
SARS-CoV-2 Proteins Interact with Alpha Synuclein and Induce Lewy Body-like Pathology In Vitro


Abstract
:1. Introduction
2. Results
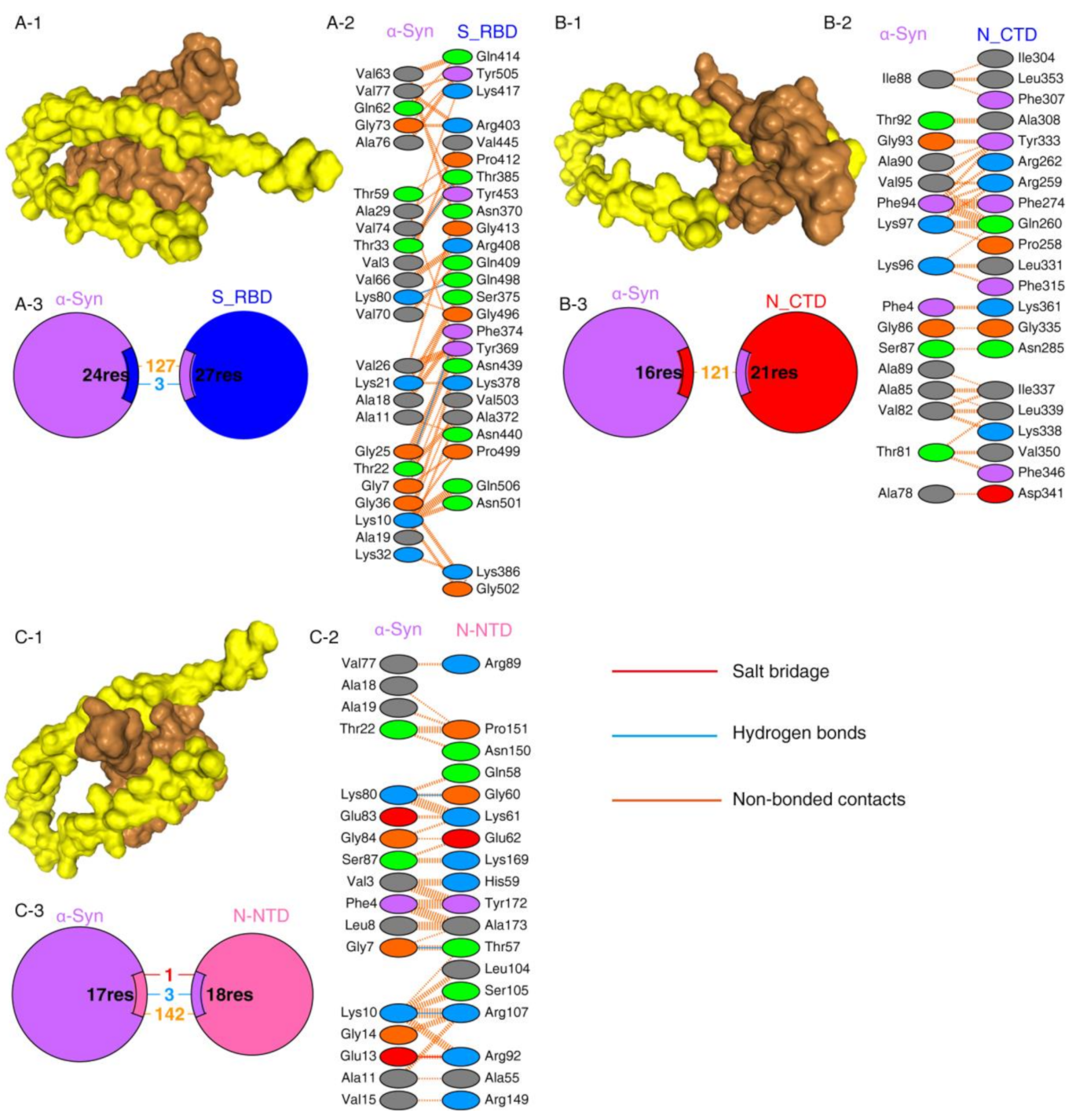
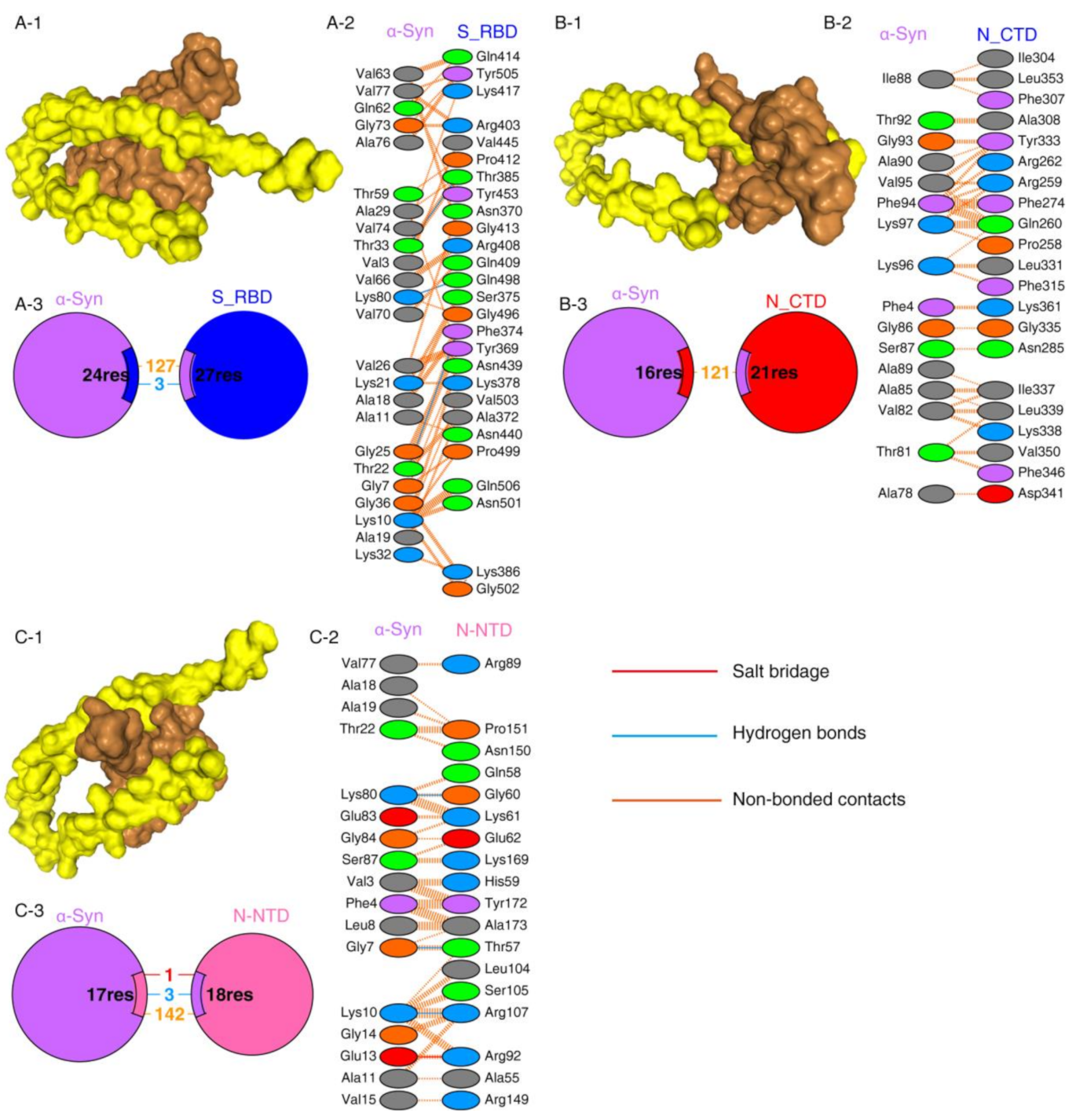
2.1. Protein–Protein Interaction between SARS-CoV-2 Proteins and α-Syn
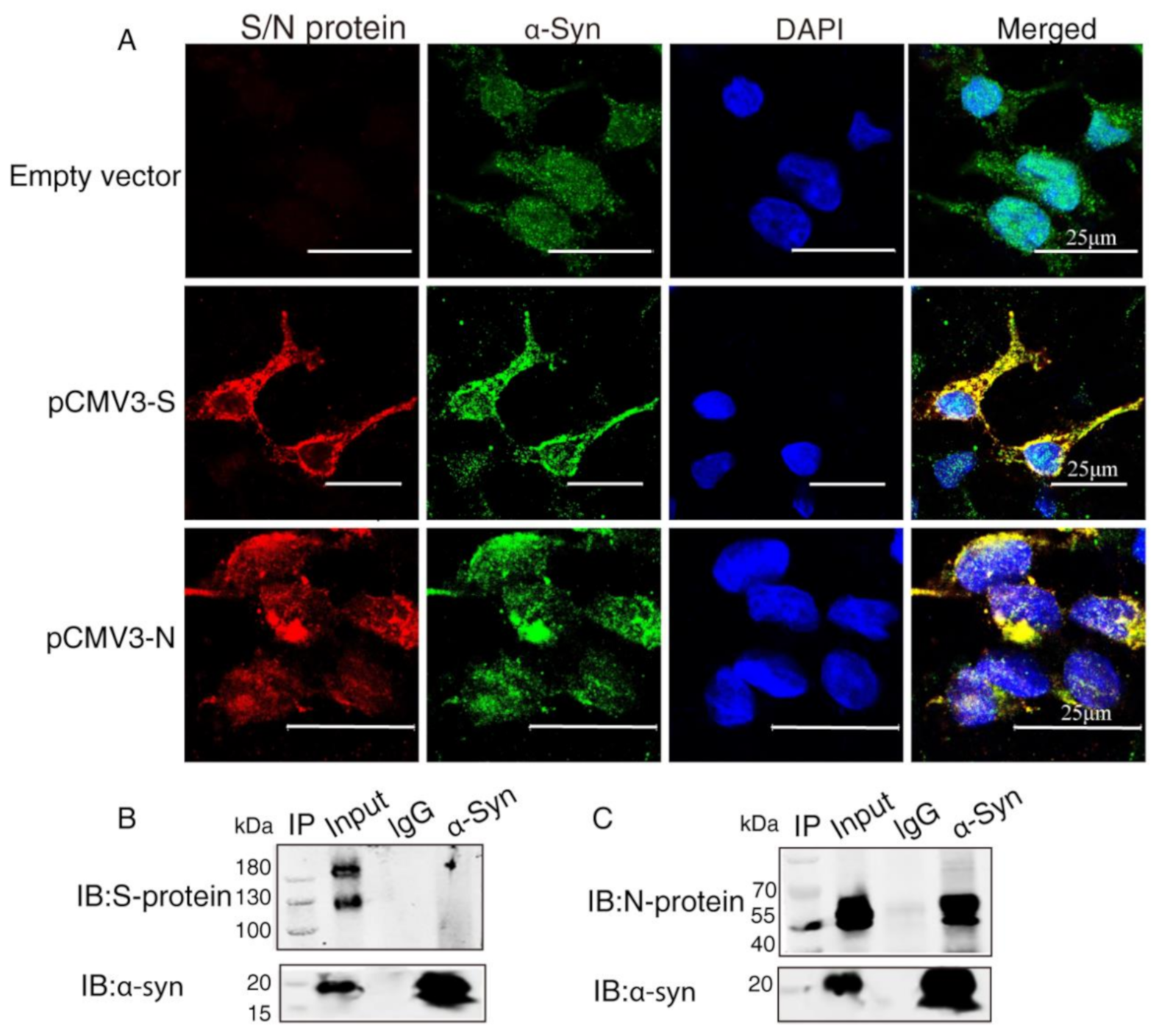
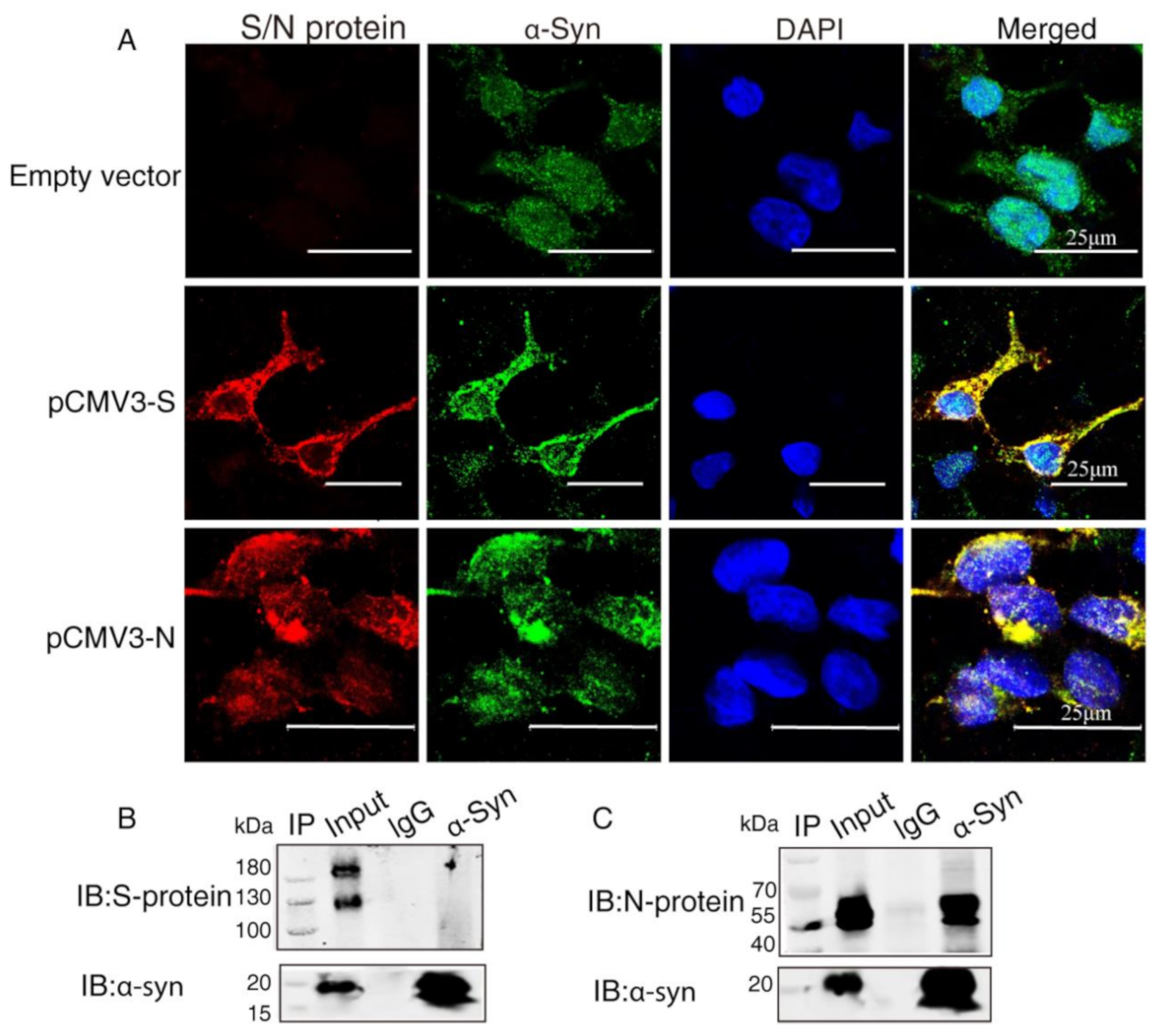
2.2. α-Syn Directly Interacts with SARS-CoV-2 Proteins in HEK293 Cells
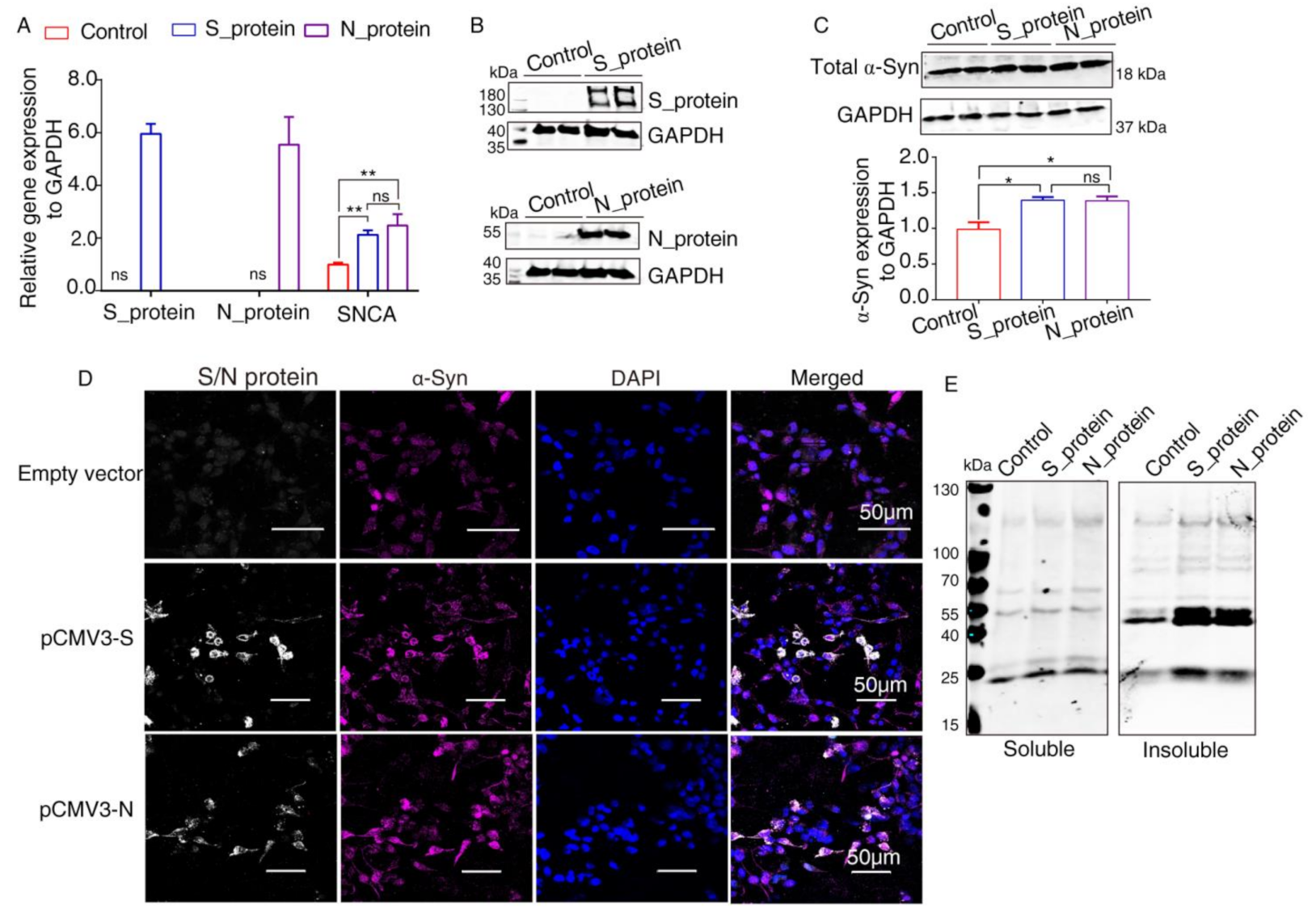
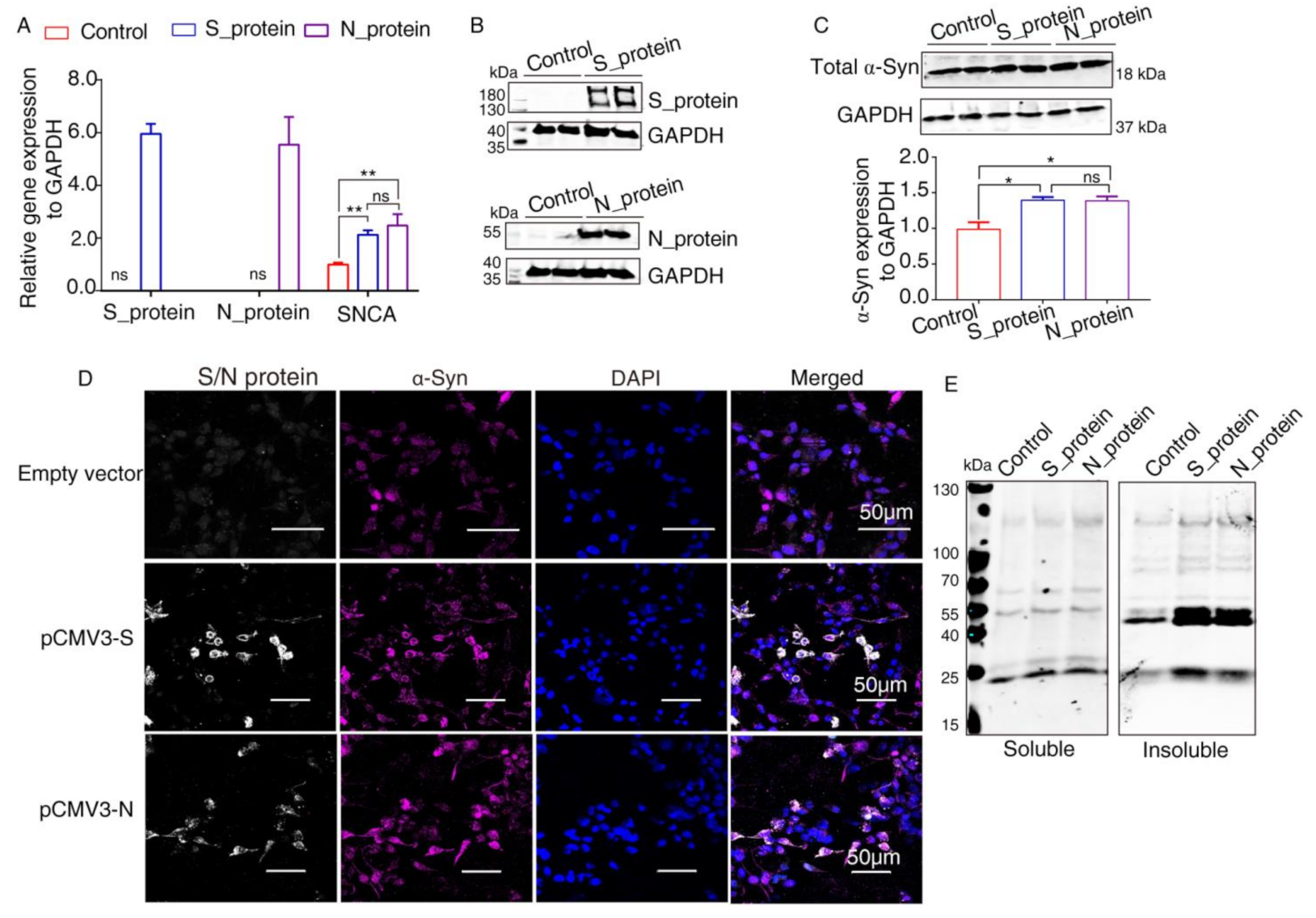
2.3. Elevated Expression of α-Syn by SARS-CoV-2 Proteins in HEK293 Cells
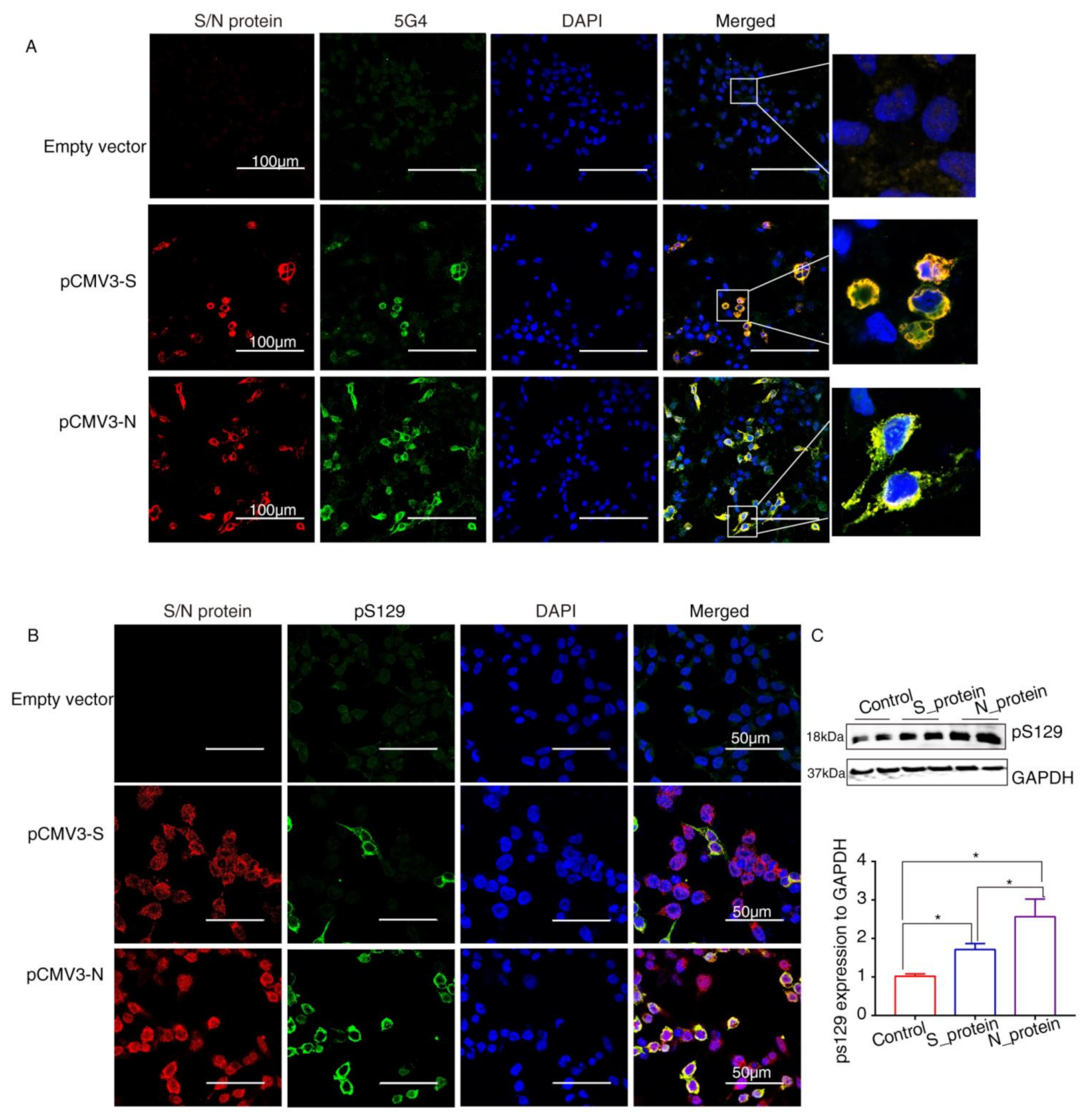
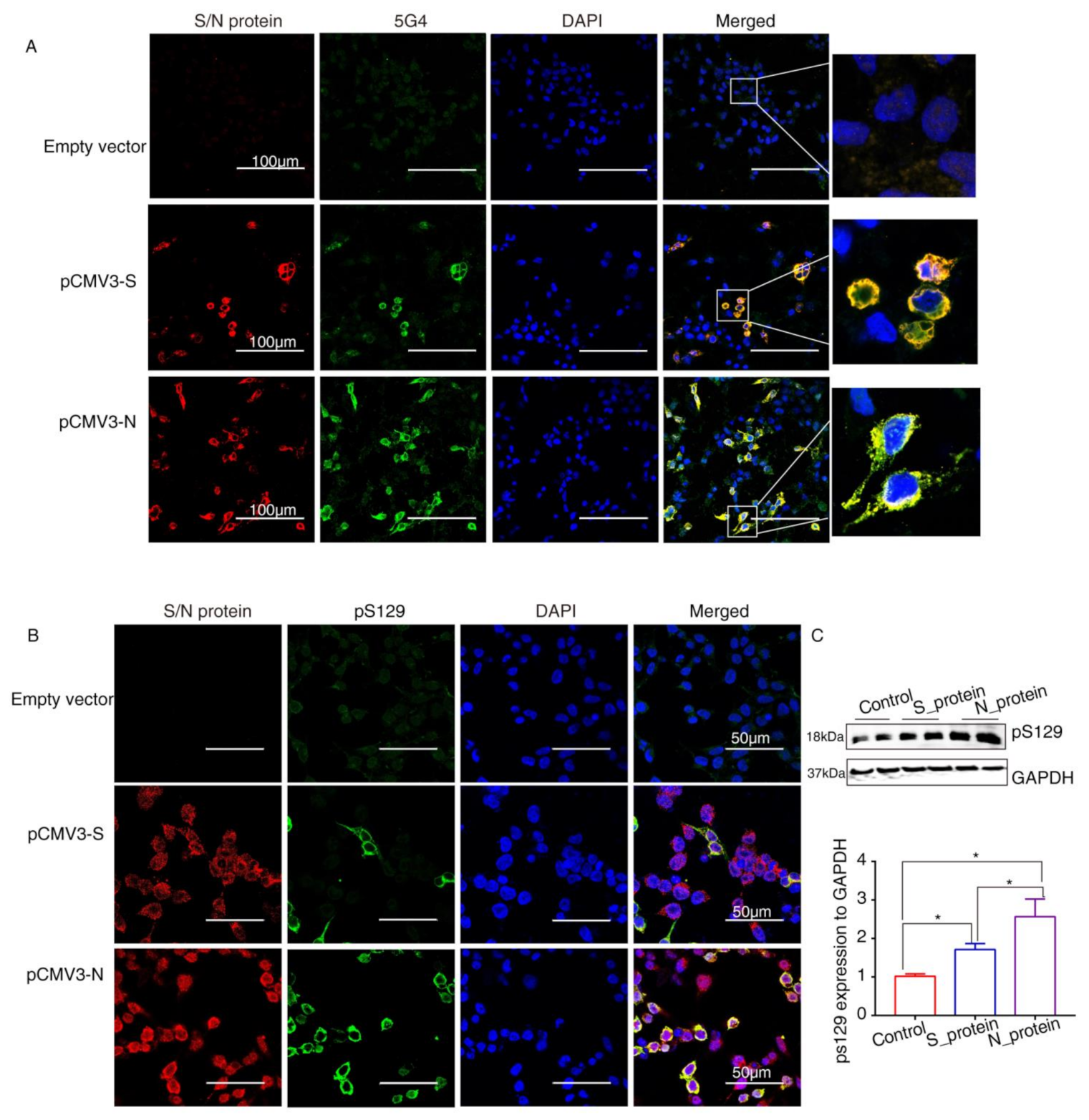
2.4. SARS-CoV-2 Proteins Caused Lewy-Like Pathology in HEK293 Cells Overexpressing α-Syn
3. Discussion
4. Materials and Methods
4.1. Protein–Protein Docking
4.2. Cell Culture and Transfection
4.3. RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
4.4. Western Blot Analysis
4.5. Co-Immunoprecipitation Assay (Co-IP)
4.6. Confocal Immunofluorescence Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PD | Parkinson’s disease |
| α-Syn | Alpha-synuclein |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| COVID-19 | 2019 novel coronavirus disease |
| LBs | Lewy bodies |
| N | nucleocapsid |
| S | spike |
| RBD | receptor-binding domain |
| ACE2 | angiotensin-converting enzyme 2 |
| NTD | N-terminal domain |
| CTD | C-terminal domain |
| PNS | peripheral nervous systems |
| CNS | central nervous systems |
| △G | binding free energy scores |
| Kd | dissociation constant |
| qRT-PCR | Quantitative real-time PCR |
| pS129 | phosphorylation at Ser129 |
| HKE29 | human kidney 293 |
References
- Shahid, Z.; Kalayanamitra, R.; McClafferty, B.; Kepko, D.; Ramgobin, D.; Patel, R.; Aggarwal, C.S.; Vunnam, R.; Sahu, N.; Bhatt, D.; et al. COVID-19 and Older Adults: What We Know. J. Am. Geriatr. Soc. 2020, 68, 926–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, B.; Kurtishi, A.; Vazquez-Jimenez, G.R.; Møller, S.G. The Intersection of Parkinson’s Disease, Viral Infections, and COVID-19. Mol. Neurobiol. 2021, 58, 4477–4486. [Google Scholar] [CrossRef] [PubMed]
- Chaná-Cuevas, P.; Salles-Gándara, P.; Rojas-Fernandez, A.; Salinas-Rebolledo, C.; Milán-Solé, A. The Potential Role of SARS-CoV-2 in the Pathogenesis of Parkinson’s Disease. Front. Neurol. 2020, 11, 1044. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Mittal, S.; Roy, R. Parkinson’s Disease and the COVID-19 Pandemic: A Review Article on the Association between SARS-CoV-2 and α-Synucleinopathy. J. Mov. Disord. 2021, 14, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Conte, C. Possible Link between SARS-CoV-2 Infection and Parkinson’s Disease: The Role of Toll-Like Receptor 4. Int. J. Mol. Sci. 2021, 22, 7135. [Google Scholar] [CrossRef] [PubMed]
- Faber, I.; Brandão, P.R.P.; Menegatti, F.; Carvalho Bispo, D.D.; Maluf, F.B.; Cardoso, F. Coronavirus Disease 2019 and Parkinsonism: A Non-post-encephalitic Case. Movement Disord. 2020, 35, 1721–1722. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Guerrero, A.; Laespada-Garcia, M.I.; Gomez-Grande, A.; Ruiz-Ortiz, M.; Blanco-Palmero, V.A.; Azcarate-Diaz, F.J.; Rabano-Suarez, P.; Alvarez-Torres, E.; de Fuenmayor-Fernandez, D.L.H.C.; Vega, P.D.; et al. Acute hypokinetic-rigid syndrome following SARS-CoV-2 infection. Neurology 2020, 95, e2109–e2118. [Google Scholar] [CrossRef]
- Rao, A.R.; Hidayathullah, S.M.; Hegde, K.; Adhikari, P. Parkinsonism: An emerging post COVID sequelae. IDCases 2022, 27, e1388. [Google Scholar] [CrossRef]
- Guedj, E.; Million, M.; Dudouet, P.; Tissot-Dupont, H.; Bregeon, F.; Cammilleri, S.; Raoult, D. 18F-FDG brain PET hypometabolism in post-SARS-CoV-2 infection: Substrate for persistent/delayed disorders? Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 592–595. [Google Scholar] [CrossRef]
- Cohen, M.E.; Eichel, R.; Steiner-Birmanns, B.; Janah, A.; Ioshpa, M.; Bar-Shalom, R.; Paul, J.J.; Gaber, H.; Skrahina, V.; Bornstein, N.M.; et al. A case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol. 2020, 19, 804–805. [Google Scholar] [CrossRef]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wang, T.; Jiang, T.; Huang, P.; Wang, Y.; Xiao, Q.; Liu, J.; Chen, S. Clinical Profile of Chinese Long-Term Parkinson’s Disease Survivors With 10 Years of Disease Duration and Beyond. Aging Dis. 2018, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Idrees, D.; Kumar, V. SARS-CoV-2 spike protein interactions with amyloidogenic proteins: Potential clues to neurodegeneration. Biochem. Bioph. Res. Commun. 2021, 554, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Y.; Zhao, R.; Gao, L.J.; Gao, X.F.; Wang, D.P.; Cao, J.M. SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. Front. Cell Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Xia, X. Domains and Functions of Spike Protein in SARS-CoV-2 in the Context of Vaccine Design. Viruses 2021, 13, 109. [Google Scholar] [CrossRef]
- Zeng, W.; Liu, G.; Ma, H.; Zhao, D.; Yang, Y.; Liu, M.; Mohammed, A.; Zhao, C.; Yang, Y.; Xie, J.; et al. Biochemical characterization of SARS-CoV-2 nucleocapsid protein. Biochem. Biophys. Res. Commun. 2020, 527, 618–623. [Google Scholar] [CrossRef]
- Perdikari, T.M.; Murthy, A.C.; Ryan, V.H.; Watters, S.; Naik, M.T.; Fawzi, N.L. SARS-CoV-2 nucleocapsid protein phase-separates with RNA and with human hnRNPs. EMBO J. 2020, 39, e106478. [Google Scholar] [CrossRef]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Yan, Y.; Huang, S.Y. Modeling Protein-Protein or Protein-DNA/RNA Complexes Using the HDOCK Webserver. Methods Mol. Biol. 2020, 2165, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Beatman, E.L.; Massey, A.; Shives, K.D.; Burrack, K.S.; Chamanian, M.; Morrison, T.E.; Beckham, J.D. Alpha-Synuclein Expression Restricts RNA Viral Infections in the Brain. J. Virol. 2015, 90, 2767–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Arawaka, S.; Sato, H.; Sasaki, A.; Koyama, S.; Kato, T. Mechanisms underlying extensive Ser129-phosphorylation in alpha-synuclein aggregates. Acta Neuropathol. Commun. 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Yamada, T.; Nakajima, S.; Nakajima, K.; Yamamoto, T.; Okada, H. The substantia nigra is a major target for neurovirulent influenza A virus. J. Exp. Med. 1995, 181, 2161–2169. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T. Viral etiology of Parkinson’s disease: Focus on influenza A virus. Parkinsonism Relat. Disord. 1996, 2, 113–121. [Google Scholar] [CrossRef]
- Limphaibool, N.; Iwanowski, P.; Holstad, M.; Kobylarek, D.; Kozubski, W. Infectious Etiologies of Parkinsonism: Pathomechanisms and Clinical Implications. Front. Neurol. 2019, 10, 652. [Google Scholar] [CrossRef] [PubMed]
- Cilia, R.; Bonvegna, S.; Straccia, G.; Andreasi, N.G.; Elia, A.E.; Romito, L.M.; Devigili, G.; Cereda, E.; Eleopra, R. Effects of COVID-19 on Parkinson’s Disease Clinical Features: A Community-Based Case-Control Study. Mov. Disord. 2020, 35, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Antonini, A.; Leta, V.; Teo, J.; Chaudhuri, K.R. Outcome of Parkinson’s Disease Patients Affected by COVID-19. Mov. Disord. 2020, 35, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Puelles, V.G.; Lutgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020, 383, 590–592. [Google Scholar] [CrossRef] [PubMed]
- Niazkar, H.R.; Zibaee, B.; Nasimi, A.; Bahri, N. The neurological manifestations of COVID-19: A review article. Neurol. Sci. 2020, 41, 1667–1671. [Google Scholar] [CrossRef] [PubMed]
- Semerdzhiev, S.A.; Fakhree, M.A.A.; Segers-Nolten, I.; Blum, C.; Claessens, M.M.A.E. Interactions between SARS-CoV-2 N-Protein and α-Synuclein Accelerate Amyloid Formation. ACS Chem. Neurosci. 2022, 13, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell Biology and Pathophysiology of α-Synuclein. CSH Perspect. Med. 2018, 8, a24091. [Google Scholar] [CrossRef]
- Marreiros, R.; Muller-Schiffmann, A.; Trossbach, S.V.; Prikulis, I.; Hansch, S.; Weidtkamp-Peters, S.; Moreira, A.R.; Sahu, S.; Soloviev, I.; Selvarajah, S.; et al. Disruption of cellular proteostasis by H1N1 influenza A virus causes alpha-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2020, 117, 6741–6751. [Google Scholar] [CrossRef]
- Jang, H.; Boltz, D.; Sturm-Ramirez, K.; Shepherd, K.R.; Jiao, Y.; Webster, R.; Smeyne, R.J. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 14063–14068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Palmero, V.A.; Azcárate-Díaz, F.J.; Ruiz-Ortiz, M.; Laespada-García, M.I.; Rábano-Suárez, P.; Méndez-Guerrero, A.; Aramendi-Ramos, M.; Eguiburu, J.L.; Pérez-Rivilla, A.; Marchán-López, A.; et al. Serum and CSF alpha-synuclein levels do not change in COVID-19 patients with neurological symptoms. J. Neurol. 2021, 268, 3116–3124. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and Dynamics of Micelle-bound Human α-Synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [Green Version]
- Chandra, S.; Chen, X.; Rizo, J.; Jahn, R.; Sudhof, T.C. A broken alpha-helix in folded alpha-Synuclein. J. Biol. Chem. 2003, 278, 15313–15318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S. HDOCK: A web server for protein–protein and protein–DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Huang, S. Pushing the accuracy limit of shape complementarity for protein-protein docking. BMC Bioinform. 2019, 20, 696. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Zou, X. An iterative knowledge-based scoring function for protein-protein recognition. Proteins Struct. Funct. Bioinform. 2008, 72, 557–579. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jablonska, J.; Pravda, L.; Varekova, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Yugandhar, K.; Gromiha, M.M. Protein-protein binding affinity prediction from amino acid sequence. Bioinformatics 2014, 30, 3583–3589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, T.; Wu, Z.; Luo, H.; Lu, S.; Ma, K. Injection of α-syn-98 Aggregates into the Brain Triggers α-Synuclein Pathology and an Inflammatory Response. Front. Mol. Neurosci. 2019, 12, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Gao, J.; Du, T.; Tang, D.; Chen, N.; Yuan, Y.; Ma, K. Alpha-synuclein is highly prone to distribution in the hippocampus and midbrain in tree shrews, and its fibrils seed Lewy body-like pathology in primary neurons. Exp. Gerontol. 2019, 116, 37–45. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein–Protein Complex | Docking Score | ΔG (kcal mol−1) | Kd (M) |
|---|---|---|---|
| N-CTD-α-Syn | −252.56 | −8.63 | 4.72 × 10−07 M |
| N-NTD-α-Syn | −209.03 | −9.09 | 2.17 × 10−07 M |
| S1_RBD-α-Syn | −243.43 | −10.68 | 1.46 ×10−08 M |
| S1_RBD-ACE2 | −291.07 | −12.95 | 3.17 × 10−10 M |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.; Zhang, X.; Huang, Z.; Ma, K. SARS-CoV-2 Proteins Interact with Alpha Synuclein and Induce Lewy Body-like Pathology In Vitro. Int. J. Mol. Sci. 2022, 23, 3394. https://doi.org/10.3390/ijms23063394
Wu Z, Zhang X, Huang Z, Ma K. SARS-CoV-2 Proteins Interact with Alpha Synuclein and Induce Lewy Body-like Pathology In Vitro. International Journal of Molecular Sciences. 2022; 23(6):3394. https://doi.org/10.3390/ijms23063394
Chicago/Turabian StyleWu, Zhengcun, Xiuao Zhang, Zhangqiong Huang, and Kaili Ma. 2022. "SARS-CoV-2 Proteins Interact with Alpha Synuclein and Induce Lewy Body-like Pathology In Vitro" International Journal of Molecular Sciences 23, no. 6: 3394. https://doi.org/10.3390/ijms23063394
APA StyleWu, Z., Zhang, X., Huang, Z., & Ma, K. (2022). SARS-CoV-2 Proteins Interact with Alpha Synuclein and Induce Lewy Body-like Pathology In Vitro. International Journal of Molecular Sciences, 23(6), 3394. https://doi.org/10.3390/ijms23063394