumatoid Arthritis in Shanghai, China. Front. Cell. Infect. Microbiol. 2019, 9, 369. [Google Scholar] [CrossRef] [Green Version]
- Trentham, D.E.; Townes, A.S.; Kang, A.H. Autoimmunity to type II collagen an experimental model of arthritis. J. Exp. Med. 1977, 146, 857–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtenay, J.S.; Dallman, M.J.; Dayan, A.D.; Martin, A.; Mosedale, B. Immunisation against heterologous type II collagen induces arthritis in mice. Nature 1980, 283, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Knight, B.; Katz, D.R.; Isenberg, D.A.; Ibrahim, M.A.; Le Page, S.; Hutchings, P.; Schwartz, R.S.; Cooke, A. Induction of adjuvant arthritis in mice. Clin. Exp. Immunol. 1992, 90, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Ditzel, H.J. The K/BxN mouse: A model of human inflammatory arthritis. Trends Mol. Med. 2004, 10, 40–45. [Google Scholar] [CrossRef]
- Hsieh, W.S.; Kung, C.C.; Huang, S.L.; Lin, S.C.; Sun, W.H. TDAG8, TRPV1, and ASIC3 involved in establishing hyperalgesic priming in experimental rheumatoid arthritis. Sci. Rep. 2017, 7, 8870. [Google Scholar] [CrossRef] [Green Version]
- Barton, N.J.; McQueen, D.S.; Thomson, D.; Gauldie, S.D.; Wilson, A.W.; Salter, D.M.; Chessell, I.P. Attenuation of experimental arthritis in TRPV1R knockout mice. Exp. Mol. Pathol. 2006, 81, 166–170. [Google Scholar] [CrossRef]
- Christensen, B.N.; Kochukov, M.; McNearney, T.A.; Taglialatela, G.; Westlund, K.N. Proton-sensing G protein-coupled receptor mobilizes calcium in human synovial cells. Am. J. Physiol. Cell Physiol. 2005, 289, C601–C608. [Google Scholar] [CrossRef] [Green Version]
- Ikeuchi, M.; Kolker, S.J.; Burnes, L.A.; Walder, R.Y.; Sluka, K.A. Role of ASIC3 in the primary and secondary hyperalgesia produced by joint inflammation in mice. Pain 2008, 137, 662–669. [Google Scholar] [CrossRef] [Green Version]
- Dai, S.P.; Hsieh, W.S.; Chen, C.H.; Lu, Y.H.; Huang, H.S.; Chang, D.M.; Huang, S.L.; Sun, W.H. TDAG8 deficiency reduces satellite glial number and pro-inflammatory macrophage number to relieve rheumatoid arthritis disease severity and chronic pain. J. Neuroinflam. 2020, 17, 170. [Google Scholar] [CrossRef]
- Kung, C.C.; Dai, S.P.; Chiang, H.; Huang, H.S.; Sun, W.H. Temporal expression patterns of distinct cytokines and M1/M2 macrophage polarization regulate rheumatoid arthritis progression. Mol. Biol. Rep. 2020, 47, 3423–3437. [Google Scholar] [CrossRef]
- Cortes, A.; Hadler, J.; Pointon, J.P.; Robinson, P.C.; Karaderi, T.; Leo, P.; Cremin, K.; Pryce, K.; Harris, J.; Lee, S.; et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat. Genet. 2013, 45, 730–738. [Google Scholar] [PubMed] [Green Version]
- Al-Mossawi, M.H.; Chen, L.; Fang, H.; Ridley, A.; de Wit, J.; Yager, N.; Hammitzsch, A.; Pulyakhina, I.; Fairfax, B.P.; Simone, D.; et al. Unique transcriptome signatures and GM-CSF expression in lymphocytes from patients with spondyloarthritis. Nat. Commun. 2017, 8, 1510. [Google Scholar] [CrossRef]
- Gaublomme, J.T.; Yosef, N.; Lee, Y.; Gertner, R.S.; Yang, L.V.; Wu, C.; Pandolfi, P.P.; Mak, T.; Satija, R.; Shalek, A.K.; et al. Single-Cell Genomics Unveils Critical Regulators of Th17 Cell Pathogenicity. Cell 2015, 163, 1400–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, S.; Bao, D.; Doyle-Meyers, L.; Dufour, J.; Wu, Y.; Liu, Y.Z.; Ling, B. Alterations of the gut bacterial microbiota in rhesus macaques with SIV infection and on short- or long-term antiretroviral therapy. Sci. Rep. 2020, 10, 19056. [Google Scholar] [CrossRef] [PubMed]
- Kozik, A.J.; Nakatsu, C.H.; Chun, H.; Jones-Hall, Y.L. Comparison of the fecal, cecal, and mucus microbiome in male and female mice after TNBS-induced colitis. PLoS ONE 2019, 14, e0225079. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Li, X.; Zhang, J.; Wang, C.; Lu, F. Effects of Different Diets on Microbiota in The Small Intestine Mucus and Weight Regulation in Rats. Sci. Rep. 2019, 9, 8500. [Google Scholar] [CrossRef]
- Peng, J.; Lu, X.; Xie, K.; Xu, Y.; He, R.; Guo, L.; Han, Y.; Wu, S.; Dong, X.; Lu, Y.; et al. Dynamic Alterations in the Gut Microbiota of Collagen-Induced Arthritis Rats Following the Prolonged Administration of Total Glucosides of Paeony. Front. Cell. Infect. Microbiol. 2019, 9, 204. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Mannaa, M.; Kim, Y.; Kim, J.; Kim, G.T.; Seo, Y.S. Comparative Analysis of Fecal Microbiota Composition Between Rheumatoid Arthritis and Osteoarthritis Patients. Genes 2019, 10, 748. [Google Scholar] [CrossRef] [Green Version]
- De Luca, F.; Shoenfeld, Y. The microbiome in autoimmune diseases. Clin. Exp. Immunol. 2019, 195, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Bergot, A.S.; Giri, R.; Thomas, R. The microbiome and rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2019, 33, 101497. [Google Scholar] [CrossRef]
- Rohr, M.; Narasimhulu, C.A.; Sharma, D.; Doomra, M.; Riad, A.; Naser, S.; Parthasarathy, S. Inflammatory Diseases of the Gut. J. Med. Food 2018, 21, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Takeda, K. Role of Gut Microbiota in Rheumatoid Arthritis. J. Clin. Med. 2017, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Rogier, R.; Evans-Marin, H.; Manasson, J.; van der Kraan, P.M.; Walgreen, B.; Helsen, M.M.; van den Bersselaar, L.A.; van de Loo, F.A.; van Lent, P.L.; Abramson, S.B.; et al. Alteration of the intestinal microbiome characterizes preclinical inflammatory arthritis in mice and its modulation attenuates established arthritis. Sci. Rep. 2017, 7, 15613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Benayoun, B.A. The microbiome: An emerging key player in aging and longevity. Transl. Med. Aging 2020, 4, 103–116. [Google Scholar] [CrossRef]
- Ke, S.; Fang, S.; He, M.; Huang, X.; Yang, H.; Yang, B.; Chen, C.; Huang, L. Age-based dynamic changes of phylogenetic composition and interaction networks of health pig gut microbiome feeding in a uniformed condition. BMC Vet. Res. 2019, 15, 172. [Google Scholar] [CrossRef] [Green Version]
- Schwiertz, A.; Taras, D.; Schafer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef]
- Severijnen, A.J.; van Kleef, R.; Hazenberg, M.P.; van de Merwe, J.P. Chronic arthritis induced in rats by cell wall fragments of Eubacterium species from the human intestinal flora. Infect. Immun. 1990, 58, 523–528. [Google Scholar] [CrossRef] [Green Version]
- Bodkhe, R.; Balakrishnan, B.; Taneja, V. The role of microbiome in rheumatoid arthritis treatment. Ther. Adv. Musculoskelet. Dis. 2019, 11, 1759720X19844632. [Google Scholar] [CrossRef]
- Picchianti-Diamanti, A.; Panebianco, C.; Salemi, S.; Sorgi, M.L.; Di Rosa, R.; Tropea, A.; Sgrulletti, M.; Salerno, G.; Terracciano, F.; D’Amelio, R.; et al. Analysis of Gut Microbiota in Rheumatoid Arthritis Patients: Disease-Related Dysbiosis and Modifications Induced by Etanercept. Int. J. Mol. Sci. 2018, 19, 2938. [Google Scholar] [CrossRef] [Green Version]
- Henke, M.T.; Kenny, D.J.; Cassilly, C.D.; Vlamakis, H.; Xavier, R.J.; Clardy, J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc. Natl. Acad. Sci. USA 2019, 116, 12672–12677. [Google Scholar] [CrossRef] [Green Version]
- Nishino, K.; Nishida, A.; Inoue, R.; Kawada, Y.; Ohno, M.; Sakai, S.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Kawahara, M.; et al. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J. Gastroenterol. 2018, 53, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, M.E.; Ciccia, F.; Willner, D.; Warrington, N.; Robinson, P.C.; Gardiner, B.; Marshall, M.; Kenna, T.J.; Triolo, G.; Brown, M.A. Brief Report: Intestinal Dysbiosis in Ankylosing Spondylitis. Arthritis Rheumatol. 2015, 67, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.R.; Wasan, A.D.; Bingham, C.O., 3rd; Bathon, J.; Haythornthwaite, J.A.; Smith, M.T.; Page, G.G. Enhanced reactivity to pain in patients with rheumatoid arthritis. Arthritis Res. Ther. 2009, 11, R61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauldie, S.D.; McQueen, D.S.; Clarke, C.J.; Chessell, I.P. A robust model of adjuvant-induced chronic unilateral arthritis in two mouse strains. J. Neurosci. Methods 2004, 139, 281–291. [Google Scholar] [CrossRef]
- Chen, C.L.; Liu, F.L.; Lee, C.C.; Chen, T.C.; Ahmed Ali, A.A.; Sytwu, H.K.; Chang, D.M.; Huang, H.S. Modified salicylanilide and 3-phenyl-2H-benzo[e][1,3]oxazine-2,4(3H)-dione derivatives as novel inhibitors of osteoclast differentiation and bone resorption. J. Med. Chem. 2014, 57, 8072–8085. [Google Scholar] [CrossRef]
- Biesbroek, G.; Sanders, E.A.; Roeselers, G.; Wang, X.; Caspers, M.P.; Trzcinski, K.; Bogaert, D.; Keijser, B.J. Deep sequencing analyses of low density microbial communities: Working at the boundary of accurate microbiota detection. PLoS ONE 2012, 7, e32942. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.R.; Neff, N.F.; Kalisky, T.; Dalerba, P.; Treutlein, B.; Rothenberg, M.E.; Mburu, F.M.; Mantalas, G.L.; Sim, S.; Clarke, M.F.; et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods 2014, 11, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewski, M.; Proietti, C.; Ellis, J.J.; Hasan, S.; Brion, M.J.; Berger, B.; Krause, L. Calypso: A user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics 2017, 33, 782–783. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Body Site | RA | Time a | Sequences b | Complete Datasets | Resampled Datasets | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Faith_pd | Shannon | Pielou_e | N | Faith_pd | Shannon | Pielou_e | |||||
| CT1-CC | Cecum Content | No | 0 | 74,635 | 253 | 15.17 | 6.76 | 0.85 | 252 | 15.16 | 6.76 | 0.85 |
| CT2-CC | Cecum Content | No | 0 | 40,683 | 213 | 13.24 | 6.83 | 0.88 | 213 | 13.24 | 6.83 | 0.88 |
| CT3-CC | Cecum Content | No | 0 | 44,222 | 230 | 15.47 | 6.82 | 0.87 | 230 | 15.47 | 6.82 | 0.87 |
| CT4-CC | Cecum Content | No | 12 | 32,782 | 213 | 17.99 | 6.75 | 0.87 | 213 | 17.99 | 6.75 | 0.87 |
| CT5-CC | Cecum Content | No | 12 | 36,207 | 211 | 17.82 | 6.64 | 0.86 | 211 | 17.82 | 6.64 | 0.86 |
| CT6-CC | Cecum Content | No | 12 | 40,825 | 230 | 14.54 | 6.77 | 0.86 | 230 | 14.54 | 6.77 | 0.86 |
| CT1-CM | Cecum Mucosa | No | 0 | 89,676 | 335 | 18.95 | 7.00 | 0.84 | 331 | 18.68 | 7.00 | 0.84 |
| CT2-CM | Cecum Mucosa | No | 0 | 86,444 | 312 | 17.83 | 6.85 | 0.83 | 311 | 17.82 | 6.85 | 0.83 |
| CT3-CM | Cecum Mucosa | No | 0 | 48,210 | 237 | 16.39 | 6.98 | 0.88 | 236 | 16.34 | 6.97 | 0.88 |
| CT4-CM | Cecum Mucosa | No | 0 | 45,748 | 218 | 16.04 | 6.86 | 0.88 | 218 | 16.04 | 6.86 | 0.88 |
| CT5-CM | Cecum Mucosa | No | 0 | 44,071 | 246 | 18.34 | 6.93 | 0.87 | 246 | 18.34 | 6.93 | 0.87 |
| CT6-CM | Cecum Mucosa | No | 0 | 46,717 | 236 | 15.26 | 6.88 | 0.87 | 236 | 15.26 | 6.89 | 0.87 |
| CT7-CM | Cecum Mucosa | No | 12 | 34,821 | 223 | 17.59 | 6.87 | 0.88 | 223 | 17.59 | 6.88 | 0.88 |
| CT8-CM | Cecum Mucosa | No | 12 | 36,539 | 215 | 16.79 | 6.70 | 0.86 | 215 | 16.79 | 6.70 | 0.87 |
| CT9-CM | Cecum Mucosa | No | 12 | 54,317 | 335 | 22.06 | 7.40 | 0.88 | 335 | 22.06 | 7.39 | 0.88 |
| CT10-CM | Cecum Mucosa | No | 12 | 40,313 | 250 | 17.64 | 6.74 | 0.85 | 250 | 17.64 | 6.74 | 0.85 |
| CT11-CM | Cecum Mucosa | No | 12 | 43,223 | 236 | 15.62 | 6.87 | 0.87 | 236 | 15.62 | 6.87 | 0.87 |
| CT1-Feces | Feces | No | 0 | 84,733 | 280 | 16.32 | 6.87 | 0.85 | 276 | 16.26 | 6.86 | 0.85 |
| CT2-Feces | Feces | No | 0 | 33,060 | 167 | 13.06 | 6.31 | 0.85 | 167 | 13.06 | 6.31 | 0.85 |
| CT3-Feces | Feces | No | 0 | 36,225 | 203 | 14.08 | 6.72 | 0.88 | 203 | 14.08 | 6.72 | 0.88 |
| CT4-Feces | Feces | No | 0 | 56,866 | 178 | 13.77 | 5.67 | 0.76 | 178 | 13.77 | 5.67 | 0.76 |
| CT5-Feces | Feces | No | 12 | 32,045 | 143 | 12.15 | 6.13 | 0.86 | 143 | 12.15 | 6.13 | 0.86 |
| CT6-Feces | Feces | No | 12 | 36,843 | 203 | 16.31 | 6.59 | 0.86 | 203 | 16.31 | 6.60 | 0.86 |
| CT7-Feces | Feces | No | 12 | 41,521 | 206 | 15.42 | 6.58 | 0.86 | 206 | 15.42 | 6.58 | 0.86 |
| RA1-CC | Cecum Content | Yes | 4 | 38,237 | 128 | 10.95 | 5.87 | 0.84 | 128 | 10.95 | 5.87 | 0.84 |
| RA2-CC | Cecum Content | Yes | 8 | 76,017 | 251 | 14.36 | 6.86 | 0.86 | 250 | 14.36 | 6.86 | 0.86 |
| RA3-CC | Cecum Content | Yes | 12 | 34,001 | 124 | 11.42 | 5.88 | 0.85 | 124 | 11.42 | 5.88 | 0.85 |
| RA1-CM | Cecum Mucosa | Yes | 4 | 87,628 | 283 | 17.09 | 6.65 | 0.82 | 281 | 17.09 | 6.64 | 0.82 |
| RA2-CM | Cecum Mucosa | Yes | 4 | 78,404 | 229 | 15.35 | 6.52 | 0.83 | 229 | 15.35 | 6.51 | 0.83 |
| RA3-CM | Cecum Mucosa | Yes | 8 | 105,853 | 360 | 19.19 | 7.27 | 0.86 | 359 | 19.19 | 7.26 | 0.86 |
| RA4-CM | Cecum Mucosa | Yes | 8 | 102,650 | 352 | 19.60 | 7.26 | 0.86 | 351 | 19.60 | 7.26 | 0.86 |
| RA5-CM | Cecum Mucosa | Yes | 12 | 93,783 | 279 | 17.09 | 6.39 | 0.79 | 276 | 17.08 | 6.37 | 0.79 |
| RA6-CM | Cecum Mucosa | Yes | 12 | 90,629 | 299 | 17.24 | 7.04 | 0.86 | 297 | 17.23 | 7.03 | 0.86 |
| RA7-CM | Cecum Mucosa | Yes | 12 | 99,632 | 336 | 16.82 | 7.33 | 0.87 | 335 | 16.81 | 7.31 | 0.87 |
| RA8-CM | Cecum Mucosa | Yes | 12 | 84,482 | 297 | 16.11 | 7.04 | 0.86 | 297 | 16.11 | 7.04 | 0.86 |
| RA9-CM | Cecum Mucosa | Yes | 12 | 79,893 | 296 | 15.76 | 6.96 | 0.85 | 292 | 15.64 | 6.95 | 0.85 |
| RA10-CM | Cecum Mucosa | Yes | 12 | 79,591 | 295 | 16.50 | 7.08 | 0.86 | 294 | 16.47 | 7.08 | 0.86 |
| RA11-CM | Cecum Mucosa | Yes | 12 | 69,997 | 181 | 12.52 | 6.36 | 0.85 | 181 | 12.52 | 6.36 | 0.85 |
| RA12-CM | Cecum Mucosa | Yes | 12 | 69,816 | 187 | 13.41 | 6.29 | 0.83 | 187 | 13.41 | 6.30 | 0.83 |
| RA1-Feces | Feces | Yes | 1 | 76,432 | 170 | 13.83 | 5.99 | 0.81 | 170 | 13.83 | 5.98 | 0.81 |
| RA2-Feces | Feces | Yes | 1 | 57,333 | 170 | 13.15 | 5.78 | 0.78 | 170 | 13.15 | 5.77 | 0.78 |
| RA3-Feces | Feces | Yes | 2 | 69,420 | 190 | 14.01 | 6.43 | 0.85 | 190 | 14.01 | 6.42 | 0.85 |
| RA4-Feces | Feces | Yes | 3 | 53,839 | 131 | 11.52 | 5.90 | 0.84 | 130 | 11.29 | 5.90 | 0.84 |
| RA5-Feces | Feces | Yes | 4 | 64,200 | 162 | 12.78 | 5.82 | 0.79 | 162 | 12.78 | 5.83 | 0.79 |
| RA6-Feces | Feces | Yes | 4 | 45,970 | 100 | 10.28 | 5.51 | 0.83 | 100 | 10.28 | 5.50 | 0.83 |
| RA7-Feces | Feces | Yes | 4 | 54,931 | 169 | 12.50 | 5.77 | 0.78 | 169 | 12.50 | 5.78 | 0.78 |
| RA8-Feces | Feces | Yes | 5 | 53,077 | 95 | 10.07 | 5.39 | 0.82 | 95 | 10.07 | 5.39 | 0.82 |
| RA9-Feces | Feces | Yes | 6 | 57,492 | 160 | 12.81 | 5.98 | 0.82 | 160 | 12.81 | 5.98 | 0.82 |
| RA10-Feces | Feces | Yes | 7 | 55,934 | 120 | 12.36 | 5.79 | 0.84 | 120 | 12.36 | 5.78 | 0.84 |
| RA11-Feces | Feces | Yes | 8 | 69,791 | 226 | 14.58 | 6.61 | 0.85 | 226 | 14.58 | 6.62 | 0.85 |
| RA12-Feces | Feces | Yes | 8 | 52,320 | 115 | 10.93 | 5.74 | 0.84 | 115 | 10.93 | 5.73 | 0.84 |
| RA13-Feces | Feces | Yes | 8 | 57,355 | 177 | 13.04 | 5.95 | 0.80 | 177 | 13.04 | 5.93 | 0.79 |
| RA14-Feces | Feces | Yes | 9 | 56,109 | 123 | 11.87 | 5.57 | 0.80 | 123 | 11.87 | 5.56 | 0.80 |
| RA15-Feces | Feces | Yes | 10 | 47,292 | 123 | 11.06 | 5.84 | 0.84 | 123 | 11.06 | 5.84 | 0.84 |
| RA16-Feces | Feces | Yes | 11 | 44,693 | 106 | 11.32 | 5.55 | 0.83 | 106 | 11.32 | 5.54 | 0.82 |
| RA17-Feces | Feces | Yes | 12 | 87,829 | 266 | 15.15 | 7.01 | 0.87 | 265 | 15.15 | 7.00 | 0.87 |
| RA18-Feces | Feces | Yes | 12 | 59,657 | 222 | 14.21 | 6.73 | 0.86 | 222 | 14.21 | 6.74 | 0.86 |
| RA19-Feces | Feces | Yes | 12 | 46,108 | 119 | 10.88 | 5.54 | 0.80 | 119 | 10.88 | 5.53 | 0.80 |
| Taxa (Phylum) | p Value | FDR | Bonferroni | AUC | 95% CI | OR No/Yes | 95% CI | Delta | Fold-Change in Expression | Mean CT | Mean RA |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fecal sample | |||||||||||
| Spirochaetes | 0.00013 | 0.0016 | 0.0016 | 0.83 | 0.63–1 | 38 | 3.65–1005.62 | 2.15 | 159,762.9 | 1.12 | 0 |
| Patescibacteria | 0.0013 | 0.0075 | 0.014 | 0.95 | 0.84–1 | 20 | 2.39–447.71 | 1.93 | 2.56 | 1.91 | 0.75 |
| Proteobacteria | 0.0074 | 0.03 | 0.074 | 0.87 | 0.73–1 | 0.05 | 0–0.42 | 1.41 | −2.22 | 0.7 | 1.56 |
| Desulfobacterota | 0.038 | 0.12 | 0.35 | 0.79 | 0.6–0.97 | 0.09 | 0–0.68 | 1.23 | −1.59 | 1.28 | 2.04 |
| Mucus sample | |||||||||||
| Parabasalia | 0.0047 | 0.02 | 0.065 | 0.77 | 0.62–0.93 | 13.2 | 1.66–287.14 | 1.58 | 11,023.73 | 0.09 | 0 |
| Bacteroidetes | 0.0051 | 0.02 | 0.066 | 0.85 | 0.69–1 | 13.33 | 2.07–130.86 | 1.34 | 1.1 | 5.82 | 5.29 |
| Spirochaetes | 0.026 | 0.083 | 0.31 | 0.76 | 0.55–0.96 | 19.25 | 2.4–425.96 | 1.68 | 8.98 | 1.32 | 0.15 |
| Deferribacteres | 0.000067 | 0.0011 | 0.0011 | 0.95 | 0.87–1 | 0.03 | 0–0.28 | 2.16 | −2.44 | 0.63 | 1.54 |
| Campilobacterota | 0.0028 | 0.02 | 0.042 | 0.87 | 0.71–1 | 0.04 | 0–0.31 | 1.72 | −1.49 | 1.99 | 2.96 |
| Phylum | Genus | CT–RA | p Value | Genus | CT–RA | p Value |
|---|---|---|---|---|---|---|
| Firmicutes | Eubacterium_Ventriosum |  | 0.01 | Roseburia |  | 0.01 |
| Lachnospiraceae_UCG001 |  | 0.05 | Ruminococcus |  | 0.01 | |
| Monoglobus |  | 0.05 | Turicibacter |  | 0.05 | |
| Oscillospiraceae_NK4A214 |  | 0.05 | Tuzzerella |  | 0.05 | |
| Bacilli_RF39 |  | 0.05 | ||||
| Bacteroidetes | Alistipes |  | 0.05 | Odoribacter |  | 0.01 |
| Alloprevotella |  | 0.05 | Parabacteroides |  | 0.05 | |
| Rikenella |  | 0.01 | Rikenellaceae_RC9 |  | 0.05 | |
| Muribaculum |  | 0.01 | ||||
| Proteobacteria | Parasutterella |  | 0.01 | |||
| Spirochaetes | Treponema |  | 0.005 | Brachyspira |  | 0.05 |
| Patescibacteria | Candidatus_Saccharimonas |  | 0.05 | Candidatus_Arthromis |  | 0.01 |
| Phylum | Genus | Week 1–12 | Week 12 | ||||
|---|---|---|---|---|---|---|---|
| CT–RA (ICR Mice) | p Value | TDAG8KO-RA (B6 Mice) | p value | CT–RA–CCL2d (ICR Mice) | p Value | ||
| Firmicutes | Negativibacillus |  | 0.01 | NA | NA |  | 0.05 |
| Eubacterium_Brachy |  | 0.05 | NA | NA |  | 0.05 | |
| Eubacterium_Siraeum |  | 0.005 |  | 0.05 |  | 0.05 | |
| Eubacterium_Xylanophilum | NA | NA |  | 0.001 |  | 0.05 | |
| Intestinimonas |  | 0.05 | NA | NA |  | 0.05 | |
| Lachnospiraceae_A2 |  | 0.05 |  | 0.05 |  | 0.05 | |
| Anaeroplasma |  | 0.01 | NA | NA |  | 0.05 | |
| Blautia |  | 0.05 | NA | NA |  | 0.01 | |
| Lachnoclostridium |  | 0.05 |  | 0.05 | NA | NA | |
| Anaerovoracaceae_UCG001 |  | NA | NA | NA |  | 0.05 | |
| Marvinbryantia |  | 0.005 | NA | NA |  | 0.05 | |
| Clostridia | NA | NA |  | 0.05 |  | 0.05 | |
| Oscillospiraceae | NA | NA |  | 0.01 |  | 0.05 | |
| Ruminococcus | NA | NA |  | 0.05 |  | 0.05 | |
| Bacteroidetes | Paraprevotella | NA | NA |  | 0.05 |  | 0.01 |
| Alloprevotella |  | 0.05 | NA | NA |  | 0.05 | |
| Muribaculaceae |  | 0.01 | NA | NA |  | 0.05 | |
| Rikenellaceae | NA | NA |  | 0.05 |  | 0.01 | |
| Proteobacteria | Mitochondria |  | 0.01 | NA | NA |  | 0.01 |
| Pseudomonas |  | 0.01 | NA | NA |  | 0.01 | |
| Spirochaetes | Treponema |  | 0.005 | NA | NA |  | 0.01 |
| Campilobacterot | Helicobacter |  | 0.01 | NA | NA |  | 0.01 |
| Deferribacteres | Mucispirillum |  | 0.005 | NA | NA |  | 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, N.T.; Sun, W.-H.; Chen, T.-H.; Tsai, P.-C.; Chen, C.-C.; Huang, S.-L. Gut Mucosal Microbiome Is Perturbed in Rheumatoid Arthritis Mice and Partly Restored after TDAG8 Deficiency or Suppression by Salicylanilide Derivative. Int. J. Mol. Sci. 2022, 23, 3527. https://doi.org/10.3390/ijms23073527
Nguyen NT, Sun W-H, Chen T-H, Tsai P-C, Chen C-C, Huang S-L. Gut Mucosal Microbiome Is Perturbed in Rheumatoid Arthritis Mice and Partly Restored after TDAG8 Deficiency or Suppression by Salicylanilide Derivative. International Journal of Molecular Sciences. 2022; 23(7):3527. https://doi.org/10.3390/ijms23073527
Chicago/Turabian StyleNguyen, Ngoc Tuan, Wei-Hsin Sun, Tzu-Hsuan Chen, Po-Chun Tsai, Chih-Chen Chen, and Shir-Ly Huang. 2022. "Gut Mucosal Microbiome Is Perturbed in Rheumatoid Arthritis Mice and Partly Restored after TDAG8 Deficiency or Suppression by Salicylanilide Derivative" International Journal of Molecular Sciences 23, no. 7: 3527. https://doi.org/10.3390/ijms23073527
APA StyleNguyen, N. T., Sun, W.-H., Chen, T.-H., Tsai, P.-C., Chen, C.-C., & Huang, S.-L. (2022). Gut Mucosal Microbiome Is Perturbed in Rheumatoid Arthritis Mice and Partly Restored after TDAG8 Deficiency or Suppression by Salicylanilide Derivative. International Journal of Molecular Sciences, 23(7), 3527. https://doi.org/10.3390/ijms23073527