
Role of Obesity, Physical Exercise, Adipose Tissue-Skeletal Muscle Crosstalk and Molecular Advances in Barrett’s Esophagus and Esophageal Adenocarcinoma

 ,
,  , ,
, ,  , , , , and
, , , , and 
Abstract
1. Introduction
2. Epidemiology
3. The Role of Obesity in BE and EAC Development
3.1. Obesity and GERD
3.2. Role of Adipokines
3.2.1. Leptin
3.2.2. Adiponectin
3.3. The Role of Insulin Resistance
3.4. Role of Diet
4. Role of Physical Activity
Role of Adipose Tissue-Muscle Crosstalk
5. Molecular Alterations in Experimental and Clinical BE and EAC Complicated by Obesity
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, J.; Koulaouzidis, A.; Marlicz, W.; Lok, V.; Chu, C.; Ngai, C.H.; Zhang, L.; Chen, P.; Wang, S.; Yuan, J.; et al. Global Burden, Risk Factors, and Trends of Esophageal Cancer: An Analysis of Cancer Registries from 48 Countries. Cancers 2021, 13, 141. [Google Scholar] [CrossRef] [PubMed]
- Kubo, A.; Corley, D.A. Body Mass Index and Adenocarcinomas of the Esophagus or Gastric Cardia: A Systematic Review and Meta-analysis. Cancer Epidemiol. Biomark. Prev. 2006, 15, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Sharma, A.N.; Murad, M.H.; Buttar, N.S.; El-Serag, H.B.; Katzka, D.A.; Iyer, P.G. Central Adiposity is Associated with Increased Risk of Esophageal Inflammation, Metaplasia, and Adenocarcinoma: A Systematic Review and Meta–analysis. Clin. Gastroenterol. Hepatol. 2013, 11, 1399–1412.e1397. [Google Scholar] [CrossRef] [PubMed]
- Leggett, C.L.; Nelsen, E.M.; Tian, J.; Schleck, C.B.; Zinsmeister, A.R.; Dunagan, K.T.; Locke, G.R., 3rd; Wang, K.K.; Talley, N.J.; Iyer, P.G. Metabolic Syndrome as a Risk Factor for Barrett Esophagus: A Population-based Case–control Study. Mayo Clin. Proc. 2013, 88, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Rustgi, A.K.; El–Serag, H.B. Esophageal Carcinoma. N. Engl. J. Med. 2014, 371, 2499–2509. [Google Scholar] [CrossRef] [PubMed]
- Chevallier, J.M.; Chiappetta, S.; Musella, M. Obesity: Barrett’s Esophagus and Esophageal Cancer Risk. In Revisiting Barrett’s Esophagus; Springer: Berlin/Heidelberg, Germany, 2019; pp. 39–50. [Google Scholar]
- Then, E.O.; Lopez, M.; Saleem, S.; Gayam, V.; Sunkara, T.; Culliford, A.; Gaduputi, V. Esophageal Cancer: An Updated Surveillance Epidemiology and End Results Database Analysis. World J. Oncol. 2020, 11, 55–64. [Google Scholar] [CrossRef]
- Arnold, M.; Laversanne, M.; Brown, L.M.; Devesa, S.S.; Bray, F. Predicting the Future Burden of Esophageal Cancer by Histological Subtype: International Trends in Incidence up to 2030. Am. J. Gastroenterol. 2017, 112, 1247–1255. [Google Scholar] [CrossRef]
- Thrift, A.P. Global Burden and Epidemiology of Barrett Oesophagus and Oesophageal cancer. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 432–443. [Google Scholar] [CrossRef]
- El–Serag, H.B. The Epidemic of Esophageal Adenocarcinoma. Gastroenterol. Clin. N. Am. 2002, 31, 421–440. [Google Scholar] [CrossRef]
- Pohl, H.; Sirovich, B.; Welch, H.G. Esophageal Adenocarcinoma Incidence: Are We Reaching the Peak? Cancer Epidemiol. Biomark. Prev. 2010, 19, 1468–1470. [Google Scholar] [CrossRef]
- Cook, M.B.; Chow, W.H.; Devesa, S.S. Oesophageal Cancer Incidence in the United States by Race, Sex, and Histologic Type, 1977–2005. Br. J. Cancer 2009, 101, 855–859. [Google Scholar] [CrossRef] [PubMed]
- Botterweck, A.A.; Schouten, L.J.; Volovics, A.; Dorant, E.; van Den Brandt, P.A. Trends in Incidence of Adenocarcinoma of the Oesophagus and Gastric Cardia in Ten European Countries. Int. J. Epidemiol. 2000, 29, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Lieberman, D.A.; Kennedy, K.F.; Hamade, N.; Thota, P.; Parasa, S.; Gorrepati, V.S.; Bansal, A.; Gupta, N.; Gaddam, S. Increasing Prevalence of High-grade Dysplasia and Adenocarcinoma on Index Endoscopy in Barrett’s Esophagus over the Past 2 Decades: Data from a Multicenter US Consortium. Gastrointest. Endosc. 2019, 89, 257–263.e253. [Google Scholar] [CrossRef] [PubMed]
- Islami, F.; DeSantis, C.E.; Jemal, A. Incidence Trends of Esophageal and Gastric Cancer Subtypes by Race, Ethnicity, and Age in the United States, 1997–2014. Clin. Gastroenterol. Hepatol. 2019, 17, 429–439. [Google Scholar] [CrossRef]
- Zhang, Y. Epidemiology of Esophageal Cancer. World J. Gastroenterol. 2013, 19, 5598–5606. [Google Scholar] [CrossRef]
- Thrift, A.P.; Whiteman, D.C. The Incidence of Esophageal Adenocarcinoma Continues to Rise: Analysis of Period and Birth Cohort Effects on Recent Trends. Ann. Oncol. 2012, 23, 3155–3162. [Google Scholar] [CrossRef]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, Regional, and National Prevalence of Overweight and Obesity in Children and Adults During 1980–2013: A Systematic Analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- El–Serag, H.B.; Sweet, S.; Winchester, C.C.; Dent, J. Update on the Epidemiology of Gastro–oesophageal Reflux Disease: A Systematic Review. Gut 2014, 63, 871–880. [Google Scholar] [CrossRef]
- Kellerman, R.; Kintanar, T. Gastroesophageal Reflux Disease. Prim. Care Clin. Off. Pract. 2017, 44, 561–573. [Google Scholar] [CrossRef]
- Johansson, J.; Hakansson, H.O.; Mellblom, L.; Kempas, A.; Johansson, K.E.; Granath, F.; Nyren, O. Prevalence of Precancerous and Other Metaplasia in the Distal Oesophagus and Gastro–oesophageal Junction. Scand. J. Gastroenterol. 2005, 40, 893–902. [Google Scholar] [CrossRef]
- Cook, M.B.; Corley, D.A.; Murray, L.J.; Liao, L.M.; Kamangar, F.; Ye, W.; Gammon, M.D.; Risch, H.A.; Casson, A.G.; Freedman, N.D.; et al. Gastroesophageal Reflux in Relation to Adenocarcinomas of the Esophagus: A Pooled Analysis from the Barrett’s and Esophageal Adenocarcinoma Consortium (BEACON). PLoS ONE 2014, 9, e103508. [Google Scholar] [CrossRef] [PubMed]
- Gharahkhani, P.; Tung, J.; Hinds, D.; Mishra, A.; Vaughan, T.L.; Whiteman, D.C.; MacGregor, S.; Barrett’s and Esophageal Adenocarcinoma Consortium (BEACON); on behalf of the BEACON Study Investigators. Chronic Gastroesophageal Reflux Disease shares Genetic Background with Esophageal Adenocarcinoma and Barrett’s Esophagus. Hum. Mol. Genet. 2016, 25, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Lagergren, J.; Bergström, R.; Lindgren, A.; Nyrén, O. Symptomatic Gastroesophageal Reflux as a Risk Factor for Esophageal Adenocarcinoma. N. Engl. J. Med. 1999, 340, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Lagergren, J. Adenocarcinoma of Oesophagus: What Exactly is the Size of the Problem and Who is at Risk? Gut 2005, 54 (Suppl. S1), i1–i5. [Google Scholar] [CrossRef]
- Anderson, L.A.; Murphy, S.J.; Johnston, B.T.; Watson, R.; Ferguson, H.; Bamford, K.B.; Ghazy, A.; McCarron, P.; McGuigan, J.; Reynolds, J.V. Relationship between Helicobacter Pylori Infection and Gastric Atrophy and the Stages of the Oesophageal Inflammation, Metaplasia, Adenocarcinoma Sequence: Results from the FINBAR Case–control Study. Gut 2008, 57, 734–739. [Google Scholar] [CrossRef]
- Hvid–Jensen, F.; Pedersen, L.; Drewes, A.M.; Sørensen, H.T.; Funch–Jensen, P. Incidence of Adenocarcinoma Among Patients with Barrett’s Esophagus. N. Engl. J. Med. 2011, 365, 1375–1383. [Google Scholar] [CrossRef]
- Bhat, S.; Coleman, H.G.; Yousef, F.; Johnston, B.T.; McManus, D.T.; Gavin, A.T.; Murray, L.J. Risk of Malignant Progression in Barrett’s Esophagus Patients: Results from a Large Population–based Study. J. Natl. Cancer Inst. 2011, 103, 1049–1057. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef]
- Dong, J.; Buas, M.F.; Gharahkhani, P.; Kendall, B.J.; Onstad, L.; Zhao, S.; Anderson, L.A.; Wu, A.H.; Ye, W.; Bird, N.C.; et al. Determining Risk of Barrett’s Esophagus and Esophageal Adenocarcinoma Based on Epidemiologic Factors and Genetic Variants. Gastroenterology 2018, 154, 1273–1281.e1273. [Google Scholar] [CrossRef]
- Lagergren, K.; Lindam, A.; Lagergren, J. Dietary Proportions of Carbohydrates, Fat, and Protein and Risk of Oesophageal Cancer by Histological Type. PLoS ONE 2013, 8, e54913. [Google Scholar] [CrossRef]
- Schlottmann, F.; Dreifuss, N.H.; Patti, M.G. Obesity and esophageal cancer: GERD, Barrett s Esophagus, and Molecular Carcinogenic Pathways. Expert Rev. Gastroenterol. Hepatol. 2020, 14, 425–433. [Google Scholar] [CrossRef] [PubMed]
- NCD Risk Factor Collaboration (NCD-RisC). Trends in Adult Body-mass Index in 200 Countries from 1975 to 2014: A Pooled Analysis of 1698 Population-based Measurement Studies with 19· 2 Million Participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef]
- Swinburn, B.A.; Sacks, G.; Hall, K.D.; McPherson, K.; Finegood, D.T.; Moodie, M.L.; Gortmaker, S.L. The Global Obesity Pandemic: Shaped by Global Drivers and Local Environments. Lancet 2011, 378, 804–814. [Google Scholar] [CrossRef]
- Flegal, K.M.; Kit, B.K.; Orpana, H.; Graubard, B.I. Association of All–cause Mortality with Overweight and Obesity Using Standard Body Mass Index Categories: A Systematic Review and Meta–analysis. JAMA 2013, 309, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Wolk, A.; Gridley, G.; Svensson, M.; Nyren, O.; McLaughlin, J.K.; Fraumeni, J.F.; Adam, H.O. A Prospective Study of Obesity and Cancer Risk (Sweden). Cancer Causes Control 2001, 12, 13–21. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker–Thurmond, K.; Thun, M.J. Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of US Adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef]
- Furer, A.; Afek, A.; Sommer, A.; Keinan-Boker, L.; Derazne, E.; Levi, Z.; Tzur, D.; Tiosano, S.; Shina, A.; Glick, Y.; et al. Adolescent Obesity and Midlife Cancer Risk: A Population–based Cohort Study of 2.3 Million Adolescents in Israel. Lancet Diabetes Endocrinol. 2020, 8, 216–225. [Google Scholar] [CrossRef]
- Hoyo, C.; Cook, M.B.; Kamangar, F.; Freedman, N.D.; Whiteman, D.C.; Bernstein, L.; Brown, L.M.; Risch, H.A.; Ye, W.; Sharp, L.; et al. Body Mass Index in Relation to Oesophageal and Oesophagogastric Junction Adenocarcinomas: A Pooled Analysis from the International BEACON Consortium. Int. J. Epidemiol. 2012, 41, 1706–1718. [Google Scholar] [CrossRef]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass Index and Incidence of Cancer: A Systematic Review and Meta–analysis of Prospective Observational Studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Arnold, M.; Abnet, C.C.; Neale, R.E.; Vignat, J.; Giovannucci, E.L.; McGlynn, K.A.; Bray, F. Global Burden of 5 Major Types of Gastrointestinal Cancer. Gastroenterology 2020, 159, 335–349.e315. [Google Scholar] [CrossRef]
- Lagergren, J.; Mattsson, F.; Nyren, O. Gastroesophageal reflux does not alter effects of body mass index on risk of esophageal adenocarcinoma. Clin. Gastroenterol. Hepatol. 2014, 12, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Coleman, H.G.; Xie, S.H.; Lagergren, J. The Epidemiology of Esophageal Adenocarcinoma. Gastroenterology 2018, 154, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Kyrgiou, M.; Kalliala, I.; Markozannes, G.; Gunter, M.J.; Paraskevaidis, E.; Gabra, H.; Martin-Hirsch, P.; Tsilidis, K.K. Adiposity and Cancer at Major Anatomical Sites: Umbrella Review of the Literature. BMJ 2017, 356, j477. [Google Scholar] [CrossRef] [PubMed]
- Steffen, A.; Huerta, J.M.; Weiderpass, E.; Bueno-de–Mesquita, H.B.; May, A.M.; Siersema, P.D.; Kaaks, R.; Neamat-Allah, J.; Pala, V.; Panico, S. General and Abdominal Obesity and Risk of Esophageal and Gastric Adenocarcinoma in the European Prospective Investigation into Cancer and Nutrition. Int. J. Cancer 2015, 137, 646–657. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef]
- James, P.T.; Leach, R.; Kalamara, E.; Shayeghi, M. The Worldwide Obesity Epidemic. Obes. Res. 2001, 9 (Suppl. S4), 228S–233S. [Google Scholar] [CrossRef]
- Kamat, P.; Wen, S.; Morris, J.; Anandasabapathy, S. Exploring the Association Between Elevated Body Mass Index and Barrett’s Esophagus: A Systematic Review and Meta–analysis. Ann. Thorac. Surg. 2009, 87, 655–662. [Google Scholar] [CrossRef]
- Corley, D.A.; Kubo, A.; Levin, T.R.; Block, G.; Habel, L.; Zhao, W.; Leighton, P.; Quesenberry, C.; Rumore, G.J.; Buffler, P.A. Abdominal Obesity and Body Mass Index as Risk Factors for Barrett’s Esophagus. Gastroenterology 2007, 133, 34–41. [Google Scholar] [CrossRef]
- World Health Organization. Available online: https://www.who.int/health–topics/obesity#tab=tab_1 (accessed on 19 February 2022).
- Sneed, N.; Morrison, S. Body Composition Methods in Adults with Type 2 Diabetes or at Risk for T2D: A Clinical Review. Curr. Diabetes Rep. 2021, 21, 14. [Google Scholar] [CrossRef]
- Stein, D.J.; El-Serag, H.B.; Kuczynski, J.; Kramer, J.R.; Sampliner, R.E. The Association of Body Mass Index with Barrett’s Oesophagus. Aliment. Pharmacol. Ther. 2005, 22, 1005–1010. [Google Scholar] [CrossRef]
- Abdallah, J.; Maradey–Romero, C.; Lewis, S.; Perzynski, A.; Fass, R. The relationship Between Length of Barrett’s Esophagus Mucosa and Body Mass Index: 9. Am. J. Gastroenterol. ACG 2014, 109, S3. [Google Scholar] [CrossRef]
- Rothman, K.J. BMI–related Errors in the Measurement of Obesity. Int. J. Obes. 2008, 32 (Suppl. S3), S56–S59. [Google Scholar] [CrossRef] [PubMed]
- Pories, W.J.; Dohm, L.G.; Mansfield, C.J. Beyond the BMI: The Search for Better Guidelines for Bariatric Surgery. Obesity 2010, 18, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Mascie–Taylor, C.G.N.; Goto, R. Human Variation and Body Mass Index: A Review of the Universality of BMI Cut–offs, Gender and Urban-rural Differences, and Secular Changes. J. Physiol. Anthropol. 2007, 26, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.; Cai, J.; Pamuk, E.R.; Williamson, D.F.; Thun, M.J.; Wood, J.L. The Effect of Age on the Association Between Body-mass Index and Mortality. N. Engl. J. Med. 1998, 338, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, L.; Del Gobbo, V.; Bigioni, M.; Premrov, M.G.; Cianci, R.; De Lorenzo, A. Body Composition Analyses in Normal Weight Obese Women. Eur. Rev. Med. Pharmacol. Sci. 2006, 10, 191–196. [Google Scholar]
- Dulloo, A.G.; Jacquet, J.; Solinas, G.; Montani, J.P.; Schutz, Y. Body Composition Phenotypes in Pathways to Obesity and the Metabolic Syndrome. Int. J. Obes. 2010, 34 (Suppl. S2), S4–S17. [Google Scholar] [CrossRef]
- Quail, D.F.; Dannenberg, A.J. The Obese Adipose Tissue Microenvironment in Cancer Development and Progression. Nat. Rev. Endocrinol. 2019, 15, 139–154. [Google Scholar] [CrossRef]
- Cani, P.D.; Jordan, B.F. Gut Microbiota–mediated Inflammation in Obesity: A Link With Gastrointestinal Cancer. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 671–682. [Google Scholar] [CrossRef]
- Liu, B.; Giffney, H.E.; Arthur, R.S.; Rohan, T.E.; Dannenberg, A.J. Cancer Risk in Normal Weight Individuals with Metabolic Obesity: A Narrative Review. Cancer Prev. Res. 2021, 14, 509–520. [Google Scholar] [CrossRef]
- Staynor, J.; Smith, M.; Donnelly, C.; Sallam, A.; Ackland, T. DXA reference values and anthropometric screening for visceral obesity in Western Australian adults. Sci. Rep. 2020, 10, 18731. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.; Donlon, N.; Beddy, P.; Donohoe, C.; Doyle, S.; King, S.; Ravi, N.; Reynolds, J. Visceral obesity with and without metabolic syndrome: Incidence and clinical impact in esophageal adenocarcinoma treated with curative intent. Dis. Esophagus 2022, doab094. [Google Scholar] [CrossRef] [PubMed]
- Nelsen, E.; Kirihara, Y.; Takahashi, N.; Shi, Q.; Lewis, J.; Namasivayam, V.; Buttar, N.; Dunagan, K.; Prasad, G. Distribution of Body Fat and Its Influence on Esophageal Inflammation and Dysplasia in Patients With Barrett’s Esophagus. Clin. Gastroenterol. Hepatol. 2012, 10, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Paris, S.; Ekeanyanwu, R.; Jiang, Y.; Davis, D.; Spechler, S.J.; Souza, R.F. Obesity and its effects on the esophageal mucosal barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 321, G335–G343. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, Z.R.; Farrow, D.C.; Bronner, M.P.; Rosen, S.N.; Vaughan, T.L. Central Adiposity and Risk of Barrett’s Esophagus. Gastroenterology 2007, 133, 403–411. [Google Scholar] [CrossRef]
- Nguyen, T.; Khalaf, N.; Ramsey, D.; El–Serag, H.B. Statin Use Is Associated with a Decreased Risk of Barrett’s Esophagus. Gastroenterology 2014, 147, 314–323. [Google Scholar] [CrossRef]
- Beales, I.L.; Dearman, L.; Vardi, I.; Loke, Y. Reduced Rrisk of Barrett’s Esophagus in Statin Users: Case–control Study and Meta-analysis. Dig. Dis. Sci. 2016, 61, 238–246. [Google Scholar] [CrossRef]
- Kantor, E.D.; Onstad, L.; Blount, P.L.; Reid, B.J.; Vaughan, T.L. Use of Statin Medications and Risk of Esophageal Adenocarcinoma in Persons with Barrett’s Esophagus. Cancer Epidemiol. Biomark. Prev. 2012, 21, 456–461. [Google Scholar] [CrossRef]
- Beales, I.; Vardi, I.; Dearman, L.; Broughton, T. Statin Use is Associated with a Reduction in the Incidence of Esophageal Adenocarcinoma: A Case Control Study. Dis. Esophagus 2013, 26, 838–846. [Google Scholar] [CrossRef]
- Nguyen, T.; Duan, Z.; Naik, A.D.; Kramer, J.R.; El-Serag, H.B. Statin Use Reduces Risk of Esophageal Adenocarcinoma in US Veterans with Barrett’s Esophagus: A Nested Case-control Study. Gastroenterology 2015, 149, 1392–1398. [Google Scholar] [CrossRef]
- El–Serag, H.B.; Hashmi, A.; Garcia, J.; Richardson, P.; Alsarraj, A.; Fitzgerald, S.; Vela, M.; Shaib, Y.; Abraham, N.S.; Velez, M.; et al. Visceral Abdominal Obesity Measured by CT Scan is Associated With an Increased Risk of Barrett’s Oesophagus: A Case-control Study. Gut 2014, 63, 220–229. [Google Scholar] [CrossRef] [PubMed]
- El–Serag, H.B.; Kvapil, P.; Hacken–Bitar, J.; Kramer, J.R. Abdominal Obesity and the Risk of Barrett’s Esophagus. Am. J. Gastroenterol. 2005, 100, 2151–2156. [Google Scholar] [CrossRef] [PubMed]
- Kambhampati, S.; Tieu, A.H.; Luber, B.; Wang, H.; Meltzer, S.J. Risk Factors for Progression of Barrett’s Esophagus to High Grade Dysplasia and Esophageal Adenocarcinoma. Sci. Rep. 2020, 10, 4899. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, D.C.; Sadeghi, S.; Pandeya, N.; Smithers, B.M.; Gotley, D.C.; Bain, C.J.; Webb, P.M.; Green, A.C.; Australian Cancer Study. Combined Effects of Obesity, Acid Reflux and Smoking on the Risk of Adenocarcinomas of the Oesophagus. Gut 2008, 57, 173–180. [Google Scholar] [CrossRef]
- Wu, A.H.; Wan, P.; Bernstein, L. A Multiethnic Population-based Study of Smoking, Alcohol and Body Size and Risk of Adenocarcinomas of the Stomach and Esophagus (United States). Cancer Causes Control 2001, 12, 721–732. [Google Scholar] [CrossRef]
- Mariosa, D.; Carreras-Torres, R.; Martin, R.M.; Johansson, M.; Brennan, P. Commentary: What Can Mendelian Randomization Tell Us About Causes of Cancer? Int. J. Epidemiol. 2019, 48, 816–821. [Google Scholar] [CrossRef]
- Kramer, J.; Fischbach, L.; Richardson, P.; Alsarraj, A.; Fitzgerald, S.; Shaib, Y.; Abraham, N.; Velez, M.; Cole, R.; Anand, B.; et al. Waist-to-Hip Ratio, but Not Body Mass Index, Is Associated With an Increased Risk of Barrett’s Esophagus in White Men. Clin. Gastroenterol. Hepatol. 2013, 11, 373–381.e1. [Google Scholar] [CrossRef]
- Spechler, S.; Zeroogian, J.; Antonioli, D.; Wang, H.; Goyal, R. Prevalence of metaplasia at the gastro-oesophageal junction. Lancet 1994, 344, 1533–1536. [Google Scholar] [CrossRef]
- Stocks, T.; Bjorge, T.; Ulmer, H.; Manjer, J.; Haggstrom, C.; Nagel, G.; Engeland, A.; Johansen, D.; Hallmans, G.; Selmer, R.; et al. Metabolic Risk Score and Cancer Risk: Pooled Analysis of Seven Cohorts. Int. J. Epidemiol. 2015, 44, 1353–1363. [Google Scholar] [CrossRef]
- Drahos, J.; Ricker, W.; Pfeiffer, R.M.; Cook, M.B. Metabolic Syndrome and Risk of Esophageal Adenocarcinoma in Elderly Patients in the United States: An Analysis of SEER-Medicare Data. Cancer 2017, 123, 657–665. [Google Scholar] [CrossRef]
- De Ceglie, A.; Fisher, D.; Filiberti, R.; Blanchi, S.; Conio, M. Barrett’s Esophagus, Esophageal and Esophagogastric Junction Adenocarcinomas: The Role of Diet. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Kubo, A.; Levin, T.; Block, G.; Rumore, G.; Quesenberry, C.; Buffler, P.; Corley, D. Dietary Patterns and the Risk of Barrett’s Esophagus. Am. J. Epidemiol. 2008, 167, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Kramer, J.; Chen, L.; Rugge, M.; Parente, P.; Verstovsek, G.; Alsarraj, A.; El-Serag, H. Dietary Consumption of Meat, Fat, Animal Products and Advanced Glycation End-products and the Risk of Barrett’s Oesophagus. Aliment. Pharmacol. Ther. 2013, 38, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Kramer, J.; Rugge, M.; Parente, P.; Verstovsek, G.; Alsarraj, A.; El-Serag, H. Dietary Intake of Vegetables, Folate, and Antioxidants and the Risk of Barrett’s Esophagus. Cancer Causes Control 2013, 24, 1005–1014. [Google Scholar] [CrossRef]
- Ellulu, M.S.; Patimah, I.; Khazáai, H.; Rahmat, A.; Abed, Y. Obesity and Inflammation: The Linking Mechanism and the Complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef]
- Lanthier, N.; Leclercq, I.A. Adipose Tissues as Endocrine Target Organs. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 545–558. [Google Scholar] [CrossRef]
- Kredel, L.I.; Siegmund, B. Adipose-tissue and Intestinal Inflammation—Visceral Obesity and Creeping Fat. Front. Immunol. 2014, 5, 462. [Google Scholar] [CrossRef]
- Tchkonia, T.; Thomou, T.; Zhu, Y.; Karagiannides, I.; Pothoulakis, C.; Jensen, M.D.; Kirkland, J.L. Mechanisms and Metabolic Implications of Regional Differences among Fat Depots. Cell Metab. 2013, 17, 644–656. [Google Scholar] [CrossRef]
- Nam, S.Y. Obesity-Related Digestive Diseases and Their Pathophysiology. Gut Liver 2017, 11, 323–334. [Google Scholar] [CrossRef]
- Brestoff, J.R.; Artis, D. Immune Regulation of Metabolic Homeostasis in Health and Disease. Cell 2015, 161, 146–160. [Google Scholar] [CrossRef]
- Russo, L.; Lumeng, C.N. Properties and Functions of Adipose Tissue Macrophages in Obesity. Immunology 2018, 155, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Roman, S.; Agil, A.; Peran, M.; Alvaro-Galue, E.; Ruiz-Ojeda, F.J.; Fernandez-Vazquez, G.; Marchal, J.A. Brown Adipose Tissue and Novel Therapeutic Approaches to Treat Metabolic Disorders. Transl. Res. 2015, 165, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, M.; Wolfrum, C. The Origin and Definition of Brite Versus White and Classical Brown adipocytes. Adipocyte 2014, 3, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Dempersmier, J.; Sul, H.S. Shades of Brown: A Model for Thermogenic Fat. Front. Endocrinol. 2015, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Goody, D.; Pfeifer, A. MicroRNAs in Brown and Beige Fat. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 29–36. [Google Scholar] [CrossRef]
- Lee, M.W.; Lee, M.; Oh, K.J. Adipose Tissue-Derived Signatures for Obesity and Type 2 Diabetes: Adipokines, Batokines and MicroRNAs. J. Clin. Med. 2019, 8, 854. [Google Scholar] [CrossRef]
- Weidinger, C.; Ziegler, J.F.; Letizia, M.; Schmidt, F.; Siegmund, B. Adipokines and Their Role in Intestinal Inflammation. Front. Immunol. 2018, 9, 1974. [Google Scholar] [CrossRef]
- Brocco, D.; Florio, R.; De Lellis, L.; Veschi, S.; Grassadonia, A.; Tinari, N.; Cama, A. The Role of Dysfunctional Adipose Tissue in Pancreatic Cancer: A Molecular Perspective. Cancers 2020, 12, 1849. [Google Scholar] [CrossRef]
- Lagergren, J. Influence of Obesity on the Risk of Esophageal Disorders. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 340–347. [Google Scholar] [CrossRef]
- Usui, G.; Shinozaki, T.; Jinno, T.; Fujibayashi, K.; Morikawa, T.; Gunji, T.; Matsuhashi, N. Association Between Visceral Abdominal Obesity and Long-segment Barrett’s Esophagus in a Japanese Population. J. Gastroenterol. 2019, 55, 189–197. [Google Scholar] [CrossRef]
- Elliott, J.A.; Reynolds, J.V. Visceral Obesity, Metabolic Syndrome, and Esophageal Adenocarcinoma. Front. Oncol. 2021, 11, 627270. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.; Johnston, B.; Lane, A.; Harvey, I.; Donovan, J.; Nair, P.; Harvey, R. Relationship Between Body Mass and Gastro-oesophageal Reflux Symptoms: The Bristol Helicobacter Project. Int. J. Epidemiol. 2003, 32, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Cai, N.; Ji, G.-Z.; Fan, Z.-N.; Wu, Y.-F.; Zhang, F.-M.; Zhao, Z.-F.; Xu, W.; Liu, Z. Association Between Body Mass Index and Erosive Esophagitis: A Meta-analysis. World J. Gastroenterol. WJG 2012, 18, 2545–2553. [Google Scholar] [CrossRef] [PubMed]
- Patti, M.G.; Schlottmann, F.; Farrell, T.M. Pathophysiology of Gastroesophageal Reflux Disease in Obese Patients. In The Perfect Sleeve Gastrectomy; Springer: Berlin/Heidelberg, Germany, 2020; pp. 169–176. [Google Scholar]
- El-Serag, H.B.; Thrift, A.P. Obesity and Gastroesophageal Reflux Disease. In The Esophagus; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2021; pp. 624–632. [Google Scholar] [CrossRef]
- Mathus-Vliegen, E.M.; Tygat, G.N. Gastro-oesophageal Reflux in Obese Subjects: Influence of Overweight, Weight Loss and Chronic Gastric Balloon Distension. Scand. J. Gastroenterol. 2002, 37, 1246–1252. [Google Scholar] [CrossRef] [PubMed]
- Mathus-Vliegen, L.M.; Tytgat, G.N. Twenty-four-hour pH Measurements in Morbid Obesity: Effects of Massive Overweight, Weight Loss and Gastric Distension. Eur. J. Gastroenterol. Hepatol. 1996, 8, 635–640. [Google Scholar]
- Mathus-Vliegen, E.M.; van Weeren, M.; van Eerten, P.V. Los Function and Obesity: The Impact of Untreated Obesity, Weight loss, and Chronic Gastric Balloon Distension. Digestion 2003, 68, 161–168. [Google Scholar] [CrossRef]
- Ness-Jensen, E.; Lindam, A.; Lagergren, J.; Hveem, K. Weight Loss and Reduction in Gastroesophageal Reflux. A Prospective Population-based Cohort Study: The HUNT study. Am. J. Gastroenterol. 2013, 108, 376–382. [Google Scholar] [CrossRef]
- Singh, M.; Lee, J.; Gupta, N.; Gaddam, S.; Smith, B.K.; Wani, S.B.; Sullivan, D.K.; Rastogi, A.; Bansal, A.; Donnelly, J.E. Weight Loss Can Lead to Resolution of Gastroesophageal Reflux Disease Symptoms: A Prospective Intervention Trial. Obesity 2013, 21, 284–290. [Google Scholar] [CrossRef]
- Suter, M. Gastroesophageal Reflux Disease, Obesity, and Roux-en-Y Gastric Bypass: Complex Relationship-a Narrative Review. Obes. Surg. 2020, 30, 3178–3187. [Google Scholar] [CrossRef]
- Herbella, F.A.; Sweet, M.P.; Tedesco, P.; Nipomnick, I.; Patti, M.G. Gastroesophageal Reflux Disease and Obesity. Pathophysiology and Implications for Treatment. J. Gastrointest. Surg. 2007, 11, 286–290. [Google Scholar] [CrossRef]
- Cote-Daigneault, J.; Leclerc, P.; Joubert, J.; Bouin, M. High Prevalence of Esophageal Dysmotility in Asymptomatic Obese Patients. Can. J. Gastroenterol. Hepatol. 2014, 28, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.H.; Kuper, M.; Konigsrainer, A.; Brucher, B. Transient Lower Esophageal Sphincter Relaxation in Morbid Obesity. Obes. Surg. 2009, 19, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.Y.; McColl, K.E. Pathophysiology of Gastroesophageal Reflux Disease. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.E.; Rubenstein, J.H. Presentation and Epidemiology of Gastroesophageal Reflux Disease. Gastroenterology 2018, 154, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.J.; Ma, W.; Hirschowitz, B.I. Association of Obesity with Hiatal Hernia and Esophagitis. Am. J. Gastroenterol. 1999, 94, 2840–2844. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Tran, T.; Richardson, P.; Ergun, G. Anthropometric Correlates of Intragastric Pressure. Scand. J. Gastroenterol. 2006, 41, 887–891. [Google Scholar] [CrossRef]
- Del Grande, L.D.M.; Herbella, F.A.M.; Katayama, R.C.; Lima, W.G.; Patti, M.G. Transdiaphragmatic Pressure Gradient (TPG) Has a Central Role in the Pathophysiology of Gastroesophageal Reflux Disease (GERD) in the Obese and it Correlates with Abdominal Circumference but not with Body Mass Index (BMI). Obes. Surg. 2020, 30, 1424–1428. [Google Scholar] [CrossRef]
- Lechien, J.R.; Bobin, F.; Muls, V.; Horoi, M.; Thill, M.-P.; Dequanter, D.; Rodriguez, A.; Saussez, S. Patients with Acid, High-fat and Low-protein Diet Have Higher Laryngopharyngeal Reflux Episodes at the Impedance-pH Monitoring. Eur. Arch. Otorhinolaryngol. 2020, 277, 511–520. [Google Scholar] [CrossRef]
- Wu, P.; Zhao, X.H.; Ai, Z.S.; Sun, H.H.; Chen, Y.; Jiang, Y.X.; Tong, Y.L.; Xu, S.C. Dietary Intake and Risk for Reflux Esophagitis: A Case-control Study. Gastroenterol. Res. Pract. 2013, 2013, 691026. [Google Scholar] [CrossRef]
- Zhang, M.; Hou, Z.K.; Huang, Z.B.; Chen, X.L.; Liu, F.B. Dietary and Lifestyle Factors Related to Gastroesophageal Reflux Disease: A Systematic Review. Ther. Clin. Risk Manag. 2021, 17, 305–323. [Google Scholar] [CrossRef]
- Ahmed, S.; Jamil, S.; Shaikh, H.; Abbasi, M. Effects of Life Style Factors on the Symptoms of Gastro Esophageal Reflux Disease: A Cross Sectional Study in a Pakistani Population. Pak. J. Med. Sci. 2020, 36, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Piesman, M.; Hwang, I.; Maydonovitch, C.; Wong, R.K. Nocturnal Reflux Episodes Following the Administration of a Standardized Meal. Does Timing Matter? Am. J. Gastroenterol. ACG 2007, 102, 2128–2134. [Google Scholar] [CrossRef] [PubMed]
- Jarosz, M.; Taraszewska, A. Risk Factors for Gastroesophageal Reflux Disease: The Role of Diet. Gastroenterol. Rev. Prz. Gastroenterol. 2014, 9, 297–301. [Google Scholar] [CrossRef]
- Fox, M.; Barr, C.; Nolan, S.; Lomer, M.; Anggiansah, A.; Wong, T. The Effects of Dietary Fat and Calorie Density on Esophageal Acid Exposure and Reflux Symptoms. Clin. Gastroenterol. Hepatol. 2007, 5, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, E.; Suzuki, H.; Sugino, Y.; Iida, T.; Nishizawa, T.; Masaoka, T.; Hosoda, H.; Kangawa, K.; Hibi, T. Decreased Levels of Adiponectin in Obese Patients with Gastroesophageal Reflux Evaluated by Videoesophagography: Possible Relationship Between Gastroesophageal Reflux and Metabolic Syndrome. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. S2), S216–S221. [Google Scholar] [CrossRef]
- Abdelkader, N.A.; Montasser, I.F.; Bioumy, E.E.; Saad, W.E. Impact of Anthropometric Measures and Serum Leptin on Severity of Gastroesophageal Reflux Disease. Dis. Esophagus 2015, 28, 691–698. [Google Scholar] [CrossRef]
- Livzan, M.A.; Lapteva, I.V.; Krolevets, T.S.; Kiselev, I.E. Specific Features of Gastroesophageal Reflux Disease Associated with Obesity and Overweight. Ter Arkh. 2016, 88, 21–27. [Google Scholar] [CrossRef]
- Livzan, M.A.; Lapteva, I.V.; Krolevets, T.S. Gastroesophageal Refluxed Disease in Persons with Obesity and Leptin Resistance. Eksp Klin Gastroenterol. 2015, 11–16. [Google Scholar] [PubMed]
- Thomas, S.J.; Almers, L.; Schneider, J.; Graham, J.E.; Havel, P.J.; Corley, D.A. Ghrelin and Leptin Have a Complex Relationship with Risk of Barrett’s Esophagus. Dig. Dis. Sci. 2016, 61, 70–79. [Google Scholar] [CrossRef]
- Rubenstein, J.H.; Kao, J.Y.; Madanick, R.D.; Zhang, M.; Wang, M.; Spacek, M.B.; Donovan, J.L.; Bright, S.D.; Shaheen, N.J. Association of Adiponectin Multimers with Barrett’s Oesophagus. Gut 2009, 58, 1583–1589. [Google Scholar] [CrossRef]
- Greer, K.B.; Falk, G.W.; Bednarchik, B.; Li, L.; Chak, A. Associations of Serum Adiponectin and Leptin With Barrett’s Esophagus. Clin. Gastroenterol. Hepatol. 2015, 13, 2265–2272. [Google Scholar] [CrossRef] [PubMed]
- Tseng, P.H.; Yang, W.S.; Liou, J.M.; Lee, Y.C.; Wang, H.P.; Lin, J.T.; Wu, M.S. Associations of Circulating Gut Hormone and Adipocytokine Levels with the Spectrum of Gastroesophageal Reflux Disease. PLoS ONE 2015, 10, e0141410. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.Y.; Choi, I.J.; Ryu, K.H.; Park, B.J.; Kim, Y.W.; Kim, H.B.; Kim, J.S. The Effect of Abdominal Visceral Fat, Circulating Inflammatory Cytokines, and Leptin Levels on Reflux Esophagitis. J. Neurogastroenterol. Motil. 2015, 21, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Asanuma, K.; Ara, N.; Iijima, K.; Hatta, W.; Hamada, S.; Asano, N.; Koike, T.; Imatani, A.; Masamune, A.; et al. Leptin Aggravates Reflux Esophagitis by Increasing Tissue Levels of Macrophage Migration Inhibitory Factor in Rats. Tohoku J. Exp. Med. 2018, 245, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Watabe, K.; Hamasaki, T.; Umeda, M.; Furubayashi, A.; Kinoshita, K.; Kishida, O.; Fujimoto, T.; Yamada, A.; Tsukamoto, Y.; et al. Association of Low Serum Adiponectin Levels with Erosive Esophagitis in Men: An Analysis of 2405 Subjects Undergoing Physical Check-ups. J. Gastroenterol. 2011, 46, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, J.A.; Harrison, R.F.; Perry, I.; Balkwill, F.; Tselepis, C. Barrett’s Metaplasia. Lancet 2000, 356, 2079–2085. [Google Scholar] [CrossRef]
- Schottenfeld, D.; Beebe-Dimmer, J. Chronic Inflammation: A Common and Important Factor in the Pathogenesis of Neoplasia. CA Cancer J. Clin. 2006, 56, 69–83. [Google Scholar] [CrossRef]
- Okayasu, I.; Ichinoe, M.; Yoshida, T. Proposal for an Organ-Specific Chronic Inflammation–Remodeling–Carcinoma Sequence. Gastrointest. Disord. 2019, 1, 341–357. [Google Scholar] [CrossRef]
- Maury, E.; Brichard, S.M. Adipokine Dysregulation, Adipose Tissue Inflammation and Metabolic Syndrome. Mol. Cell. Endocrinol. 2010, 314, 1–16. [Google Scholar] [CrossRef]
- Van Meijel, R.L.; Blaak, E.E.; Goossens, G.H. Adipose Tissue Metabolism and Inflammation in Obesity. In Mechanisms and Manifestations of Obesity in Lung Disease; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–22. [Google Scholar]
- Garcia, J.M.; Splenser, A.E.; Kramer, J.; Alsarraj, A.; Fitzgerald, S.; Ramsey, D.; El-Serag, H.B. Circulating Inflammatory Cytokines and Adipokines are Associated with Increased Risk of Barrett’s Esophagus: A Case-control Study. Clin. Gastroenterol. Hepatol. 2014, 12, 229–238.e223. [Google Scholar] [CrossRef]
- Chandar, A.K.; Devanna, S.; Lu, C.; Singh, S.; Greer, K.; Chak, A.; Iyer, P.G. Association of Serum Levels of Adipokines and Insulin With Risk of Barrett’s Esophagus: A Systematic Review and Meta-Analysis. Clin. Gastroenterol. Hepatol. 2015, 13, 2241–2255.e4. [Google Scholar] [CrossRef] [PubMed]
- Aloor, S.; Tamariz, L.; Deshpande, A.R.; Sussman, D.A. Sa1865 Barrett’s Esophagus and Serum Adipokines: A Meta-Analysis. Gastroenterology 2014, 146, S-315. [Google Scholar] [CrossRef]
- Nam, S.Y. Circulating Inflammatory Cytokines Are Associated With the Risk of Barrett’s Esophagus in Western Persons. J. Neurogastroenterol. Motil. 2014, 20, 558–559. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yoon, Y.S.; Kwon, A.R.; Lee, Y.K.; Oh, S.W. Circulating Adipokines and Risk of Obesity Related Cancers: A Systematic Review and Meta-analysis. Obes. Res. Clin. Pract. 2019, 13, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Diakowska, D.; Markocka-Maczka, K.; Nienartowicz, M.; Rosinczuk, J.; Krzystek-Korpacka, M. Assessment of Apelin, Apelin Receptor, Resistin, and Adiponectin Levels in the Primary Tumor and Serum of Patients with Esophageal Squamous Cell Carcinoma. Adv. Clin. Exp. Med. 2019, 28, 671–678. [Google Scholar] [CrossRef]
- Khandekar, M.J.; Cohen, P.; Spiegelman, B.M. Molecular Mechanisms of Cancer Development in Obesity. Nat. Rev. Cancer 2011, 11, 886–895. [Google Scholar] [CrossRef]
- Roberts, D.L.; Dive, C.; Renehan, A.G. Biological Mechanisms Linking Obesity and Cancer Risk: New Perspectives. Annu. Rev. Med. 2010, 61, 301–316. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, E.H.; Hahm, K.B. Oxidative Stress in Inflammation-based Gastrointestinal Tract Diseases: Challenges and Opportunities. J. Gastroenterol. Hepatol. 2012, 27, 1004–1010. [Google Scholar] [CrossRef]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose Tissue and Adipocytes Support Tumorigenesis and Metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef]
- Quante, M.; Bhagat, G.; Abrams, J.A.; Marache, F.; Good, P.; Lee, M.D.; Lee, Y.; Friedman, R.; Asfaha, S.; Dubeykovskaya, Z.; et al. Bile Acid and Inflammation Activate Gastric Cardia Stem Cells in a Mouse Model of Barrett-like Metaplasia. Cancer Cell 2012, 21, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Beales, I.L.P.; Garcia-Morales, C.; Ogunwobi, O.O.; Mutungi, G. Adiponectin Inhibits Leptin-induced Oncogenic Signalling in Oesophageal Cancer Cells by Activation of PTP1B. Mol. Cell. Endocrinol. 2014, 382, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Mokrowiecka, A.; Daniel, P.; Jasinska, A.; Pietruczuk, M.; Pawlowski, M.; Szczesniak, P.; Orszulak-Michalak, D.; Malecka-Panas, E. Serum Adiponectin, Resistin, Leptin Concentration and Central Adiposity Parameters in Barrett’s Esophagus Patients with and without Intestinal Metaplasia in Comparison to Healthy Controls and Patients with GERD. Hepatogastroenterology 2011, 59, 2395–2399. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, L.; Long, E.; Beales, I.L. Pathophysiological Mechanisms Linking Obesity and Esophageal Adenocarcinoma. World J. Gastrointest. Pathophysiol. 2014, 5, 534–549. [Google Scholar] [CrossRef]
- Rubenstein, J.H.; Dahlkemper, A.; Kao, J.Y.; Zhang, M.; Morgenstern, H.; McMahon, L.; Inadomi, J.M. A Pilot Study of the Association of Low Plasma Adiponectin and Barrett’s Esophagus. Am. J. Gastroenterol. 2008, 103, 1358–1364. [Google Scholar] [CrossRef][Green Version]
- Fakhraldeen, M.; Mostafa, H.S.; Abdulwahab, G.A. Adiponectin Multimers in Patients with Barrett’s Oesophagus. Egypt. J. Hosp. Med. 2018, 39, 268–274. [Google Scholar] [CrossRef]
- Yildirim, A.; Bilici, M.; Cayir, K.; Yanmaz, V.; Yildirim, S.; Tekin, S.B. Serum Adiponectin Levels in Patients with Esophageal Cancer. Jpn. J. Clin. Oncol. 2009, 39, 92–96. [Google Scholar] [CrossRef]
- Duggan, C.; Onstad, L.; Hardikar, S.; Blount, P.L.; Reid, B.J.; Vaughan, T.L. Association Between Markers of Obesity and Progression from Barrett’s Esophagus to Esophageal Adenocarcinoma. Clin. Gastroenterol. Hepatol. 2013, 11, 934–943. [Google Scholar] [CrossRef]
- Thompson, O.M.; Beresford, S.A.; Kirk, E.A.; Bronner, M.P.; Vaughan, T.L. Serum Leptin and Adiponectin Levels and Risk of Barrett’s Esophagus and Intestinal Metaplasia of the Gastroesophageal Junction. Obesity 2010, 18, 2204–2211. [Google Scholar] [CrossRef]
- Allott, E.H.; Lysaght, J.; Cathcart, M.C.; Donohoe, C.L.; Cummins, R.; McGarrigle, S.A.; Kay, E.; Reynolds, J.V.; Pidgeon, G.P. MMP9 Expression in Oesophageal Adenocarcinoma is Upregulated with Visceral Obesity and is Associated with Poor Tumour Differentiation. Mol. Carcinog. 2013, 52, 144–154. [Google Scholar] [CrossRef]
- Sinha, M.K. Human Leptin: The Hormone of Adipose Tissue. Eur. J. Endocrinol. 1997, 136, 461–464. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fasshauer, M.; Bluher, M. Adipokines in Health and Disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Mokrowiecka, A.; Sokolowska, M.; Luczak, E.; Dudojc, M.; Wieczfinska, J.; Kacprzak, D.; Wierzchniewska-Lawska, A.; Pawliczak, R.; Malecka-Panas, E. Adiponectin and Leptin Receptors Expression in Barrett’s Esophagus and Normal Squamous Epithelium in Relation to Central Obesity Status. J. Physiol. Pharmacol. 2013, 64, 193–199. [Google Scholar] [PubMed]
- Howard, J.M.; Cathcart, M.C.; Healy, L.; Beddy, P.; Muldoon, C.; Pidgeon, G.P.; Reynolds, J.V. Leptin and Adiponectin Receptor Expression in Oesophageal Cancer. Br. J. Surg. 2014, 101, 643–652. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pai, R.; Lin, C.; Tran, T.; Tarnawski, A. Leptin Activates STAT and ERK2 Pathways and Induces Gastric Cancer Cell Proliferation. Biochem. Biophys. Res. Commun. 2005, 331, 984–992. [Google Scholar] [CrossRef]
- Dieudonne, M.N.; Machinal-Quelin, F.; Serazin-Leroy, V.; Leneveu, M.C.; Pecquery, R.; Giudicelli, Y. Leptin Mediates a Proliferative Response in Human MCF7 Breast Cancer Cells. Biochem. Biophys. Res. Commun. 2002, 293, 622–628. [Google Scholar] [CrossRef]
- Ogunwobi, O.O.; Beales, I.L. The Anti-apoptotic and Growth Stimulatory Actions of Leptin in Human Colon Cancer Cells Involves Activation of JNK Mitogen Activated Protein Kinase, JAK2 and PI3 Kinase/Akt. Int. J. Colorectal Dis. 2007, 22, 401–409. [Google Scholar] [CrossRef]
- Ogunwobi, O.O.; Mutungi, G.; Beales, I.L. Leptin Stimulates Proliferation and Inhibits Apoptosis in Barrett’s Esophageal Adenocarcinoma Cells by Cyclooxygenase-2-dependent, Prostaglandin-E2-mediated Transactivation of the Epidermal Growth Factor Receptor and c-Jun NH2-terminal Kinase Activation. Endocrinology 2006, 147, 4505–4516. [Google Scholar] [CrossRef]
- Francois, F.; Roper, J.; Goodman, A.J.; Pei, Z.; Ghumman, M.; Mourad, M.; de Perez, A.Z.; Perez-Perez, G.I.; Tseng, C.H.; Blaser, M.J. The Association of Gastric Leptin with Oesophageal Inflammation and Metaplasia. Gut 2008, 57, 16–24. [Google Scholar] [CrossRef]
- Ogunwobi, O.O.; Beales, I.L. Leptin Stimulates the Proliferation of Human Oesophageal Adenocarcinoma Cells via HB-EGF and Tgfalpha Mediated Transactivation of the Epidermal Growth Factor Receptor. Br. J. Biomed. Sci. 2008, 65, 121–127. [Google Scholar] [CrossRef]
- Kendall, B.J.; Macdonald, G.A.; Hayward, N.K.; Prins, J.B.; Brown, I.; Walker, N.; Pandeya, N.; Green, A.C.; Webb, P.M.; Whiteman, D.C.; et al. Leptin and the Risk of Barrett’s Oesophagus. Gut 2008, 57, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.H.; Morgenstern, H.; McConell, D.; Scheiman, J.M.; Schoenfeld, P.; Appelman, H.; McMahon, L.F., Jr.; Kao, J.Y.; Metko, V.; Zhang, M.; et al. Associations of Diabetes Mellitus, Insulin, Leptin, and Ghrelin with Gastroesophageal Reflux and Barrett’s Esophagus. Gastroenterology 2013, 145, 1237–1244.e5. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Valles, A.; Inoue, W.; Rummel, C.; Luheshi, G.N. Obesity, Adipokines and Neuroinflammation. Neuropharmacology 2015, 96, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Kitayama, J.; Kazama, S.; Hiramatsu, T.; Hatano, K.; Nagawa, H. Plasma Adiponectin and Gastric Cancer. Clin. Cancer Res. 2005, 11, 466–472. [Google Scholar] [PubMed]
- Gonullu, G.; Kahraman, H.; Bedir, A.; Bektas, A.; Yucel, I. Association Between Adiponectin, Resistin, Insulin Resistance, and Colorectal Tumors. Int. J. Colorectal Dis. 2010, 25, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Stampfer, M.J.; Mucci, L.; Rifai, N.; Qiu, W.; Kurth, T.; Ma, J. A 25-year Prospective Study of Plasma Adiponectin and Leptin Concentrations and Prostate Cancer Risk and Survival. Clin. Chem. 2010, 56, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Ogunwobi, O.O.; Beales, I.L. Globular Adiponectin, Acting via Adiponectin Receptor-1, Inhibits Leptin-stimulated Oesophageal Adenocarcinoma Cell Proliferation. Mol. Cell. Endocrinol. 2008, 285, 43–50. [Google Scholar] [CrossRef]
- Samani, A.A.; Yakar, S.; LeRoith, D.; Brodt, P. The Role of the IGF System in Cancer Growth and Metastasis: Overview and Recent Insights. Endocr. Rev. 2007, 28, 20–47. [Google Scholar] [CrossRef]
- Iravani, S.; Zhang, H.Q.; Yuan, Z.Q.; Cheng, J.Q.; Karl, R.C.; Jove, R.; Coppola, D. Modification of Insulin-like Growth Factor 1 Receptor, c-Src, and Bcl-XL Protein Expression During the Progression of Barrett’s Neoplasia. Hum. Pathol. 2003, 34, 975–982. [Google Scholar] [CrossRef]
- Doyle, S.L.; Donohoe, C.L.; Finn, S.P.; Howard, J.M.; Lithander, F.E.; Reynolds, J.V.; Pidgeon, G.P.; Lysaght, J. IGF-1 and its Receptor in Esophageal Cancer: Association with Adenocarcinoma and Visceral Obesity. Am. J. Gastroenterol. 2012, 107, 196–204. [Google Scholar] [CrossRef]
- Donohoe, C.L.; Doyle, S.L.; McGarrigle, S.; Cathcart, M.C.; Daly, E.; O’Grady, A.; Lysaght, J.; Pidgeon, G.P.; Reynolds, J.V. Role of the Insulin-like Growth Factor 1 Axis and Visceral Adiposity in Oesophageal Adenocarcinoma. Br. J. Surg. 2012, 99, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Arcidiacono, D.; Dedja, A.; Giacometti, C.; Fassan, M.; Nucci, D.; Francia, S.; Fabris, F.; Zaramella, A.; Gallagher, E.J.; Cassaro, M.; et al. Hyperinsulinemia Promotes Esophageal Cancer Development in a Surgically-Induced Duodeno-Esophageal Reflux Murine Model. Int. J. Mol. Sci. 2018, 19, 1198. [Google Scholar] [CrossRef] [PubMed]
- Clark, G.W.; Smyrk, T.C.; Mirvish, S.S.; Anselmino, M.; Yamashita, Y.; Hinder, R.A.; DeMeester, T.R.; Birt, D.F. Effect of Gastroduodenal Juice and Dietary fat on the Development of Barrett’s Esophagus and Esophageal Neoplasia: An Experimental Rat Model. Ann. Surg. Oncol. 1994, 1, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Mukaisho, K.; Sugihara, H.; Araki, Y.; Yamamoto, G.; Hattori, T. High Animal-fat Intake Changes the Bile-acid Composition of Bile Juice and Enhances the Development of Barrett’s Esophagus and Esophageal Adenocarcinoma in a Rat Duodenal-contents Reflux Model. Cancer Sci. 2007, 98, 1683–1688. [Google Scholar] [CrossRef]
- Molendijk, J.; Nguyen, T.-M.-T.; Brown, I.; Mohamed, A.; Lim, Y.; Barclay, J.; Hodson, M.P.; Hennessy, T.P.; Krause, L.; Morrison, M. Chronic High-Fat Diet Induces Early Barrett’s Esophagus in Mice through Lipidome Remodeling. Biomolecules 2020, 10, 776. [Google Scholar] [CrossRef]
- Fowler, A.J.; Richer, A.L.; Bremner, R.M.; Inge, L.J. A High-fat Diet is Associated with Altered Adipokine Production and a More Aggressive Esophageal Adenocarcinoma Phenotype In Vivo. J. Thorac. Cardiovasc. Surg. 2015, 149, 1185–1191. [Google Scholar] [CrossRef]
- Munch, N.S.; Fang, H.Y.; Ingermann, J.; Maurer, H.C.; Anand, A.; Kellner, V.; Sahm, V.; Wiethaler, M.; Baumeister, T.; Wein, F.; et al. High-Fat Diet Accelerates Carcinogenesis in a Mouse Model of Barrett’s Esophagus via Interleukin 8 and Alterations to the Gut Microbiome. Gastroenterology 2019, 157, 492–506.e492. [Google Scholar] [CrossRef]
- Kaakoush, N.O.; Morris, M.J. TheOoesophageal Microbiome: An Unexplored Link in Obesity-associated Oesophageal Adenocarcinoma. FEMS Microbiol. Ecol. 2016, 92, fiw161. [Google Scholar] [CrossRef]
- Kaakoush, N.O.; Lecomte, V.; Maloney, C.A.; Morris, M.J. Cross-talk Among Metabolic Parameters, Esophageal Microbiota, and Host Gene Expression Following Chronic Exposure to an Obesogenic Diet. Sci. Rep. 2017, 7, 45753. [Google Scholar] [CrossRef]
- Blackett, K.L.; Siddhi, S.S.; Cleary, S.; Steed, H.; Miller, M.H.; Macfarlane, S.; Macfarlane, G.T.; Dillon, J.F. Oesophageal Bacterial Biofilm Changes in Gastro-oesophageal Reflux Disease, Barrett’s and Oesophageal Carcinoma: Association or Causality? Aliment. Pharmacol. Ther. 2013, 37, 1084–1092. [Google Scholar] [CrossRef]
- Okereke, I.C.; Miller, A.L.; Jupiter, D.C.; Hamilton, C.F.; Reep, G.L.; Krill, T.; Andersen, C.R.; Pyles, R.B. Microbiota Detection Patterns Correlate With Presence and Severity of Barrett’s Esophagus. Front. Cell. Infect. Microbiol. 2021, 11, 555072. [Google Scholar] [CrossRef] [PubMed]
- Nardone, G.; Compare, D.; Rocco, A. A Microbiota-centric View of Diseases of the Upper Gastrointestinal Tract. Lancet Gastroenterol. Hepatol. 2017, 2, 298–312. [Google Scholar] [CrossRef]
- Kaakoush, N.O.; Castano-Rodriguez, N.; Man, S.M.; Mitchell, H.M. Is Campylobacter to Esophageal Adenocarcinoma as Helicobacter is to Gastric Adenocarcinoma? Trends Microbiol. 2015, 23, 455–462. [Google Scholar] [CrossRef]
- Snider, E.J.; Compres, G.; Freedberg, D.E.; Khiabanian, H.; Nobel, Y.R.; Stump, S.; Uhlemann, A.C.; Lightdale, C.J.; Abrams, J.A. Alterations to the Esophageal Microbiome Associated with Progression from Barrett’s Esophagus to Esophageal Adenocarcinoma. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1687–1693. [Google Scholar] [CrossRef] [PubMed]
- Snider, E.J.; Freedberg, D.E.; Abrams, J.A. Potential Role of the Microbiome in Barrett’s Esophagus and Esophageal Adenocarcinoma. Dig. Dis. Sci. 2016, 61, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Gall, A.; Fero, J.; McCoy, C.; Claywell, B.C.; Sanchez, C.A.; Blount, P.L.; Li, X.; Vaughan, T.L.; Matsen, F.A.; Reid, B.J.; et al. Bacterial Composition of the Human Upper Gastrointestinal Tract Microbiome Is Dynamic and Associated with Genomic Instability in a Barrett’s Esophagus Cohort. PLoS ONE 2015, 10, e0129055. [Google Scholar] [CrossRef]
- So, B.; Kim, H.J.; Kim, J.; Song, W. Exercise-induced Myokines in Health and Metabolic Diseases. Integr. Med. Res. 2014, 3, 172–179. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Saltin, B. Evidence for Prescribing Exercise as Therapy in Chronic Disease. Scand. J. Med. Sci. Sports 2006, 16 (Suppl. S1), 3–63. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Saltin, B. Exercise as Medicine—Evidence for Prescribing Exercise as Therapy in 26 Different Chronic Diseases. Scand. J. Med. Sci. Sports 2015, 25 (Suppl. S3), 1–72. [Google Scholar] [CrossRef]
- Moore, S.C.; Lee, I.M.; Weiderpass, E.; Campbell, P.T.; Sampson, J.N.; Kitahara, C.M.; Keadle, S.K.; Arem, H.; de Gonzalez, A.B.; Hartge, P.; et al. Association of Leisure-Time Physical Activity With Risk of 26 Types of Cancer in 1.44 Million Adults. JAMA Intern. Med. 2016, 176, 816–825. [Google Scholar] [CrossRef]
- Pedersen, L.; Idorn, M.; Olofsson, G.H.; Lauenborg, B.; Nookaew, I.; Hansen, R.H.; Johannesen, H.H.; Becker, J.C.; Pedersen, K.S.; Dethlefsen, C.; et al. Voluntary Running Suppresses Tumor Growth Through Epinephrine- and IL-6-Dependent NK Cell Mobilization and Redistribution. Cell Metab. 2016, 23, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Hojman, P.; Dethlefsen, C.; Brandt, C.; Hansen, J.; Pedersen, L.; Pedersen, B.K. Exercise-induced Muscle-derived Cytokines Inhibit Mammary Cancer Cell Growth. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E504–E510. [Google Scholar] [CrossRef] [PubMed]
- Falk, G.W.; Jacobson, B.C.; Riddell, R.H.; Rubenstein, J.H.; El-Zimaity, H.; Drewes, A.M.; Roark, K.S.; Sontag, S.J.; Schnell, T.G.; Leya, J.; et al. Barrett’s Esophagus: Prevalence-incidence and Etiology-origins. Ann. N. Y. Acad. Sci. 2011, 1232, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Johnsen, R.; Ye, W.; Hveem, K.; Lagergren, J. Lifestyle Related Risk Factors in the Aetiology of Gastro-oesophageal Reflux. Gut 2004, 53, 1730–1735. [Google Scholar] [CrossRef]
- Nocon, M.; Labenz, J.; Willich, S. Lifestyle Factors and Symptoms of Gastro-oesophageal Reflux—A Population-based Study. Aliment. Pharmacol. Ther. 2006, 23, 169–174. [Google Scholar] [CrossRef]
- Zheng, Z.; Nordenstedt, H.; Pedersen, N.L.; Lagergren, J.; Ye, W. Lifestyle Factors and Risk for Symptomatic Gastroesophageal Reflux in Monozygotic Twins. Gastroenterology 2007, 132, 87–95. [Google Scholar] [CrossRef]
- Sise, A.; Friedenberg, F.K. A Comprehensive Review of Gastroesophageal Reflux Disease and Obesity. Obes. Rev. 2008, 9, 194–203. [Google Scholar] [CrossRef]
- Schmidt, M.; Ankerst, D.P.; Chen, Y.; Wiethaler, M.; Slotta-Huspenina, J.; Becker, K.F.; Horstmann, J.; Kohlmayer, F.; Lehmann, A.; Linkohr, B.; et al. Epidemiologic Risk Factors in a Comparison of a Barrett Esophagus Registry (BarrettNET) and a Case-Control Population in Germany. Cancer Prev. Res. 2020, 13, 377–384. [Google Scholar] [CrossRef]
- Behrens, G.; Jochem, C.; Keimling, M.; Ricci, C.; Schmid, D.; Leitzmann, M.F. The Association Between Physical Activity and Gastroesophageal Cancer: Systematic Review and Meta-analysis. Eur. J. Epidemiol. 2014, 29, 151–170. [Google Scholar] [CrossRef]
- Singh, S.; Devanna, S.; Varayil, J.E.; Murad, M.H.; Iyer, P.G. Physical Activity is Associated with Reduced Risk of Esophageal cancer, particularly esophageal adenocarcinoma: A systematic review and meta-analysis. BMC Gastroenterol. 2014, 14, 101. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, C.; Li, Y. Physical Activity and Risks of Esophageal and Gastric Cancers: A Meta-analysis. PLoS ONE 2014, 9, e88082. [Google Scholar] [CrossRef] [PubMed]
- Balbuena, L.; Casson, A.G. Physical Activity, Obesity and Risk for Esophageal Adenocarcinoma. Future Oncol. 2009, 5, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Leitzmann, M.F.; Koebnick, C.; Freedman, N.D.; Park, Y.; Ballard-Barbash, R.; Hollenbeck, A.; Schatzkin, A.; Abnet, C.C. Physical Activity and Esophageal and Gastric Carcinoma in a Large Prospective Study. Am. J. Prev. Med. 2009, 36, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Vigen, C.; Bernstein, L.; Wu, A.H. Occupational Physical Activity and Risk of Adenocarcinomas of the Esophagus and Stomach. Int. J. Cancer 2006, 118, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Huerta, J.M.; Navarro, C.; Chirlaque, M.-D.; Tormo, M.-J.; Steindorf, K.; Buckland, G.; Carneiro, F.; Johnsen, N.F.; Overvad, K.; Stegger, J. Prospective Study of Physical Activity and Risk of Primary Adenocarcinomas of the Oesophagus and Stomach in the EPIC (European Prospective Investigation into Cancer and nutrition) Cohort. Cancer Causes Control 2010, 21, 657–669. [Google Scholar] [CrossRef]
- Bruunsgaard, H. Physical Activity and Modulation of Systemic Low-level Inflammation. J. Leukoc. Biol. 2005, 78, 819–835. [Google Scholar] [CrossRef]
- Mathur, N.; Pedersen, B.K. Exercise as a Mean to Control Low-grade Systemic Inflammation. Mediat. Inflamm. 2008, 2008, 109502. [Google Scholar] [CrossRef]
- Pedersen, B.K. Muscles and Their Myokines. J. Exp. Biol. 2011, 214, 337–346. [Google Scholar] [CrossRef]
- Schnyder, S.; Handschin, C. Skeletal Muscle as an Endocrine Organ: PGC-1alpha, Myokines and Exercise. Bone 2015, 80, 115–125. [Google Scholar] [CrossRef]
- Bilski, J.; Brzozowski, B.; Mazur-Bialy, A.; Sliwowski, Z.; Brzozowski, T. The Role of Physical Exercise in Inflammatory Bowel Disease. Biomed. Res. Int. 2014, 2014, 429031. [Google Scholar] [CrossRef]
- Saeidi, A.; Haghighi, M.M.; Kolahdouzi, S.; Daraei, A.; Abderrahmane, A.B.; Essop, M.F.; Laher, I.; Hackney, A.C.; Zouhal, H. The Effects of Physical Activity on Adipokines in Individuals with Overweight/Obesity Across the Lifespan: A Narrative Review. Obes. Rev. 2021, 22, e13090. [Google Scholar] [CrossRef] [PubMed]
- Indrakusuma, I.; Sell, H.; Eckel, J. Novel Mediators of Adipose Tissue and Muscle Crosstalk. Curr. Obes. Rep. 2015, 4, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, A.; Hashemy, S.I.; Azimi-Nezhad, M.; Dehghani, A.; Saeidi, J.; Mohtashami, M. The Cross-talk Between Adipokines and miRNAs in Health and Obesity-mediated Diseases. Clin. Chim. Acta 2019, 499, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Obi, P.O.; Bydak, B.; Safdar, A.; Saleem, A. Extracellular Vesicles and Circulating miRNAs—Exercise-Induced Mitigation of Obesity and Associated Metabolic Diseases. In Pathophysiology of Obesity-Induced Health Complications; Springer: Berlin/Heidelberg, Germany, 2020; pp. 59–80. [Google Scholar]
- Ge, Q.; Gerard, J.; Noel, L.; Scroyen, I.; Brichard, S.M. MicroRNAs Regulated by Adiponectin as Novel Targets for Controlling Adipose Tissue Inflammation. Endocrinology 2012, 153, 5285–5296. [Google Scholar] [CrossRef] [PubMed]
- Straub, L.G.; Scherer, P.E. Metabolic Messengers: Adiponectin. Nat. Metab. 2019, 1, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Dalamaga, M. Interplay of Adipokines and Myokines in Cancer Pathophysiology: Emerging Therapeutic Implications. World J. Exp. Med. 2013, 3, 26–33. [Google Scholar] [CrossRef]
- Friedenreich, C.M.; Orenstein, M.R. Physical Activity and Cancer Prevention: Etiologic Evidence and Biological Mechanisms. J. Nutr. 2002, 132, 3456S–3464S. [Google Scholar] [CrossRef]
- Holick, C.N.; Newcomb, P.A.; Trentham-Dietz, A.; Titus-Ernstoff, L.; Bersch, A.J.; Stampfer, M.J.; Baron, J.A.; Egan, K.M.; Willett, W.C. Physical activity and survival after diagnosis of invasive breast cancer. Cancer Epidemiol. Biomark. Prev. 2008, 17, 379–386. [Google Scholar] [CrossRef]
- Jones, L.W.; Viglianti, B.L.; Tashjian, J.A.; Kothadia, S.M.; Keir, S.T.; Freedland, S.J.; Potter, M.Q.; Moon, E.J.; Schroeder, T.; Herndon, J.E., 2nd; et al. Effect of Aerobic Exercise on Tumor Physiology in an Animal Model of Human Breast Cancer. J. Appl. Physiol. 2010, 108, 343–348. [Google Scholar] [CrossRef]
- Aoi, W.; Naito, Y.; Takagi, T.; Tanimura, Y.; Takanami, Y.; Kawai, Y.; Sakuma, K.; Hang, L.P.; Mizushima, K.; Hirai, Y.; et al. A Novel Myokine, Secreted Protein Acidic and Rich in Cysteine (SPARC), Suppresses Colon Tumorigenesis via Regular Exercise. Gut 2013, 62, 882–889. [Google Scholar] [CrossRef]
- Gannon, N.P.; Vaughan, R.A.; Garcia-Smith, R.; Bisoffi, M.; Trujillo, K.A. Effects of the Exercise-inducible Myokine Irisin on Malignant and Non-malignant Breast Epithelial Cell Behavior In Vitro. Int. J. Cancer 2015, 136, E197–E202. [Google Scholar] [CrossRef] [PubMed]
- Mazur-Bialy, A.I.; Oplawski, M.; Wypasek, E.; Zarawski, M. ID: 228: Irisin–A Newly Discovered Adipomiokine–Impairs Growth and Progression of Breast Cancer MDA-MB-231 cell line. Cytokine 2015, 76, 107. [Google Scholar] [CrossRef]
- Suat, T.; Yavuz, E.; Suleyman, S.; Bayram, Y. Is Irisin an Anticarcinogenic Peptide? Med. Sci. 2015, 4, 2172–2180. [Google Scholar]
- Moon, H.S.; Mantzoros, C.S. Regulation of Cell Proliferation and Malignant Potential by Irisin in Endometrial, Colon, Thyroid and Esophageal Cancer Cell Lines. Metabolism 2014, 63, 188–193. [Google Scholar] [CrossRef]
- Sun, Z.; Shi, K.; Yang, S.; Liu, J.; Zhou, Q.; Wang, G.; Song, J.; Li, Z.; Zhang, Z.; Yuan, W. Effect of Exosomal miRNA on Cancer Biology and Clinical Applications. Mol. Cancer 2018, 17, 147. [Google Scholar] [CrossRef]
- Dufresne, S.; Rebillard, A.; Muti, P.; Friedenreich, C.M.; Brenner, D.R. A Review of Physical Activity and Circulating miRNA Expression: Implications in Cancer Risk and Progression. Cancer Epidemiol. Biomark. Prev. 2018, 27, 11–24. [Google Scholar] [CrossRef]
- Chang, M.L.; Yang, Z.; Yang, S.S. Roles of Adipokines in Digestive Diseases: Markers of Inflammation, Metabolic Alteration and Disease Progression. Int. J. Mol. Sci. 2020, 21, 8308. [Google Scholar] [CrossRef]
- Chemnitzer, O.; Götzel, K.; Maurer, L.; Dietrich, A.; Eichfeld, U.; Lyros, O.; Jansen-Winkeln, B.; Hoffmeister, A.; Gockel, I.; Thieme, R. Response to TNF-α is Increasing Along with the Progression in Barrett’s Esophagus. Dig. Dis. Sci. 2017, 62, 3391–3401. [Google Scholar] [CrossRef]
- Cook, M.B.; Barnett, M.J.; Bock, C.H.; Cross, A.J.; Goodman, P.J.; Goodman, G.E.; Haiman, C.A.; Khaw, K.T.; McCullough, M.L.; Newton, C.C.; et al. Prediagnostic Circulating Markers of Inflammation and Risk of Oesophageal Adenocarcinoma: A Study within the National Cancer Institute Cohort Consortium. Gut 2019, 68, 960–968. [Google Scholar] [CrossRef]
- Konturek, P.C.; Burnat, G.; Rau, T.; Hahn, E.G.; Konturek, S. Effect of Adiponectin and Ghrelin on Apoptosis of Barrett’s Adenocarcinoma Cell Line. Dig. Dis. Sci. 2008, 53, 597–605. [Google Scholar] [CrossRef]
- Moodi, M.; Tavakoli, T.; Tahergorabi, Z. Crossroad between Obesity and Gastrointestinal Cancers: A Review of Molecular Mechanisms and Interventions. Int. J. Prev. Med. 2021, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Arcidiacono, D.; Zaramella, A.; Fabris, F.; Sánchez-Rodríguez, R.; Nucci, D.; Fassan, M.; Nardi, M.; Benna, C.; Cristofori, C.; Morbin, T.; et al. Insulin/IGF-1 Signaling Is Downregulated in Barrett’s Esophagus Patients Undergoing a Moderate Calorie and Protein Restriction Program: A Randomized 2-Year Trial. Nutrients 2021, 13, 3638. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.M.; Beddy, P.; Ennis, D.; Keogan, M.; Pidgeon, G.P.; Reynolds, J.V. Associations between Leptin and Adiponectin Receptor Upregulation, Visceral Obesity and Tumour Stage in Oesophageal and Junctional Adenocarcinoma. Br. J. Surg. 2010, 97, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wu, J.; Liu, D.; Shan, H.; Zhang, J. Anti-inflammatory Effect of Full-length Adiponectin and Proinflammatory Effect of Globular Adiponectin in Esophageal Adenocarcinoma Cells. Oncol. Res. 2013, 21, 15–21. [Google Scholar] [CrossRef]
- Trevellin, E.; Scarpa, M.; Carraro, A.; Lunardi, F.; Kotsafti, A.; Porzionato, A.; Saadeh, L.; Cagol, M.; Alfieri, R.; Tedeschi, U.; et al. Esophageal Adenocarcinoma and Obesity: Peritumoral Adipose Tissue Plays a Role in Lymph Node Invasion. Oncotarget 2015, 6, 11203–11215. [Google Scholar] [CrossRef]
- Hyland, P.L.; Hu, N.; Rotunno, M.; Su, H.; Wang, C.; Wang, L.; Pfeiffer, R.M.; Gherman, B.; Giffen, C.; Dykes, C.; et al. Global Changes in Gene Expression of Barrett’s Esophagus Compared to Normal Squamous Esophagus and Gastric Cardia Tissues. PLoS ONE 2014, 9, e93219. [Google Scholar] [CrossRef]


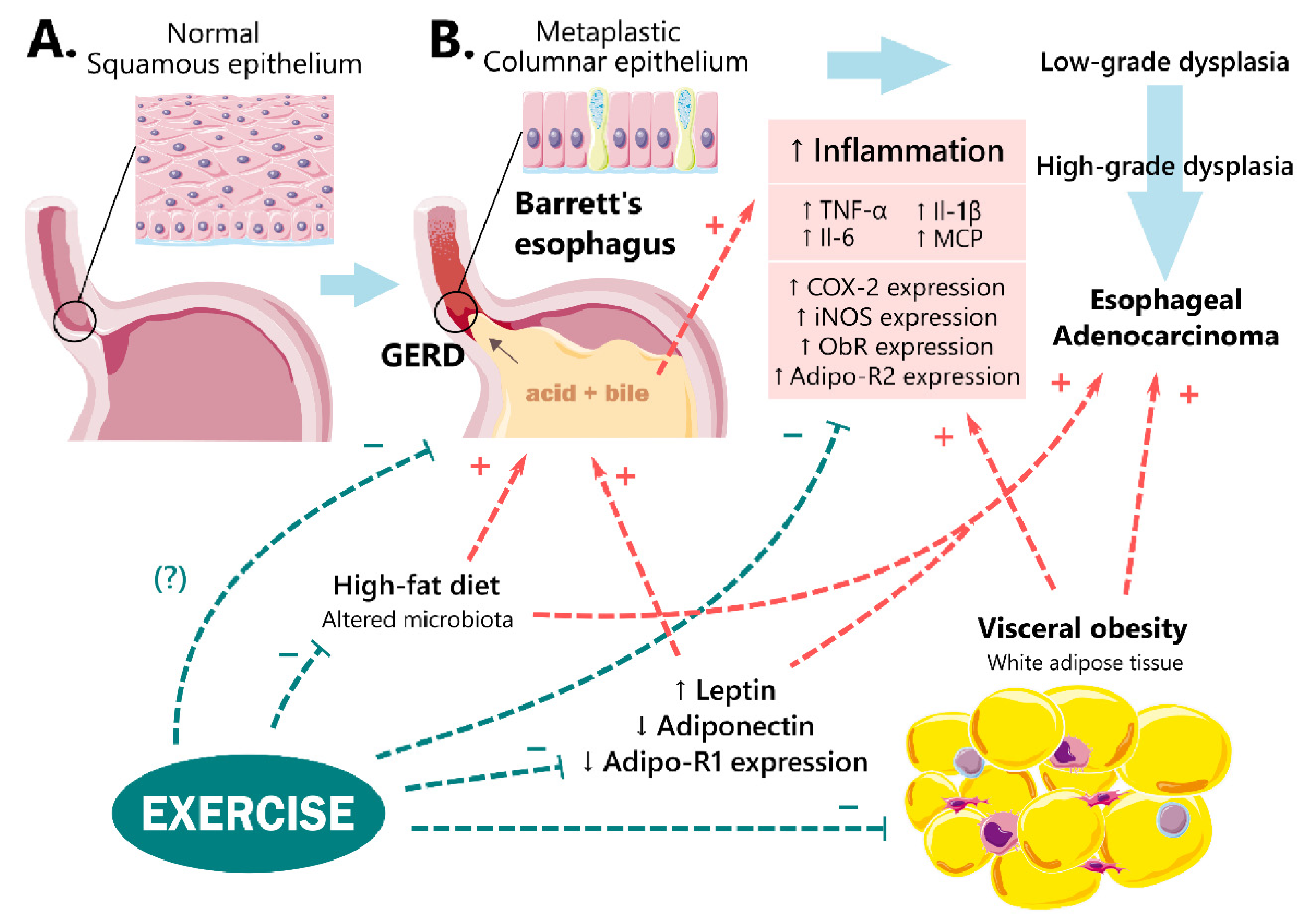{kind=link}
| Mediator | Role in BE | Role in EAC | |
|---|---|---|---|
| Adipokines | Leptin | ||
| TNF-α, IL-1β, IL-6 | |||
| Adiponectin | |||
| Myokines | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilski, J.; Pinkas, M.; Wojcik-Grzybek, D.; Magierowski, M.; Korbut, E.; Mazur-Bialy, A.; Krzysiek-Maczka, G.; Kwiecien, S.; Magierowska, K.; Brzozowski, T. Role of Obesity, Physical Exercise, Adipose Tissue-Skeletal Muscle Crosstalk and Molecular Advances in Barrett’s Esophagus and Esophageal Adenocarcinoma. Int. J. Mol. Sci. 2022, 23, 3942. https://doi.org/10.3390/ijms23073942
Bilski J, Pinkas M, Wojcik-Grzybek D, Magierowski M, Korbut E, Mazur-Bialy A, Krzysiek-Maczka G, Kwiecien S, Magierowska K, Brzozowski T. Role of Obesity, Physical Exercise, Adipose Tissue-Skeletal Muscle Crosstalk and Molecular Advances in Barrett’s Esophagus and Esophageal Adenocarcinoma. International Journal of Molecular Sciences. 2022; 23(7):3942. https://doi.org/10.3390/ijms23073942
Chicago/Turabian StyleBilski, Jan, Monika Pinkas, Dagmara Wojcik-Grzybek, Marcin Magierowski, Edyta Korbut, Agnieszka Mazur-Bialy, Gracjana Krzysiek-Maczka, Slawomir Kwiecien, Katarzyna Magierowska, and Tomasz Brzozowski. 2022. "Role of Obesity, Physical Exercise, Adipose Tissue-Skeletal Muscle Crosstalk and Molecular Advances in Barrett’s Esophagus and Esophageal Adenocarcinoma" International Journal of Molecular Sciences 23, no. 7: 3942. https://doi.org/10.3390/ijms23073942
APA StyleBilski, J., Pinkas, M., Wojcik-Grzybek, D., Magierowski, M., Korbut, E., Mazur-Bialy, A., Krzysiek-Maczka, G., Kwiecien, S., Magierowska, K., & Brzozowski, T. (2022). Role of Obesity, Physical Exercise, Adipose Tissue-Skeletal Muscle Crosstalk and Molecular Advances in Barrett’s Esophagus and Esophageal Adenocarcinoma. International Journal of Molecular Sciences, 23(7), 3942. https://doi.org/10.3390/ijms23073942