
Differential Metabotypes in Synovial Fibroblasts and Synovial Fluid in Hip Osteoarthritis Patients Support Inflammatory Responses

 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Results
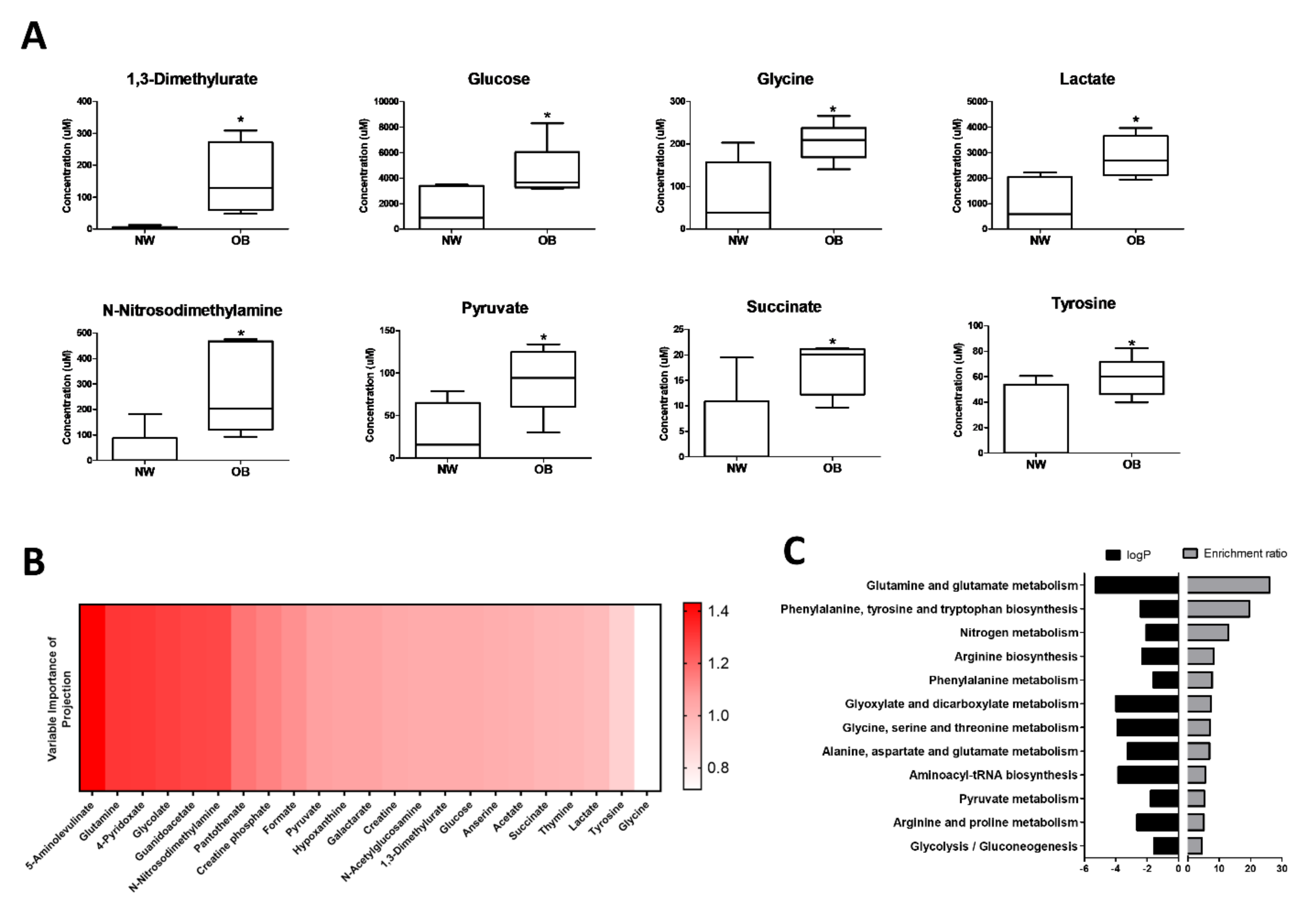
2.1. NMR Spectroscopy Reveals Differential Synovial Fluid Metabotypes in Obese and Normal Weight Hip OA Patients
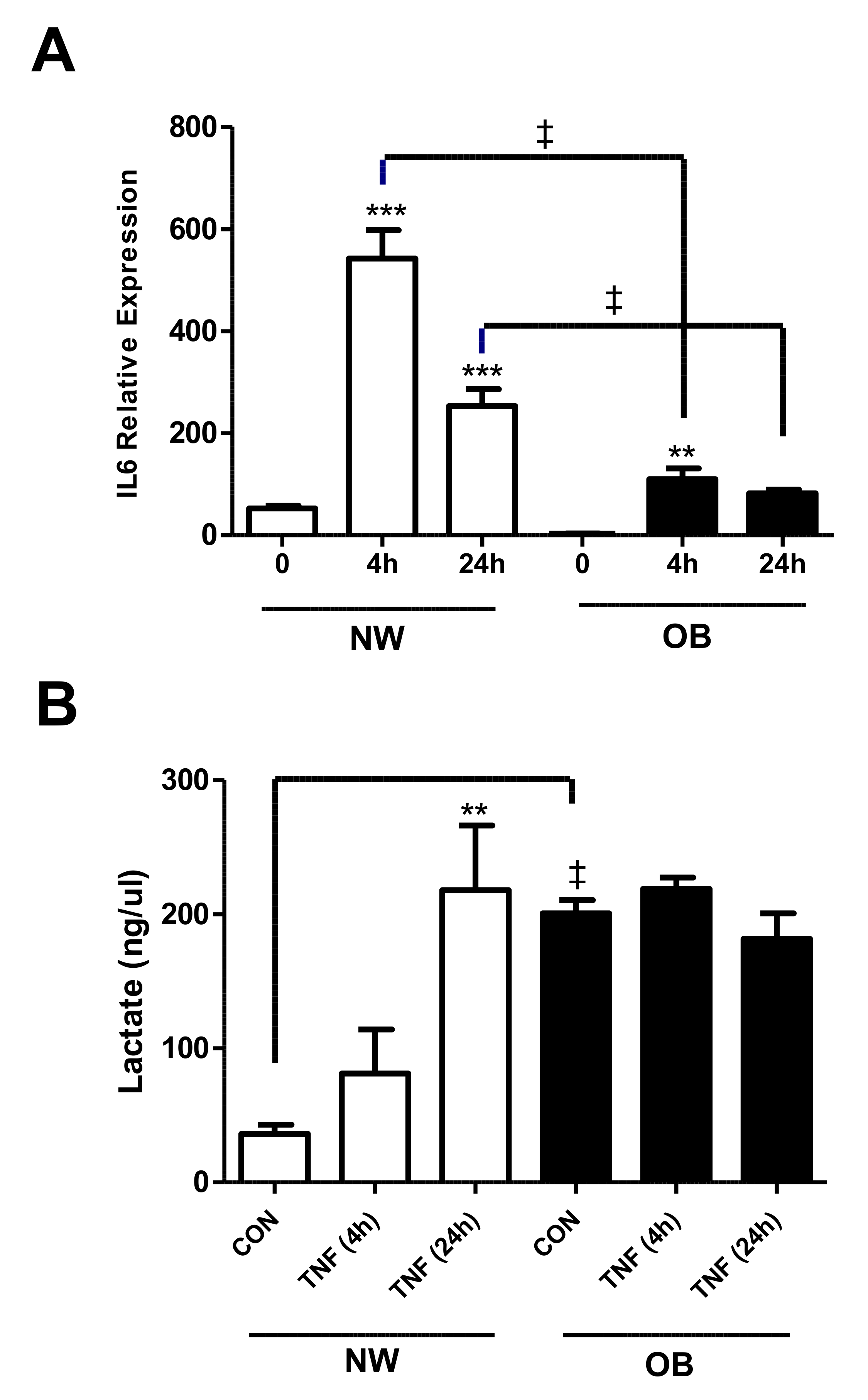
2.2. Lactate Secretion Is Greater in Obese OA Synovial Fibroblasts and Is Induced during the Inflammatory Response
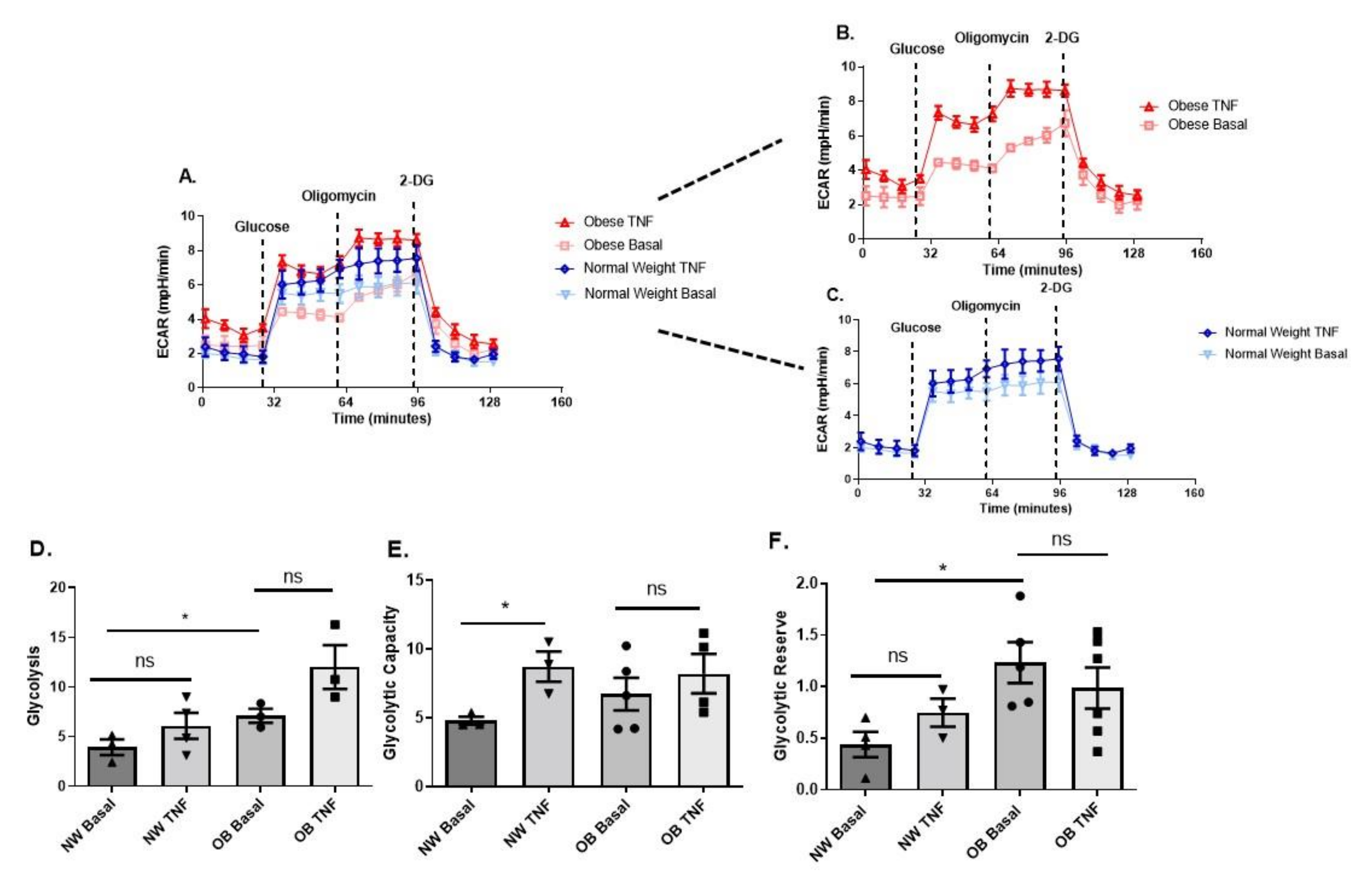
2.3. Obese OA Synovial Fibroblasts Increase Mitochondrial Respiration When Stimulated with Pro-Inflammatory Cytokine TNFα and Show Increased Aerobic Glycolysis Compared to OA Synovial Fibroblasts from Normal Weight Patients
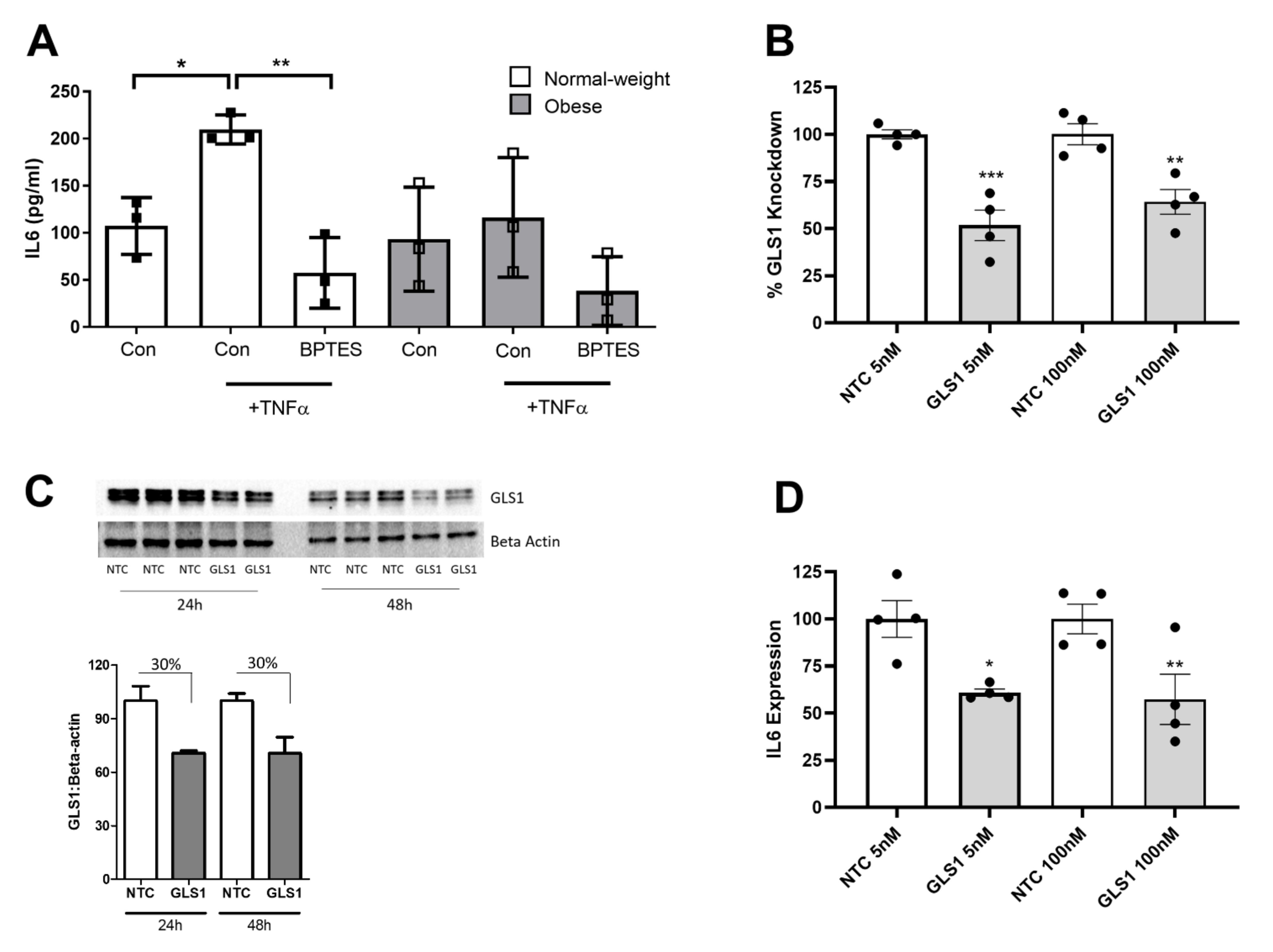
2.4. Glutaminase-1 Inhibition Attenuates the OA Synovial Fibroblast Inflammatory Response
3. Discussion
4. Materials and Methods
4.1. Patient Samples and Synovial Fibroblast Cell Culture
4.2. siRNA Transfection
4.3. Quantitative RT-PCR
4.4. ELISA
4.5. Western Blotting
4.6. Seahorse Metabolic Analysis of OA Synovial Fibroblast
4.7. Lactate Assay
4.8. NMR Spectroscopy
5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tonge, D.P.; Pearson, M.J.; Jones, S.W. The hallmarks of osteoarthritis and the potential to develop personalised disease-modifying pharmacological therapeutics. Osteoarthr. Cartil. 2014, 22, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Grassel, S.; Zaucke, F.; Madry, H. Osteoarthritis: Novel Molecular Mechanisms Increase Our Understanding of the Disease Pathology. J. Clin. Med. 2021, 10, 1938. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.W.; Brockbank, S.M.; Clements, K.M.; Le Good, N.; Campbell, D.; Read, S.J.; Needham, M.R.; Newham, P. Mitogen-activated protein kinase-activated protein kinase 2 (MK2) modulates key biological pathways associated with OA disease pathology. Osteoarthr. Cartil. 2009, 17, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.W.; Brockbank, S.M.; Mobbs, M.L.; Le Good, N.J.; Soma-Haddrick, S.; Heuze, A.J.; Langham, C.J.; Timms, D.; Newham, P.; Needham, M.R. The orphan G-protein coupled receptor RDC1: Evidence for a role in chondrocyte hypertrophy and articular cartilage matrix turnover. Osteoarthr. Cartil. 2006, 14, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Nanus, D.E.; Badoume, A.; Wijesinghe, S.N.; Halsey, A.M.; Hurley, P.; Ahmed, Z.; Botchu, R.; Davis, E.T.; Lindsay, M.A.; Jones, S.W. Synovial tissue from sites of joint pain in knee osteoarthritis patients exhibits a differential phenotype with distinct fibroblast subsets. EBioMedicine 2021, 72, 103618. [Google Scholar] [CrossRef]
- De Lange-Brokaar, B.J.; Ioan-Facsinay, A.; Yusuf, E.; Visser, A.W.; Kroon, H.M.; van Osch, G.J.; Zuurmond, A.M.; Stojanovic-Susulic, V.; Bloem, J.L.; Nelissen, R.G.; et al. Association of pain in knee osteoarthritis with distinct patterns of synovitis. Arthritis Rheumatol. 2015, 67, 733–740. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Jadon, D.R.; Bulmer, D.C.; Smith, E.S.J. Human osteoarthritic synovial fluid increases excitability of mouse dorsal root ganglion sensory neurons: An in-vitro translational model to study arthritic pain. Rheumatology 2020, 59, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Myers, S.L.; Brandt, K.D.; Ehlich, J.W.; Braunstein, E.M.; Shelbourne, K.D.; Heck, D.A.; Kalasinski, L.A. Synovial inflammation in patients with early osteoarthritis of the knee. J. Rheumatol. 1990, 17, 1662–1669. [Google Scholar]
- Prieto-Potin, I.; Largo, R.; Roman-Blas, J.A.; Herrero-Beaumont, G.; Walsh, D.A. Characterization of multinucleated giant cells in synovium and subchondral bone in knee osteoarthritis and rheumatoid arthritis. BMC Musculoskelet Disord. 2015, 16, 226. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Fang, Y.; Tan, X.; Jiang, H.; Gong, X.; Wang, X.; Hong, W.; Tu, J.; Wei, W. The emerging role of fibroblast-like synoviocytes-mediated synovitis in osteoarthritis: An update. J. Cell Mol. Med. 2020, 24, 9518–9532. [Google Scholar] [CrossRef] [PubMed]
- Philp, A.M.; Davis, E.T.; Jones, S.W. Developing anti-inflammatory therapeutics for patients with osteoarthritis. Rheumatology 2017, 56, 869–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, M.J.; Herndler-Brandstetter, D.; Tariq, M.A.; Nicholson, T.A.; Philp, A.M.; Smith, H.L.; Davis, E.T.; Jones, S.W.; Lord, J.M. IL-6 secretion in osteoarthritis patients is mediated by chondrocyte-synovial fibroblast cross-talk and is enhanced by obesity. Sci. Rep. 2017, 7, 3451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanus, D.E.; Wijesinghe, S.N.; Pearson, M.J.; Hadjicharalambous, M.R.; Rosser, A.; Davis, E.T.; Lindsay, M.A.; Jones, S.W. Regulation of the Inflammatory Synovial Fibroblast Phenotype by Metastasis-Associated Lung Adenocarcinoma Transcript 1 Long Noncoding RNA in Obese Patients With Osteoarthritis. Arthritis Rheumatol. 2020, 72, 609–619. [Google Scholar] [CrossRef]
- Farah, H.; Young, S.P.; Mauro, C.; Jones, S.W. Metabolic dysfunction and inflammatory disease: The role of stromal fibroblasts. FEBS J. 2021, 288, 5555–5568. [Google Scholar] [CrossRef]
- Yin, X.N.; Wang, J.; Cui, L.F.; Fan, W.X. Enhanced glycolysis in the process of renal fibrosis aggravated the development of chronic kidney disease. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4243–4251. [Google Scholar] [CrossRef]
- Xie, N.; Tan, Z.; Banerjee, S.; Cui, H.; Ge, J.; Liu, R.M.; Bernard, K.; Thannickal, V.J.; Liu, G. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am. J. Respir Crit. Care Med. 2015, 192, 1462–1474. [Google Scholar] [CrossRef] [Green Version]
- Volchenkov, R.; Dung Cao, M.; Elgstoen, K.B.; Goll, G.L.; Eikvar, K.; Bjorneboe, O.; Bathen, T.F.; Holen, H.L.; Kvien, T.K.; Skalhegg, B.S. Metabolic profiling of synovial tissue shows altered glucose and choline metabolism in rheumatoid arthritis samples. Scand. J. Rheumatol. 2017, 46, 160–161. [Google Scholar] [CrossRef]
- Garcia-Carbonell, R.; Divakaruni, A.S.; Lodi, A.; Vicente-Suarez, I.; Saha, A.; Cheroutre, H.; Boss, G.R.; Tiziani, S.; Murphy, A.N.; Guma, M. Critical Role of Glucose Metabolism in Rheumatoid Arthritis Fibroblast-like Synoviocytes. Arthritis Rheumatol. 2016, 68, 1614–1626. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Saegusa, J.; Sendo, S.; Okano, T.; Akashi, K.; Irino, Y.; Morinobu, A. Glutaminase 1 plays a key role in the cell growth of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 76. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.K.; Kim, S.; Hwang, J.; Kim, J.; Kim, K.H.; Cha, H.S. GC/TOF-MS-based metabolomic profiling in cultured fibroblast-like synoviocytes from rheumatoid arthritis. Jt. Bone Spine 2016, 83, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Coggon, D.; Reading, I.; Croft, P.; McLaren, M.; Barrett, D.; Cooper, C. Knee osteoarthritis and obesity. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 622–627. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Tian, W.; Wang, Y.; Rong, J.; Bao, C.; Liu, Y.; Zhao, Y.; Wang, C. Body mass index and susceptibility to knee osteoarthritis: A systematic review and meta-analysis. Jt. Bone Spine 2012, 79, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Rong, J.; Wang, Y.; Hu, F.; Bao, C.; Li, X.; Zhao, Y. The relationship between body mass index and hip osteoarthritis: A systematic review and meta-analysis. Jt. Bone Spine 2011, 78, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Oliveria, S.A.; Felson, D.T.; Cirillo, P.A.; Reed, J.I.; Walker, A.M. Body weight, body mass index, and incident symptomatic osteoarthritis of the hand, hip, and knee. Epidemiology 1999, 10, 161–166. [Google Scholar] [CrossRef]
- Philp, A.M.; Butterworth, S.; Davis, E.T.; Jones, S.W. eNAMPT Is Localised to Areas of Cartilage Damage in Patients with Hip Osteoarthritis and Promotes Cartilage Catabolism and Inflammation. Int. Mol. Sci 2021, 22, 6719. [Google Scholar] [CrossRef]
- Philp, A.M.; Collier, R.L.; Grover, L.M.; Davis, E.T.; Jones, S.W. Resistin promotes the abnormal Type I collagen phenotype of subchondral bone in obese patients with end stage hip osteoarthritis. Sci. Rep. 2017, 7, 4042. [Google Scholar] [CrossRef] [Green Version]
- Certo, M.; Marone, G.; de Paulis, A.; Mauro, C.; Pucino, V. Lactate: Fueling the fire starter. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1474. [Google Scholar] [CrossRef] [Green Version]
- Khatib-Massalha, E.; Bhattacharya, S.; Massalha, H.; Biram, A.; Golan, K.; Kollet, O.; Kumari, A.; Avemaria, F.; Petrovich-Kopitman, E.; Gur-Cohen, S.; et al. Lactate released by inflammatory bone marrow neutrophils induces their mobilization via endothelial GPR81 signaling. Nat. Commun. 2020, 11, 3547. [Google Scholar] [CrossRef]
- Pucino, V.; Certo, M.; Bulusu, V.; Cucchi, D.; Goldmann, K.; Pontarini, E.; Haas, R.; Smith, J.; Headland, S.E.; Blighe, K.; et al. Lactate Buildup at the Site of Chronic Inflammation Promotes Disease by Inducing CD4(+) T Cell Metabolic Rewiring. Cell Metab. 2019, 30, 1055–1074.e1058. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.R.; Chokesuwattanaskul, S.; Phelan, M.M.; Welting, T.J.M.; Lian, L.Y.; Peffers, M.J.; Wright, H.L. 1H NMR Metabolomics Identifies Underlying Inflammatory Pathology in Osteoarthritis and Rheumatoid Arthritis Synovial Joints. J. Proteome Res. 2018, 17, 3780–3790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic Reprogramming of Cancer Associated Fibroblasts: The Slavery of Stromal Fibroblasts. Biomed. Res. Int. 2018, 2018, 6075403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, J.; Cui, H.; Xie, N.; Banerjee, S.; Guo, S.; Dubey, S.; Barnes, S.; Liu, G. Glutaminolysis Promotes Collagen Translation and Stability via alpha-Ketoglutarate-mediated mTOR Activation and Proline Hydroxylation. Am. J. Respir. Cell Mol. Biol. 2018, 58, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.W.; Motz, K.M.; Ding, D.; Lina, I.; Murphy, M.K.; Benner, D.; Feeley, M.; Hooper, J.; Hillel, A.T. Inhibition of glutaminase to reverse fibrosis in iatrogenic laryngotracheal stenosis. Laryngoscope 2020, 130, E773–E781. [Google Scholar] [CrossRef]
- Zhang, L.; Xing, R.; Huang, Z.; Ding, L.; Zhang, L.; Li, M.; Li, X.; Wang, P.; Mao, J. Synovial Fibrosis Involvement in Osteoarthritis. Front. Med. 2021, 8, 684389. [Google Scholar] [CrossRef]
- Remst, D.F.; Blaney Davidson, E.N.; van der Kraan, P.M. Unravelling osteoarthritis-related synovial fibrosis: A step closer to solving joint stiffness. Rheumatology 2015, 54, 1954–1963. [Google Scholar] [CrossRef] [Green Version]
- Remst, D.F.; Blaney Davidson, E.N.; Vitters, E.L.; Blom, A.B.; Stoop, R.; Snabel, J.M.; Bank, R.A.; van den Berg, W.B.; van der Kraan, P.M. Osteoarthritis-related fibrosis is associated with both elevated pyridinoline cross-link formation and lysyl hydroxylase 2b expression. Osteoarthr. Cartil. 2013, 21, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Elhaj Mahmoud, D.; Kaabachi, W.; Sassi, N.; Mokhtar, A.; Ben Ammar, L.; Rekik, S.; Tarhouni, L.; Kallel-Sellami, M.; Cheour, E.; Laadhar, L. Expression of extracellular matrix components and cytokine receptors in human fibrocytes during rheumatoid arthritis. Connect. Tissue Res. 2021, 62, 720–731. [Google Scholar] [CrossRef]
- Baker, K.; Grainger, A.; Niu, J.; Clancy, M.; Guermazi, A.; Crema, M.; Hughes, L.; Buckwalter, J.; Wooley, A.; Nevitt, M.; et al. Relation of synovitis to knee pain using contrast-enhanced MRIs. Annu. Rheum. Dis. 2010, 69, 1779–1783. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, H.; Richter, W. Osteoarthritis: Cellular and molecular changes in degenerating cartilage. Prog. Histochem. Cytochem. 2006, 40, 135–163. [Google Scholar] [CrossRef]
- Glasson, S.S.; Askew, R.; Sheppard, B.; Carito, B.; Blanchet, T.; Ma, H.L.; Flannery, C.R.; Peluso, D.; Kanki, K.; Yang, Z.; et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 2005, 434, 644–648. [Google Scholar] [CrossRef]
- Croft, A.P.; Campos, J.; Jansen, K.; Turner, J.D.; Marshall, J.; Attar, M.; Savary, L.; Wehmeyer, C.; Naylor, A.J.; Kemble, S.; et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 2019, 570, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Chen, C. Body mass index and risk of knee osteoarthritis: Systematic review and meta-analysis of prospective studies. BMJ Open 2015, 5, e007568. [Google Scholar] [CrossRef] [PubMed]
- Christensen, R.; Henriksen, M.; Leeds, A.R.; Gudbergsen, H.; Christensen, P.; Sorensen, T.J.; Bartels, E.M.; Riecke, B.F.; Aaboe, J.; Frederiksen, R.; et al. Effect of weight maintenance on symptoms of knee osteoarthritis in obese patients: A twelve-month randomized controlled trial. Arthritis Care Res. 2015, 67, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Gersing, A.S.; Schwaiger, B.J.; Nevitt, M.C.; Joseph, G.B.; Chanchek, N.; Guimaraes, J.B.; Mbapte Wamba, J.; Facchetti, L.; McCulloch, C.E.; Link, T.M. Is Weight Loss Associated with Less Progression of Changes in Knee Articular Cartilage among Obese and Overweight Patients as Assessed with MR Imaging over 48 Months? Data from the Osteoarthritis Initiative. Radiology 2017, 284, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, C.; Gunther, U.L. MetaboLab—Advanced NMR data processing and analysis for metabolomics. BMC Bioinform. 2011, 12, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Obese | Normal-Weight | p-Value | ||
|---|---|---|---|---|
| Age (years) | 63 ± 4 | 72 ± 2.0 | 0.05 | |
| Female:Male (n) | 5:3 | 5:3 | ||
| BMI (kg/m2) | 33.6 ± 1.2 | 22.9 ± 0.7 | <0.0001 | |
| BMI (min, max) | 30.1, 38.6 | 20.0, 23.7 | ||
| Waist circumference (cm) | 106.5 ± 5.1 | 86.6 ± 3.2 | <0.01 | |
| Hip circumference, (cm) | 116.4 ± 3.6 | 94.4 ± 2.1 | <0.001 | |
| Waist:Hip ratio | 0.91 ± 0.03 | 0.92 ± 0.03 | 0.93 | |
| KL Grade | Median (IQR) | 4 (3–4) | 3.5 (3–4) | |
| KL3 (%) | 37.5 | 37.5 | ||
| KL4 (%) | 62.5 | 62.5 | ||
| Joint Space (mm) | 1.4 ± 0.7 | 1.8 ± 0.3 | 0.60 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farah, H.; Wijesinghe, S.N.; Nicholson, T.; Alnajjar, F.; Certo, M.; Alghamdi, A.; Davis, E.T.; Young, S.P.; Mauro, C.; Jones, S.W. Differential Metabotypes in Synovial Fibroblasts and Synovial Fluid in Hip Osteoarthritis Patients Support Inflammatory Responses. Int. J. Mol. Sci. 2022, 23, 3266. https://doi.org/10.3390/ijms23063266
Farah H, Wijesinghe SN, Nicholson T, Alnajjar F, Certo M, Alghamdi A, Davis ET, Young SP, Mauro C, Jones SW. Differential Metabotypes in Synovial Fibroblasts and Synovial Fluid in Hip Osteoarthritis Patients Support Inflammatory Responses. International Journal of Molecular Sciences. 2022; 23(6):3266. https://doi.org/10.3390/ijms23063266
Chicago/Turabian StyleFarah, Hussein, Susanne N. Wijesinghe, Thomas Nicholson, Fawzeyah Alnajjar, Michelangelo Certo, Abdullah Alghamdi, Edward T. Davis, Stephen P. Young, Claudio Mauro, and Simon W. Jones. 2022. "Differential Metabotypes in Synovial Fibroblasts and Synovial Fluid in Hip Osteoarthritis Patients Support Inflammatory Responses" International Journal of Molecular Sciences 23, no. 6: 3266. https://doi.org/10.3390/ijms23063266
APA StyleFarah, H., Wijesinghe, S. N., Nicholson, T., Alnajjar, F., Certo, M., Alghamdi, A., Davis, E. T., Young, S. P., Mauro, C., & Jones, S. W. (2022). Differential Metabotypes in Synovial Fibroblasts and Synovial Fluid in Hip Osteoarthritis Patients Support Inflammatory Responses. International Journal of Molecular Sciences, 23(6), 3266. https://doi.org/10.3390/ijms23063266