
Structural Modifications on Chalcone Framework for Developing New Class of Cholinesterase Inhibitors

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
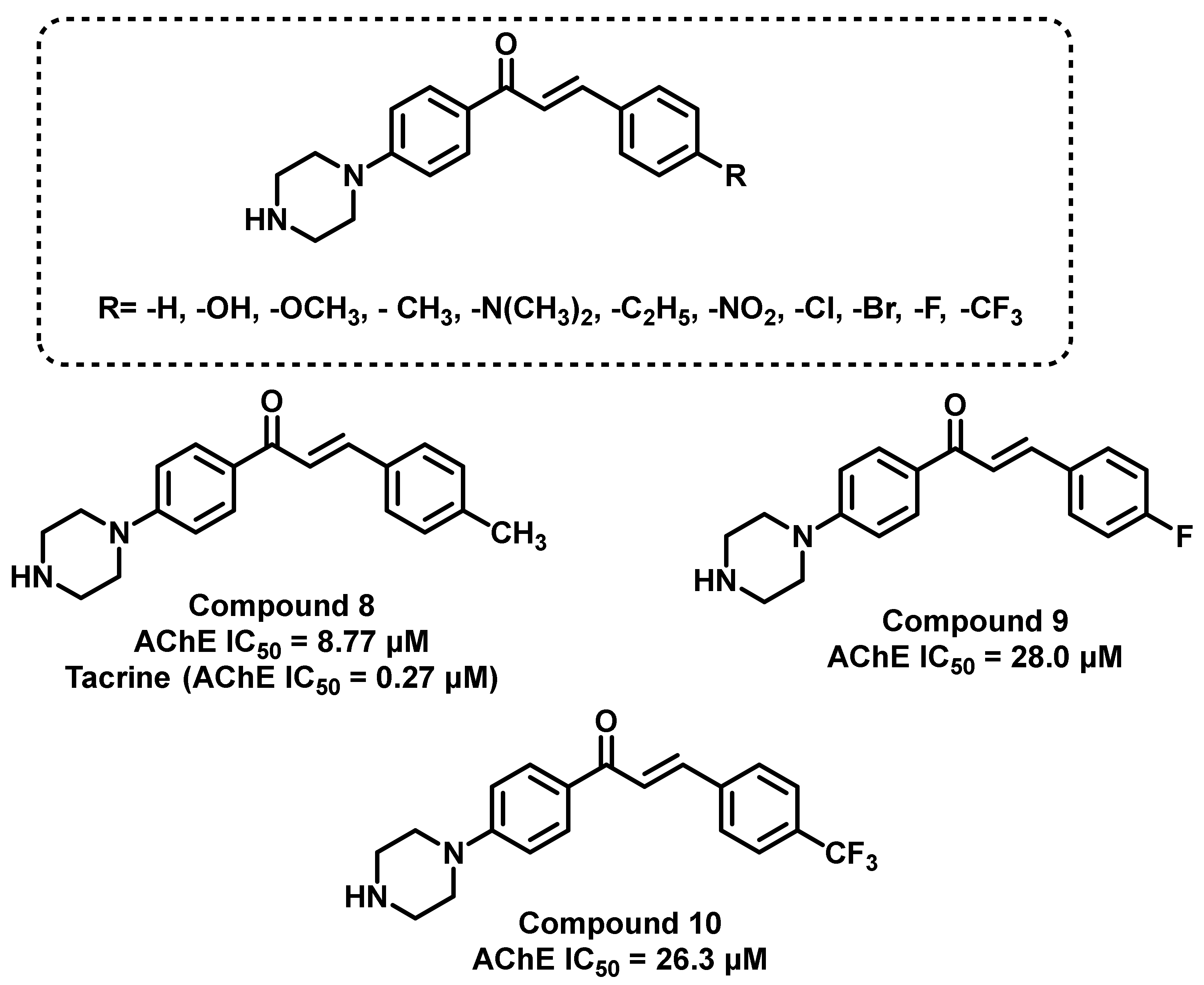{kind=link}
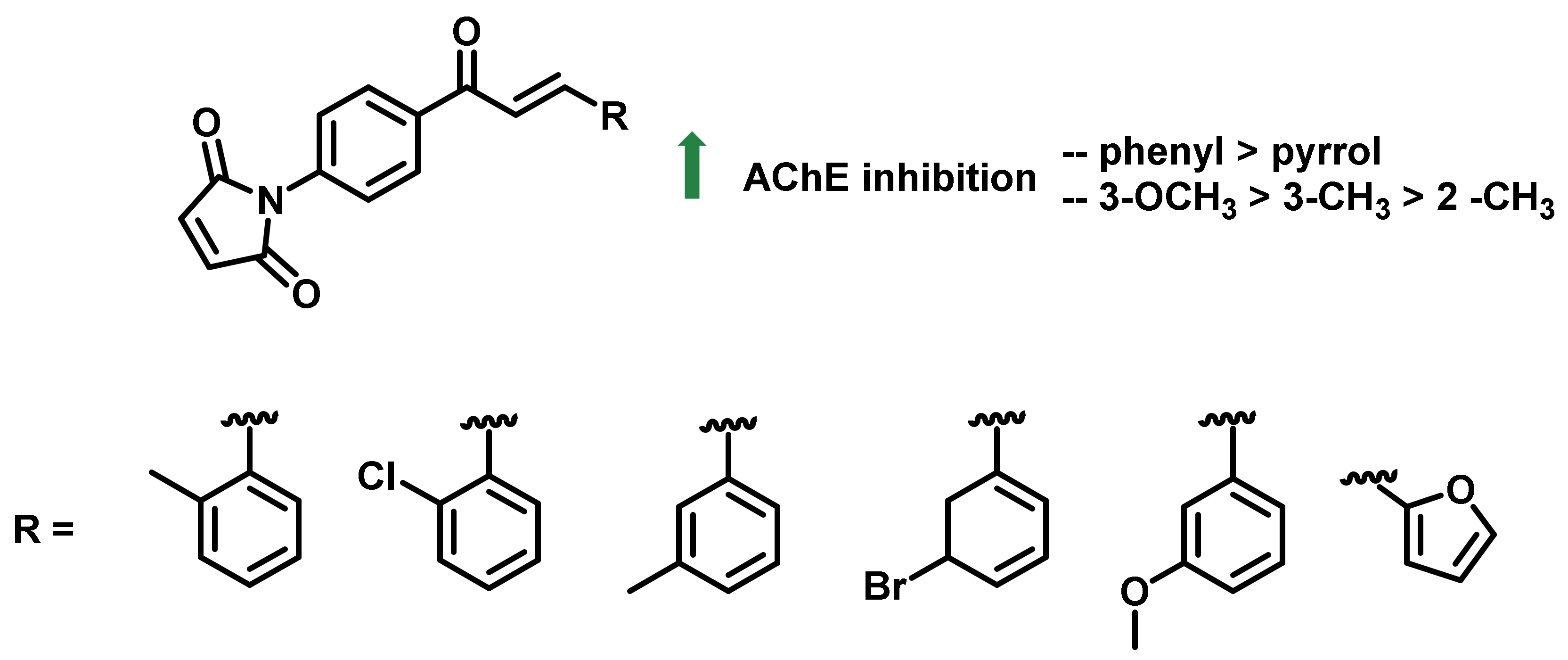{kind=link}
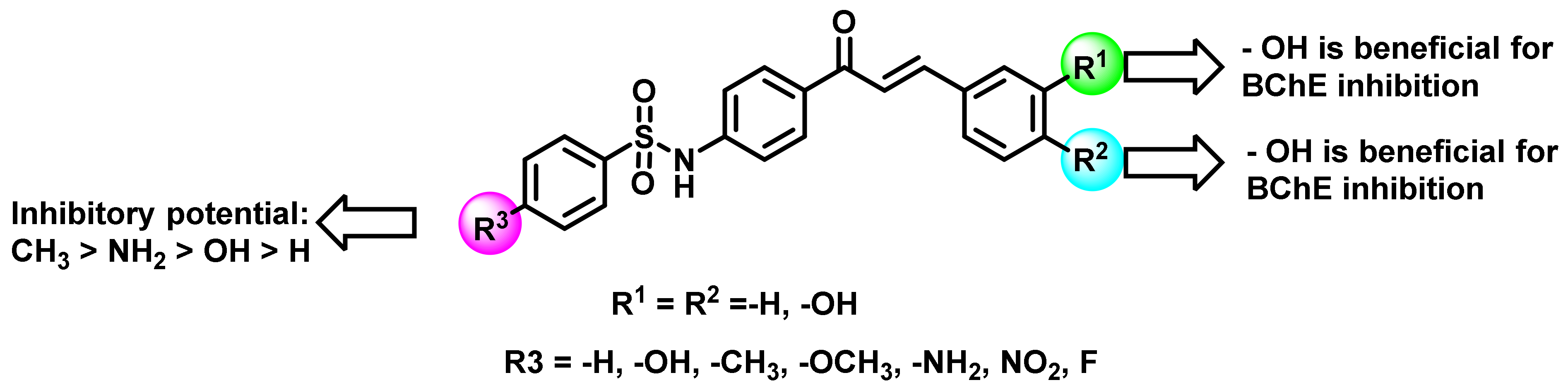{kind=link}
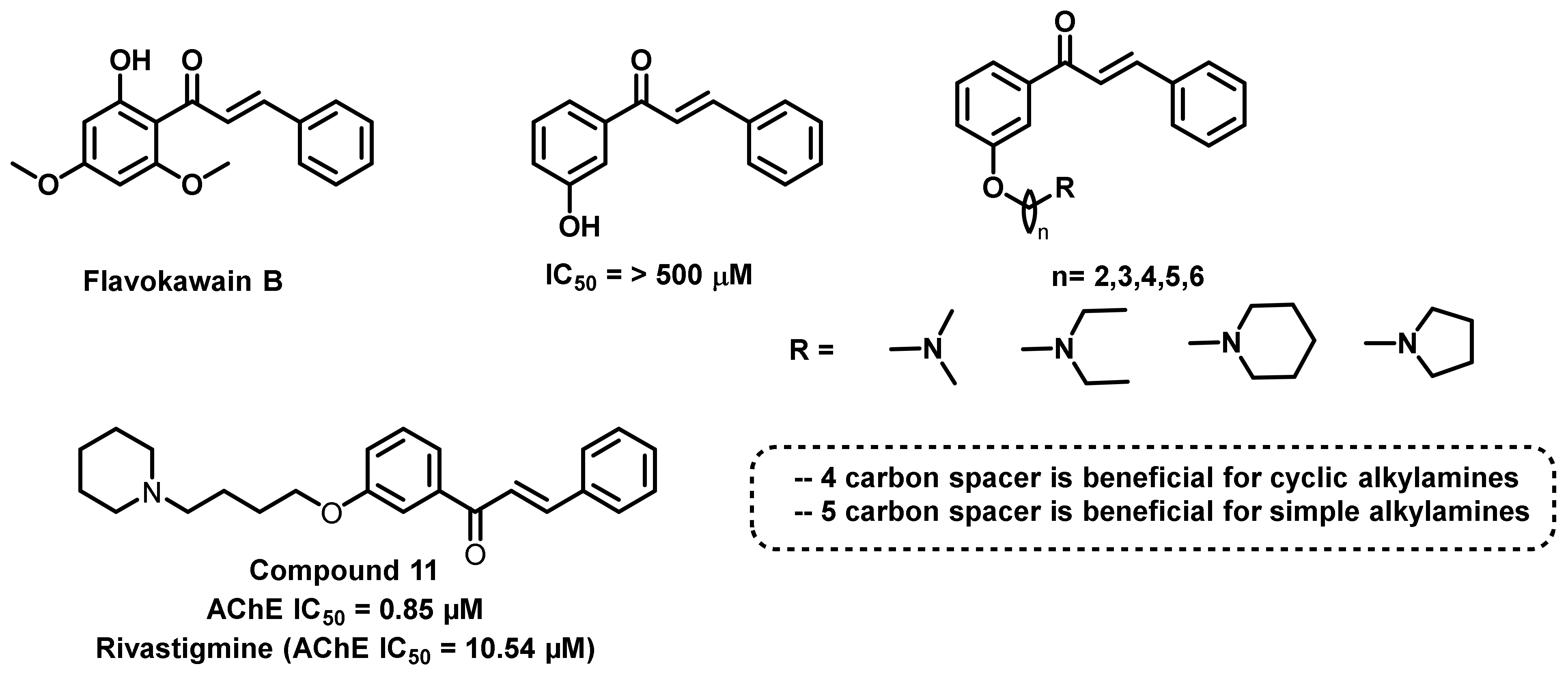{kind=link}
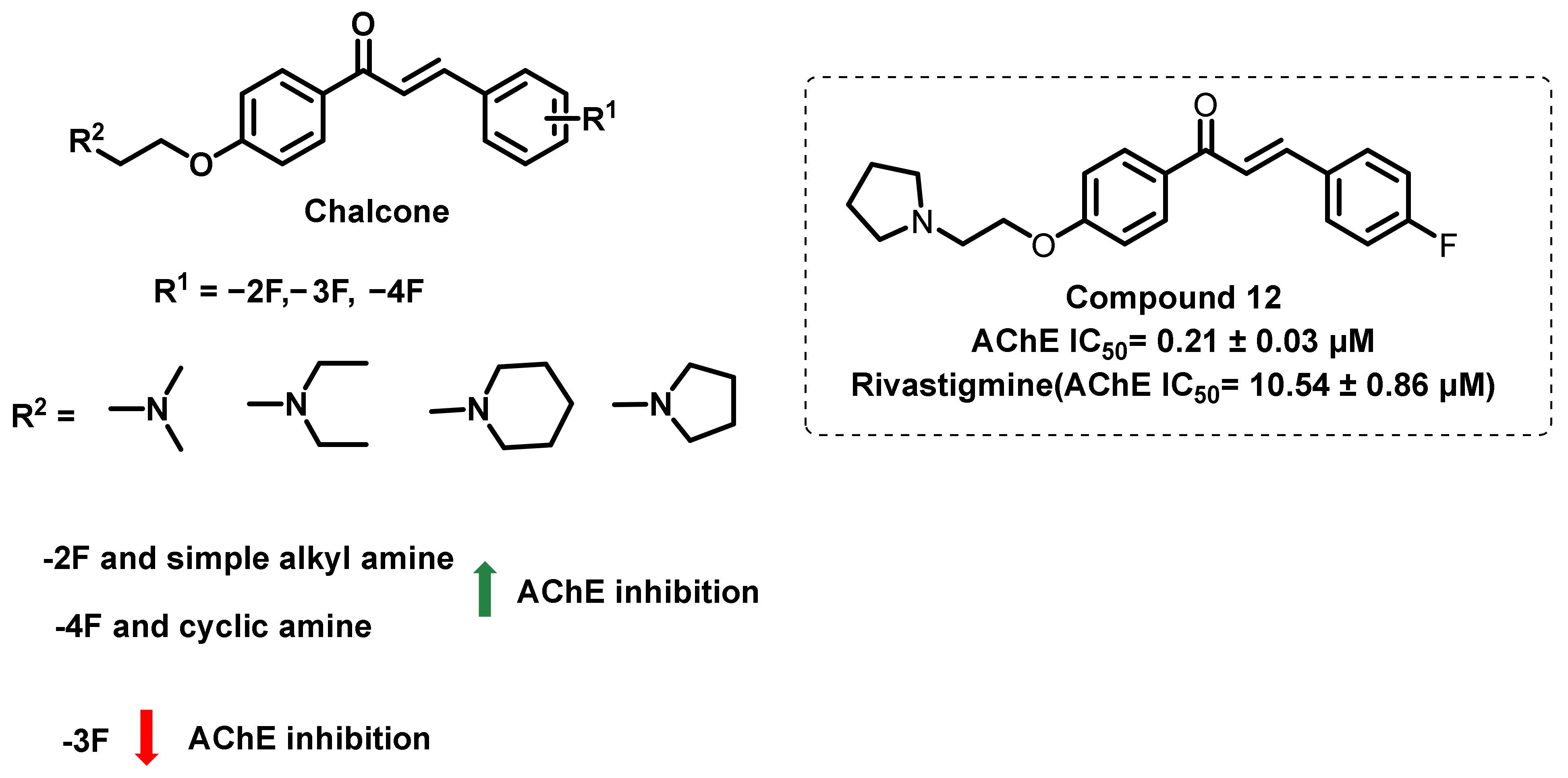{kind=link}
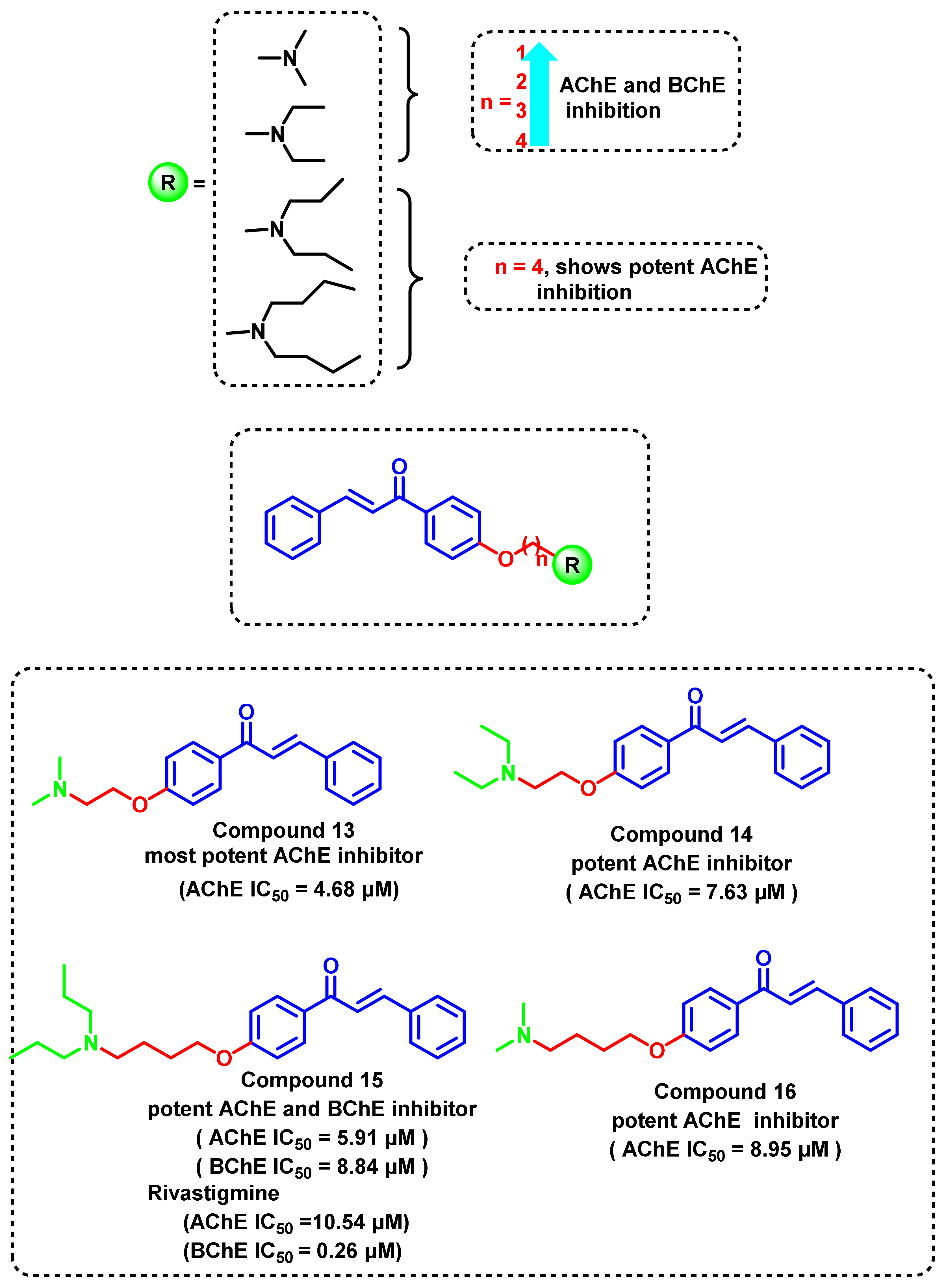{kind=link}
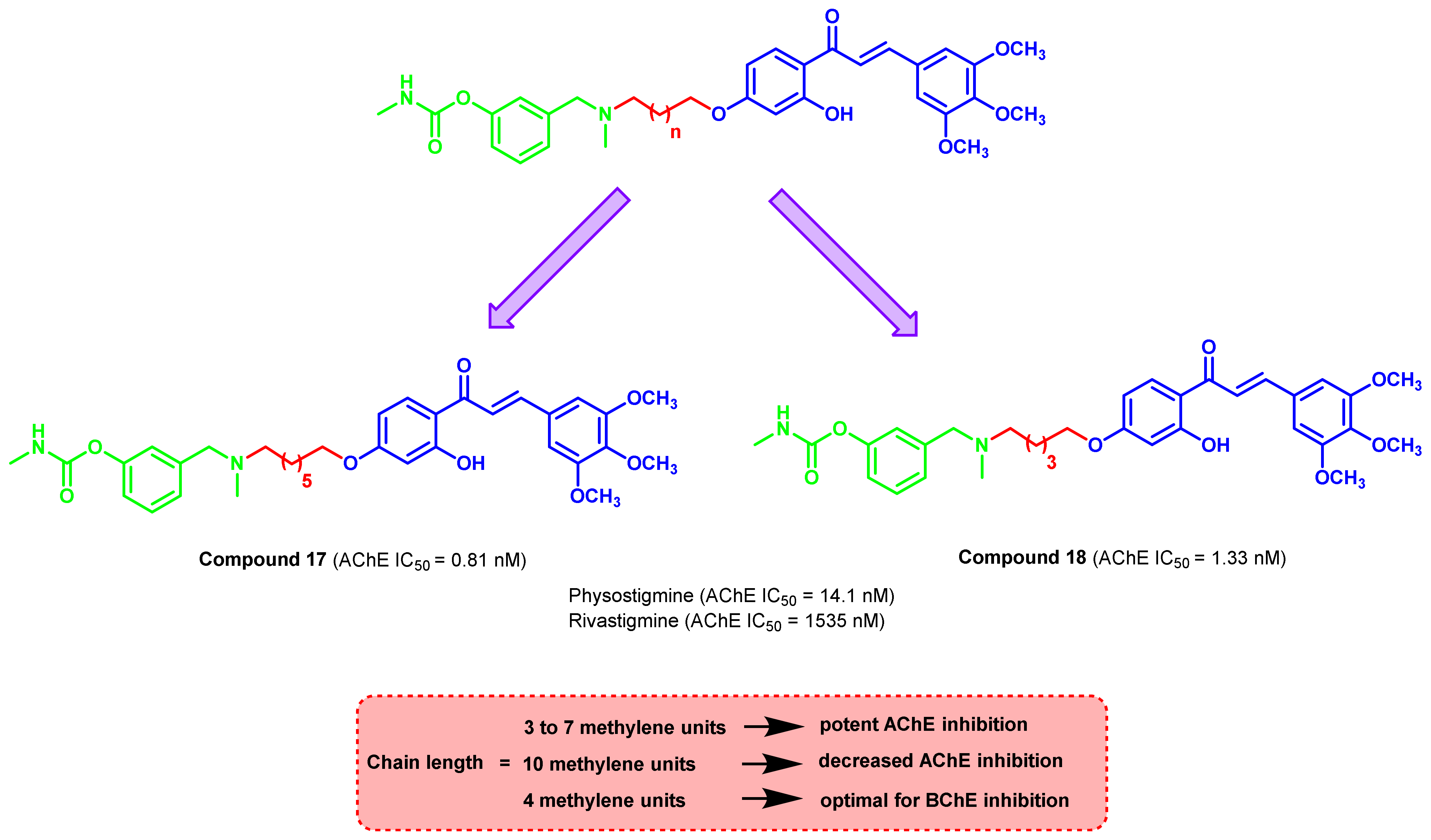{kind=link}
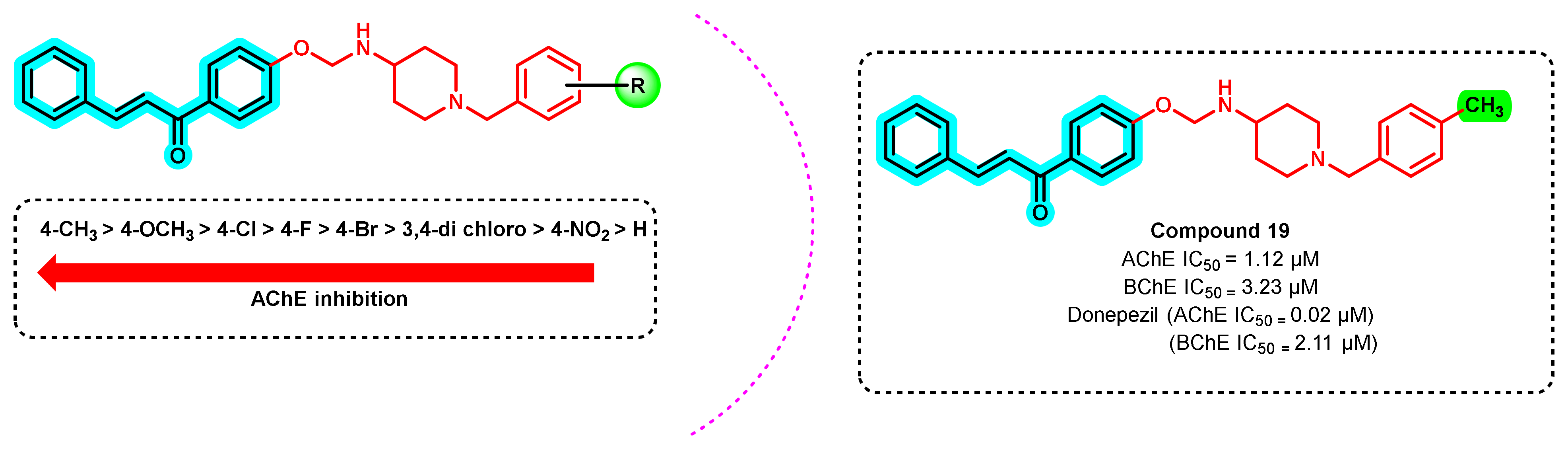{kind=link}
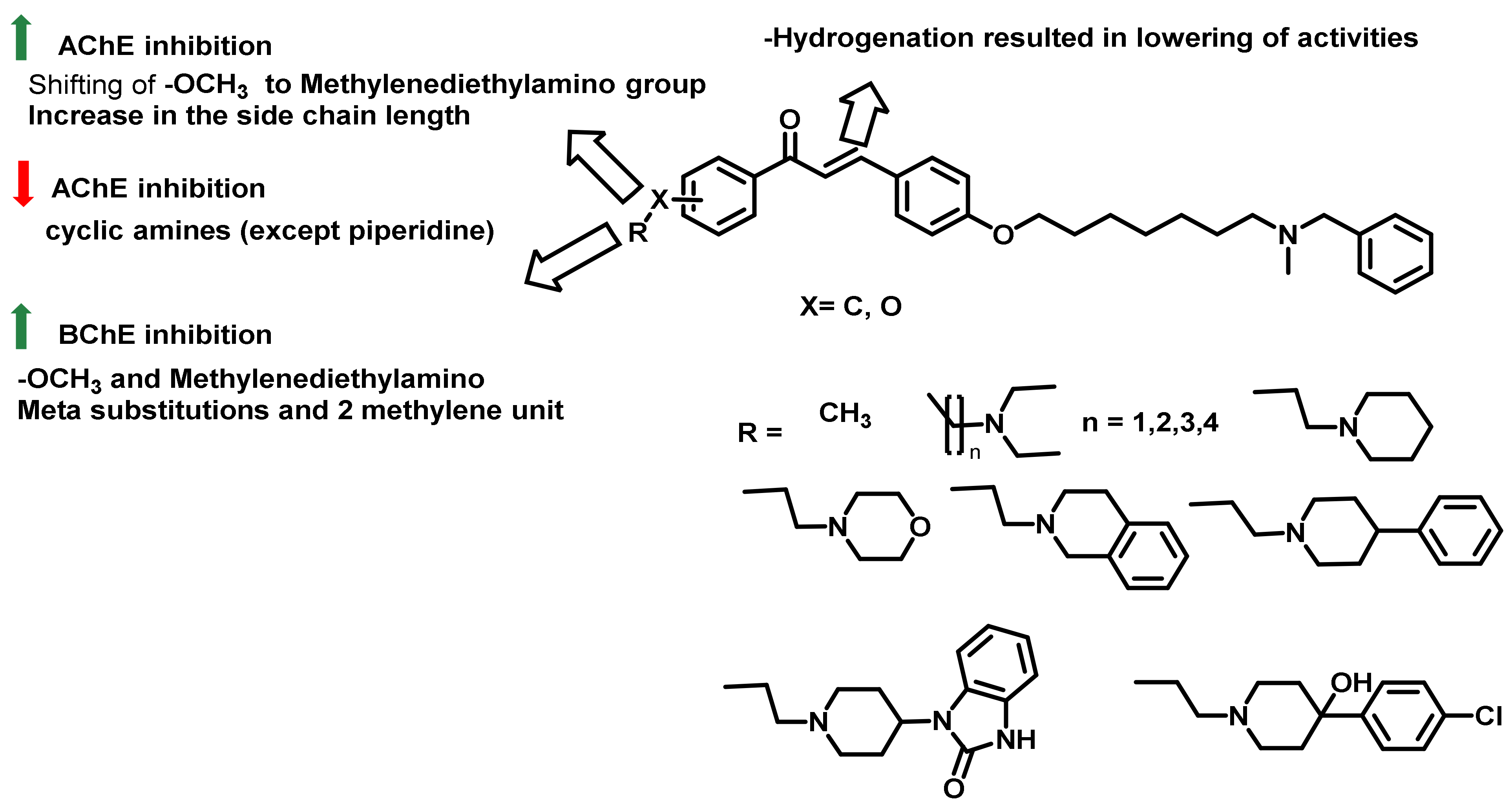{kind=link}
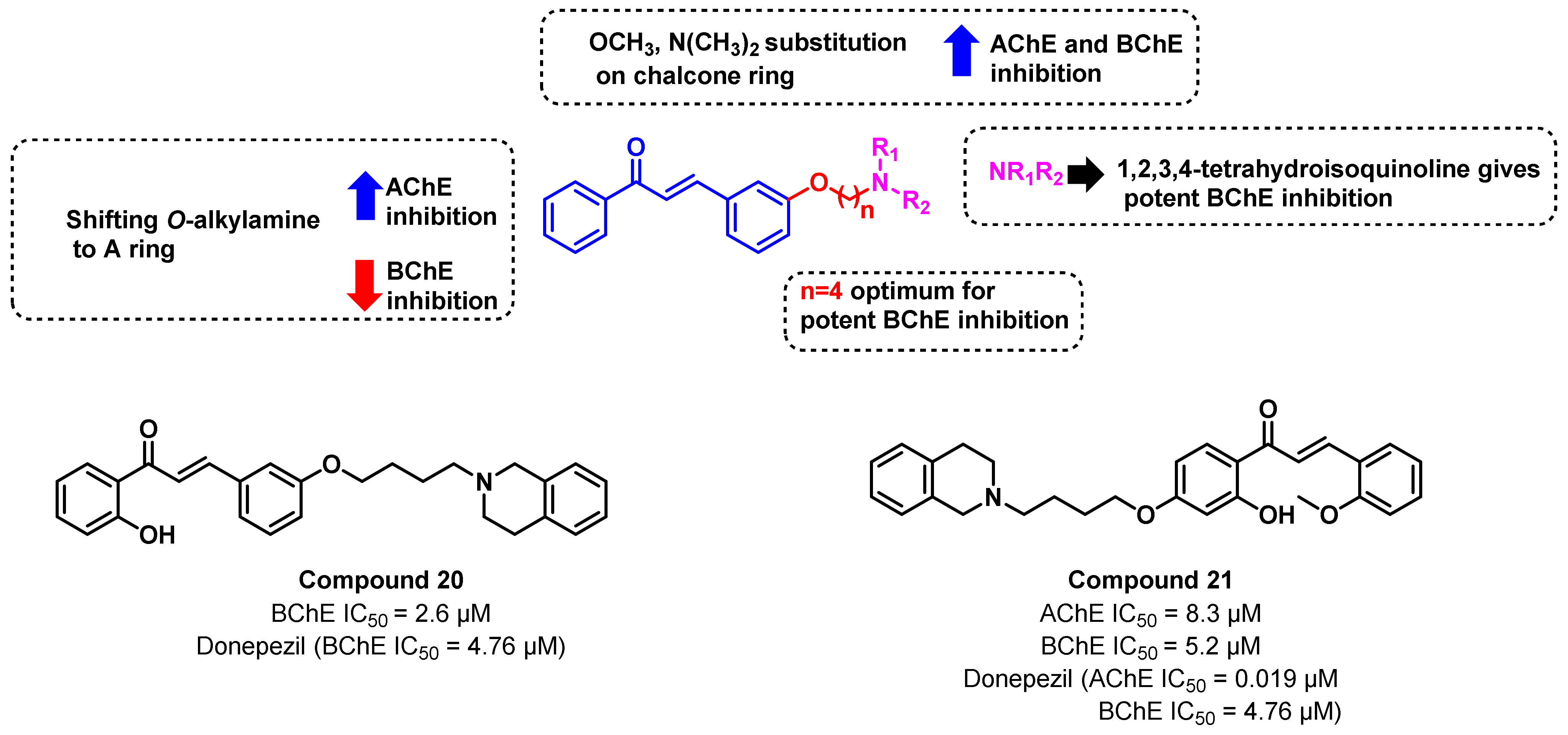{kind=link}
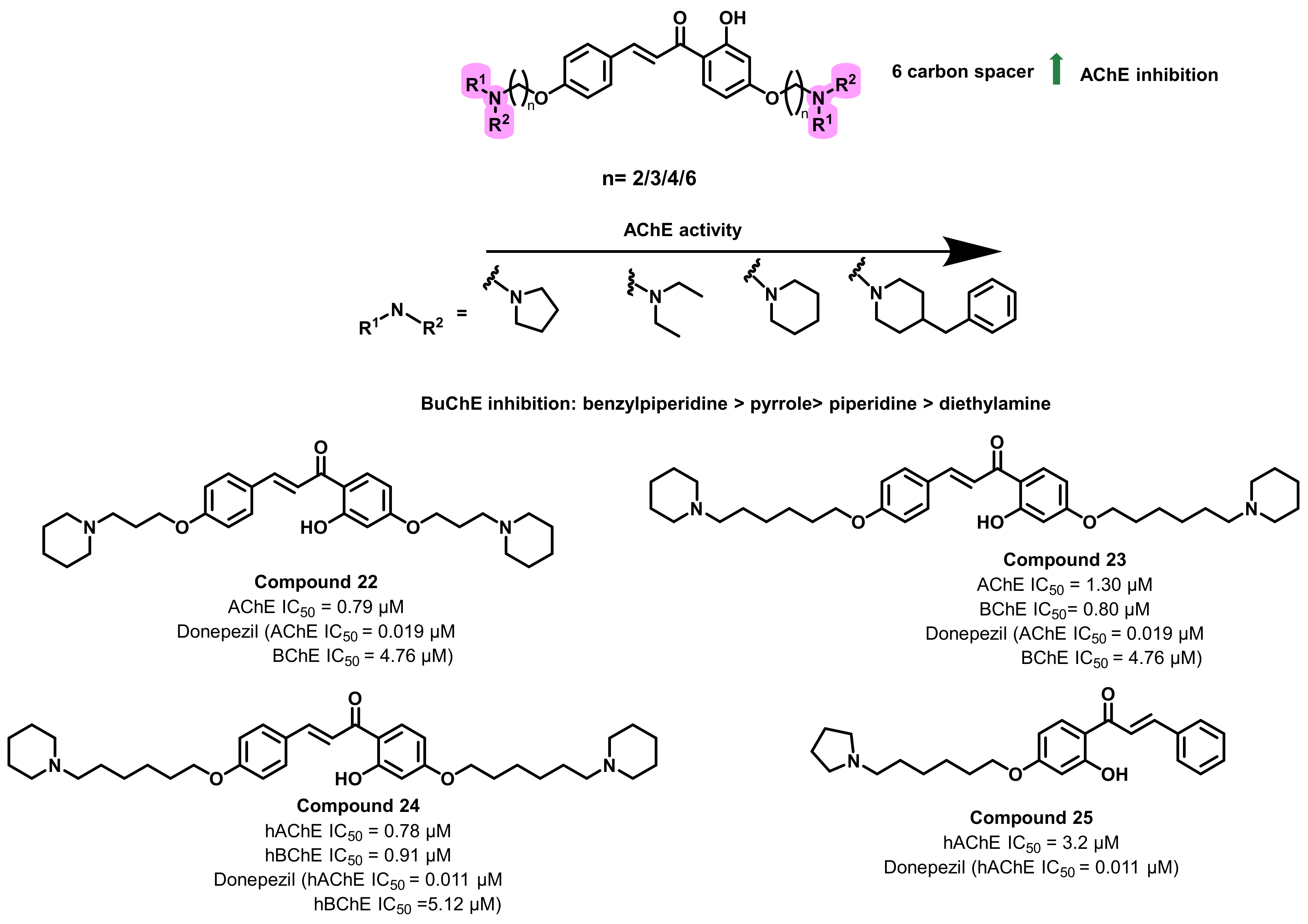{kind=link}
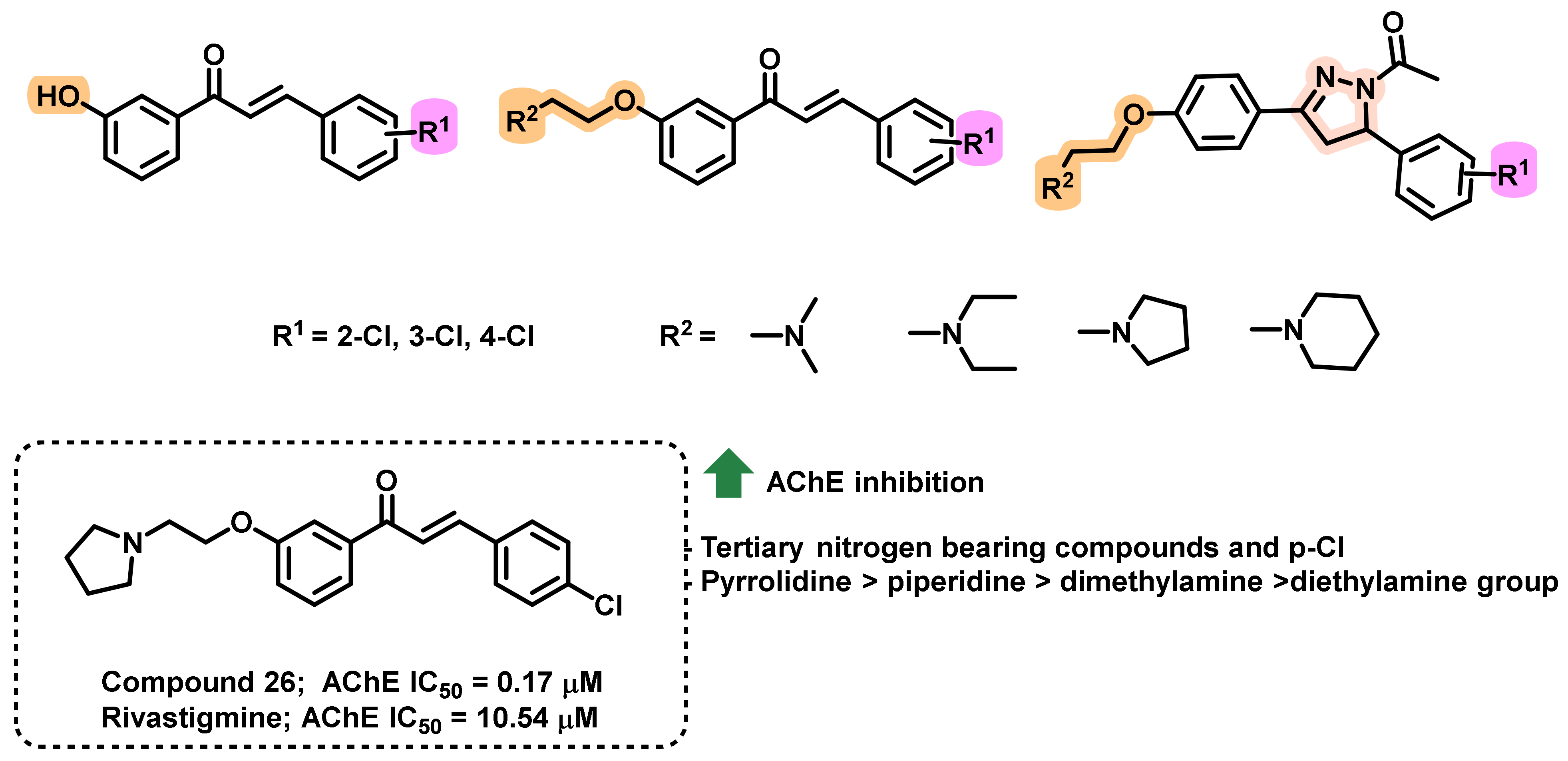{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
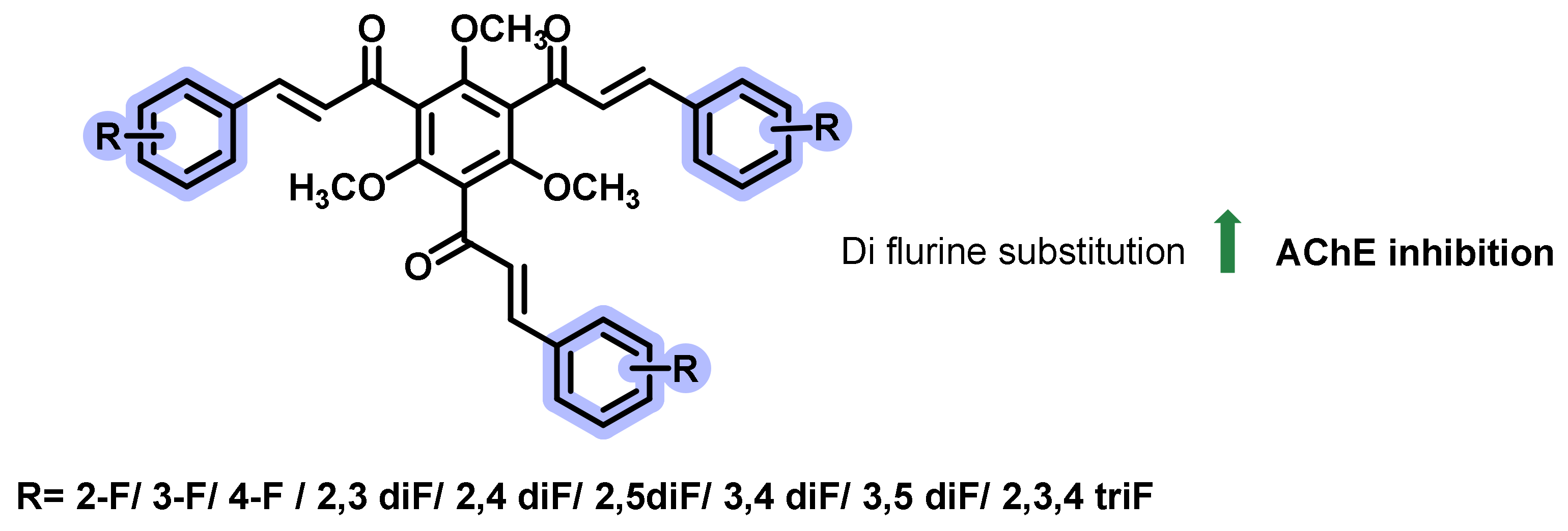{kind=link}
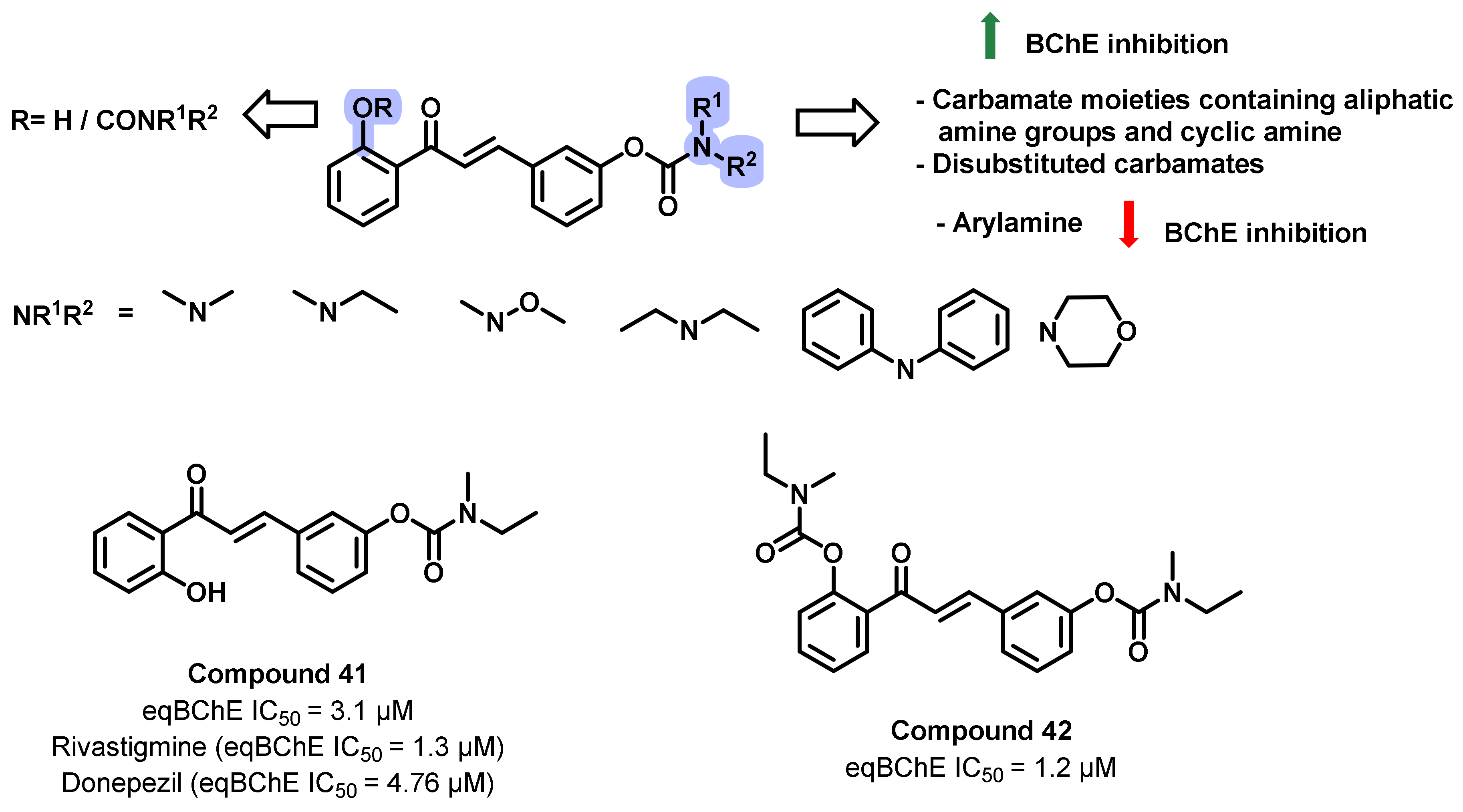{kind=link}
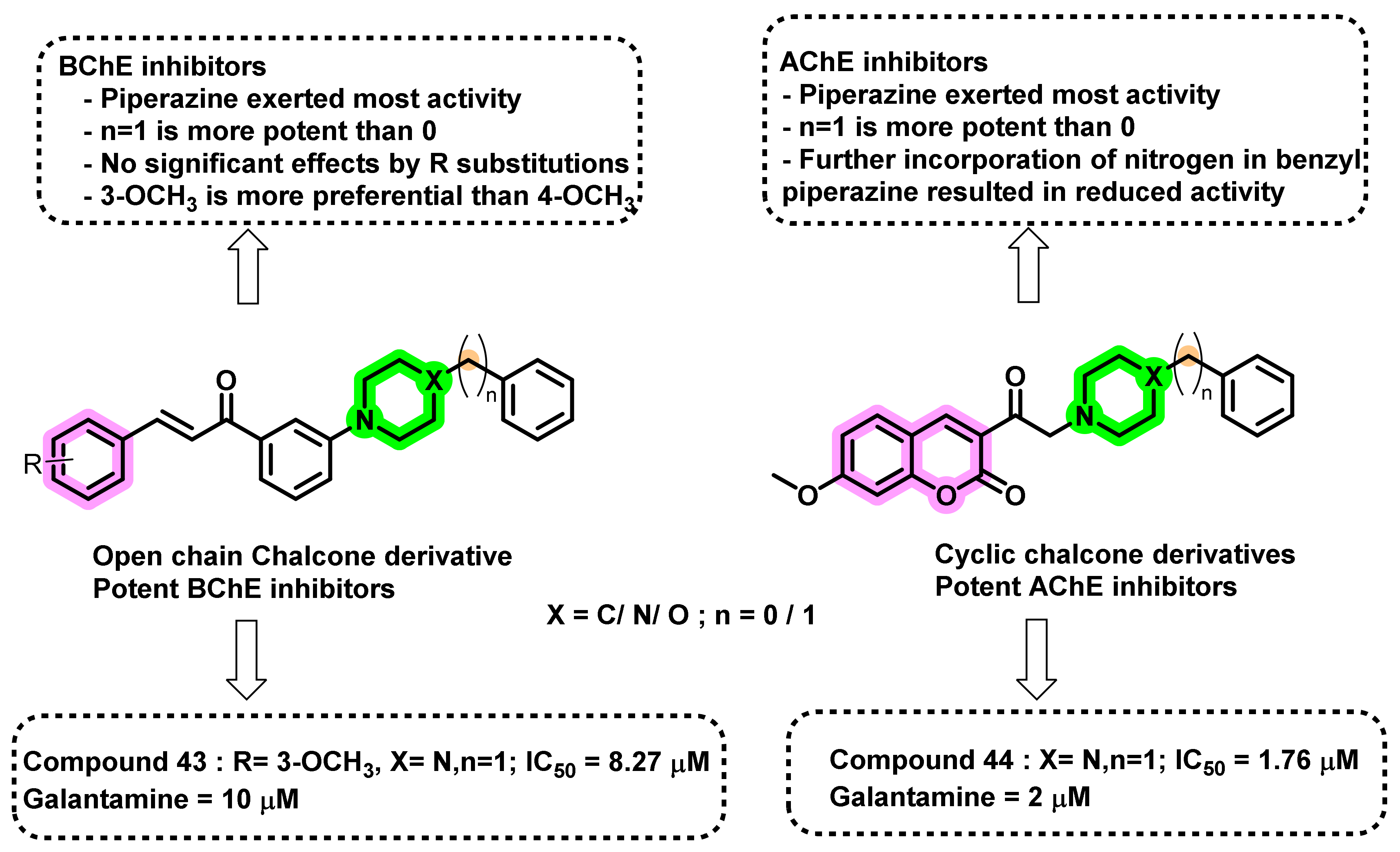{kind=link}
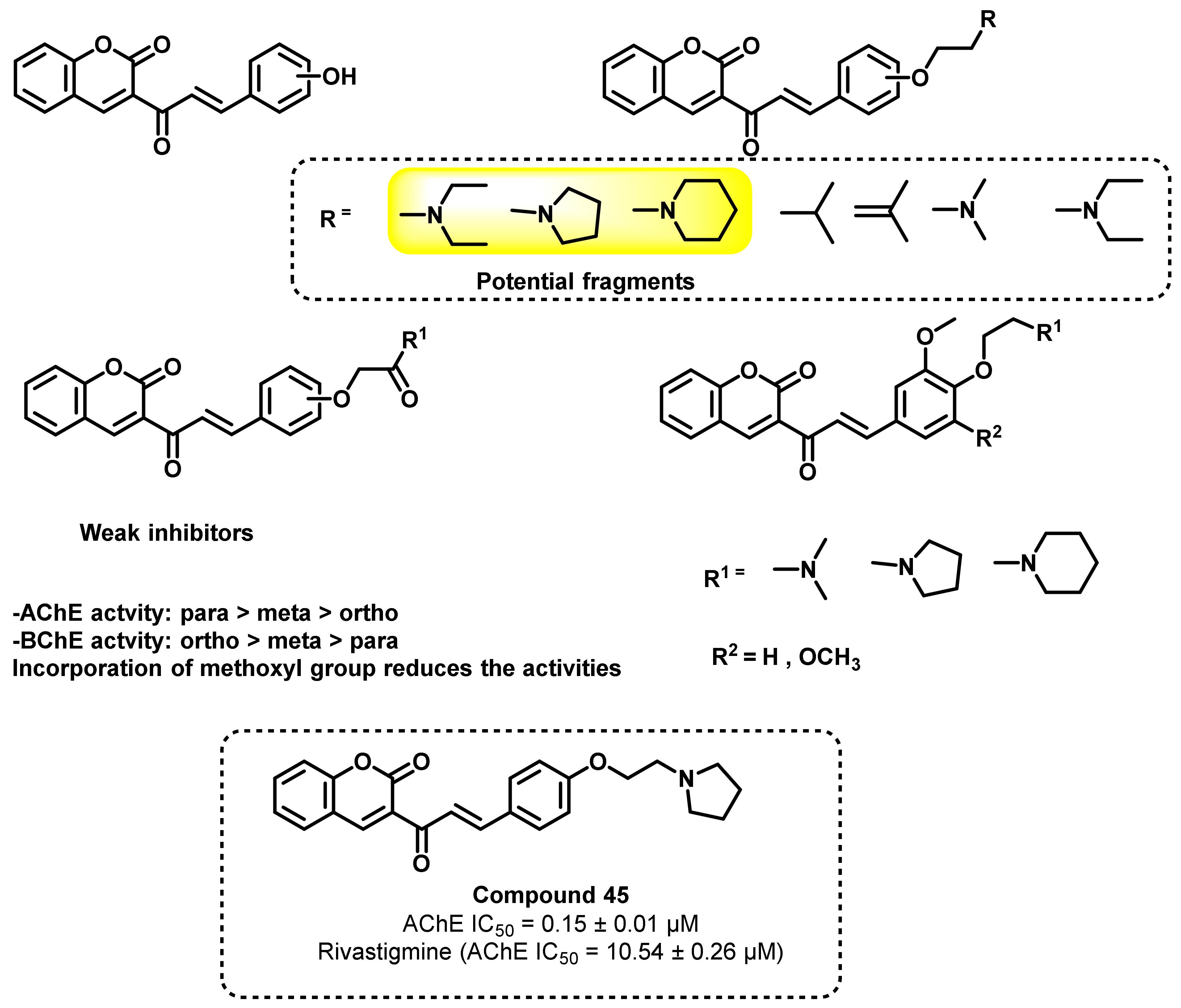{kind=link}
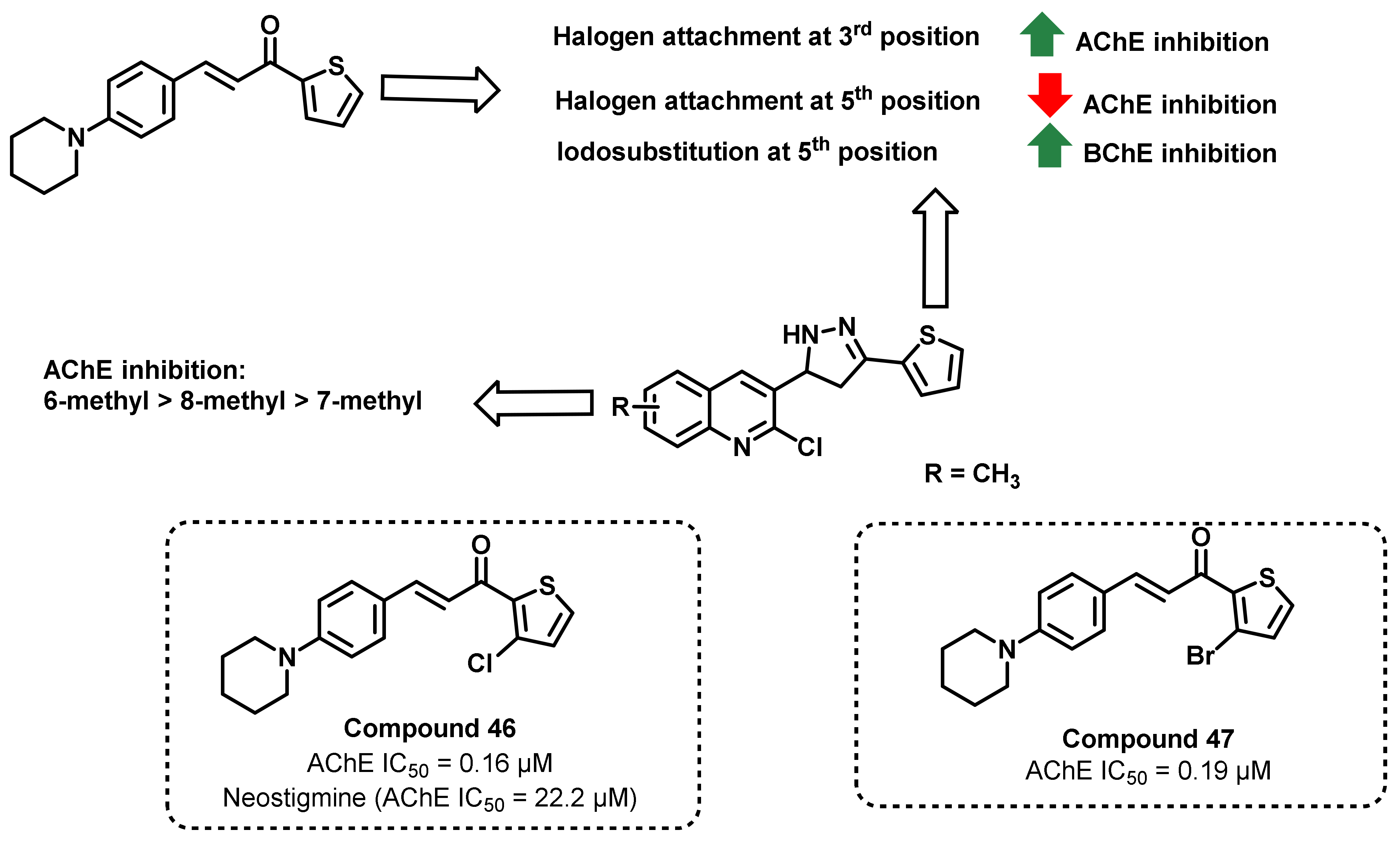{kind=link}
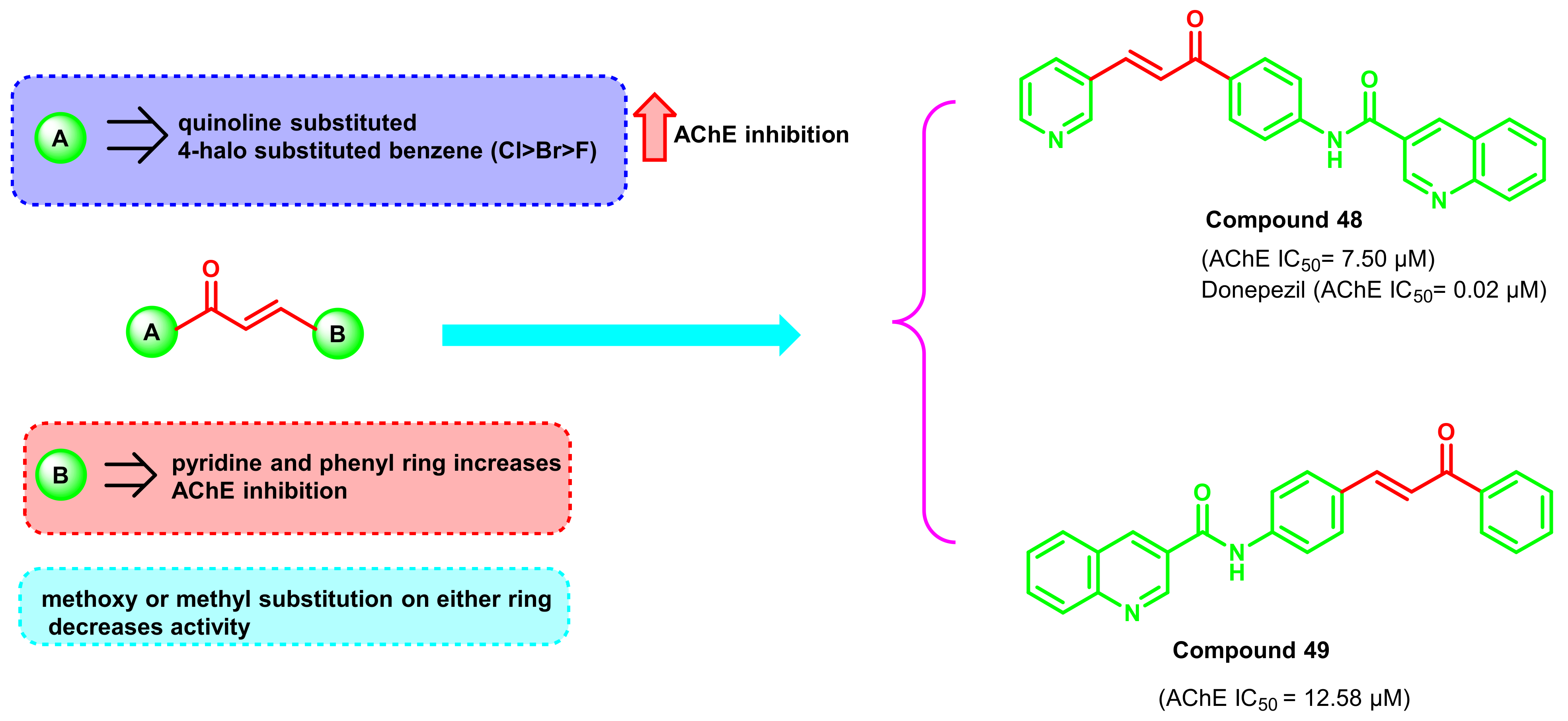{kind=link}
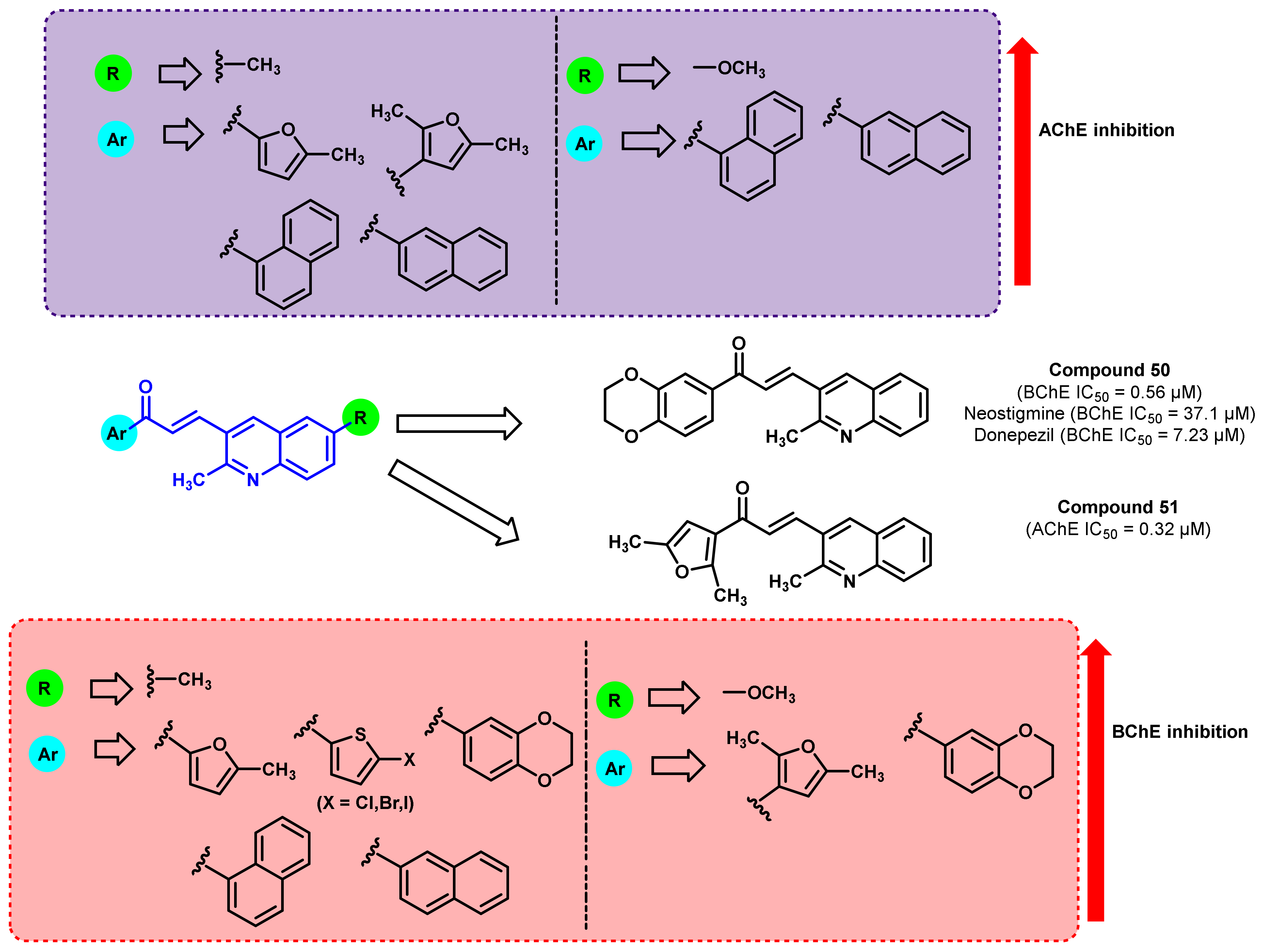{kind=link}
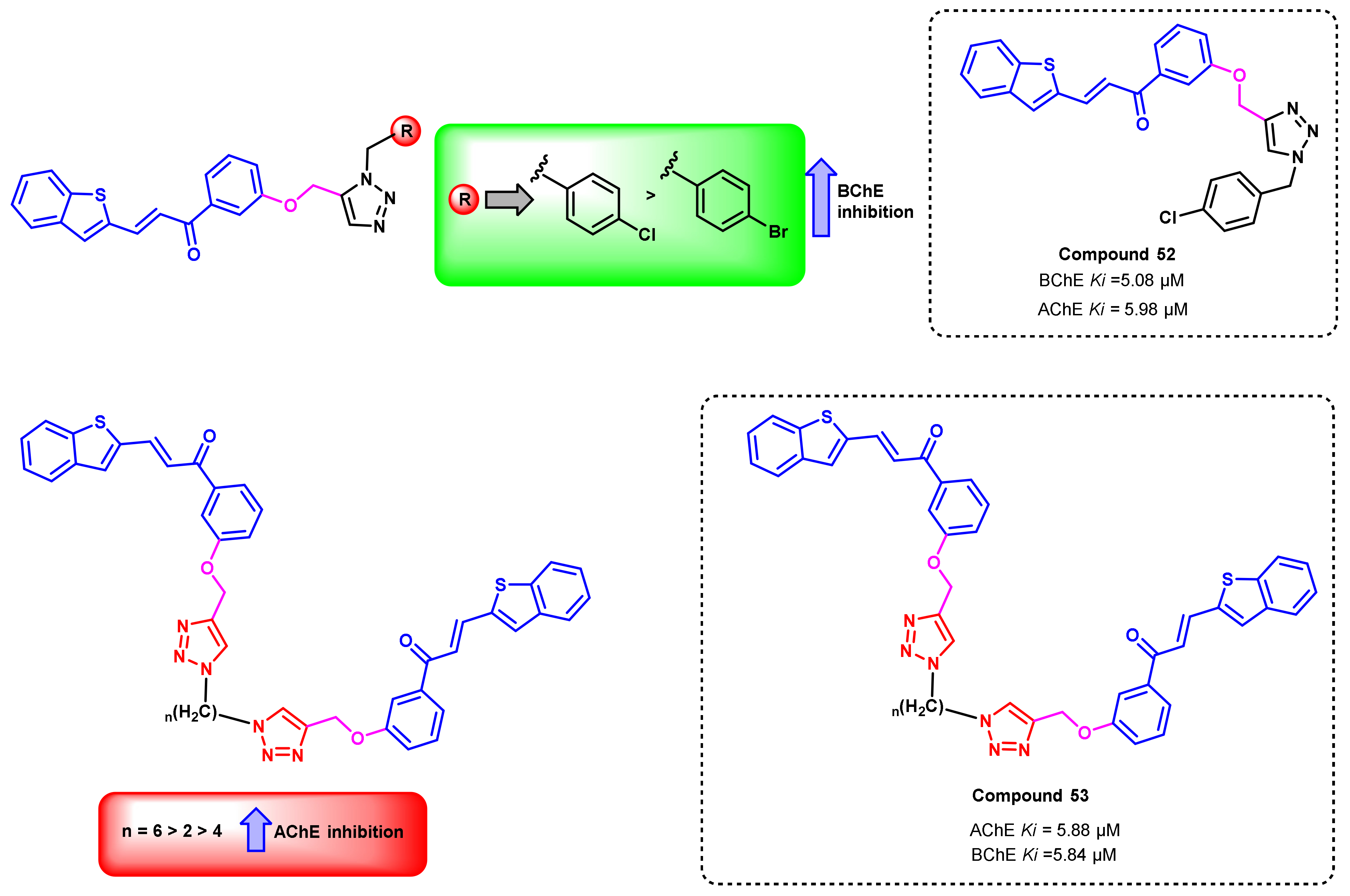{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
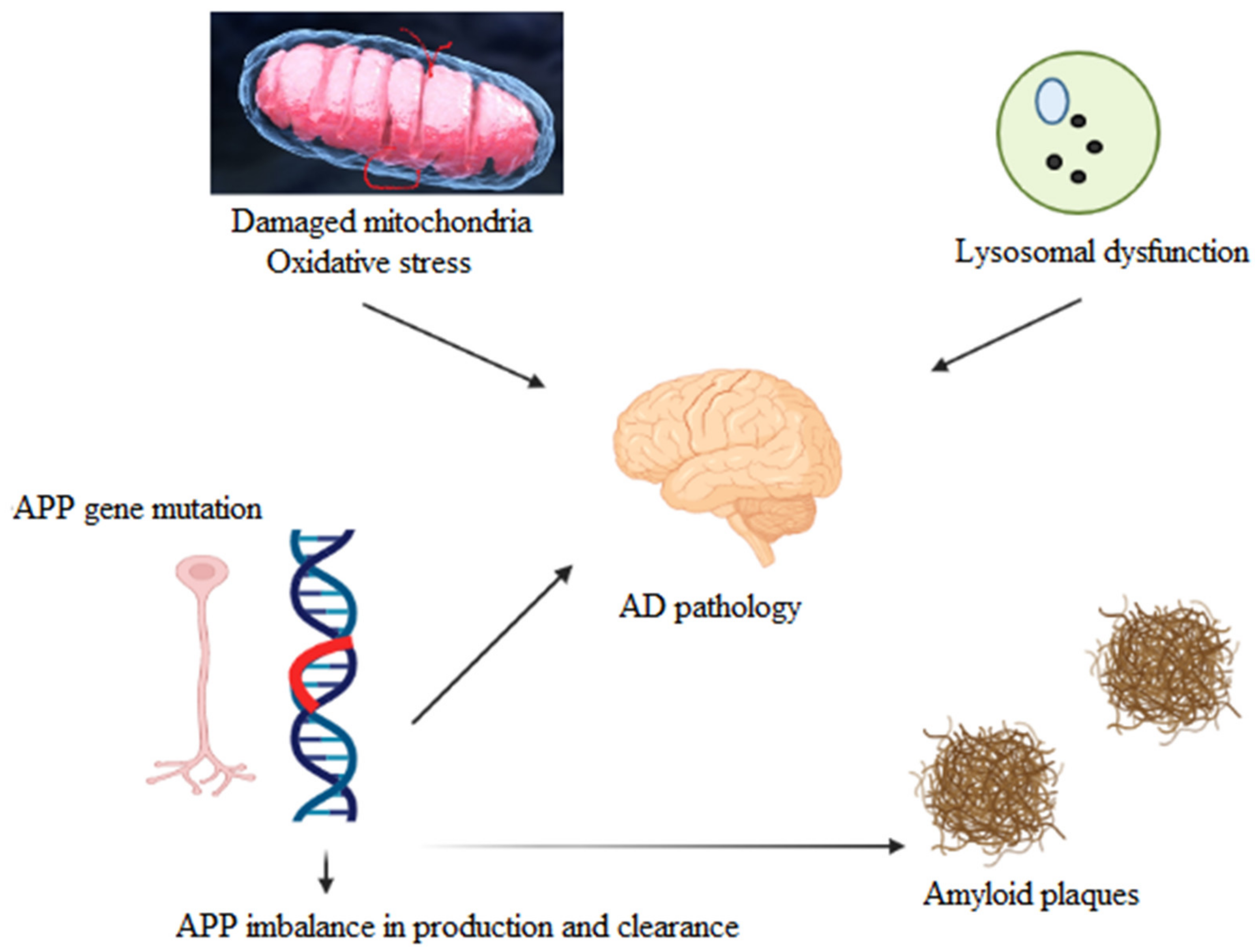
1. Introduction
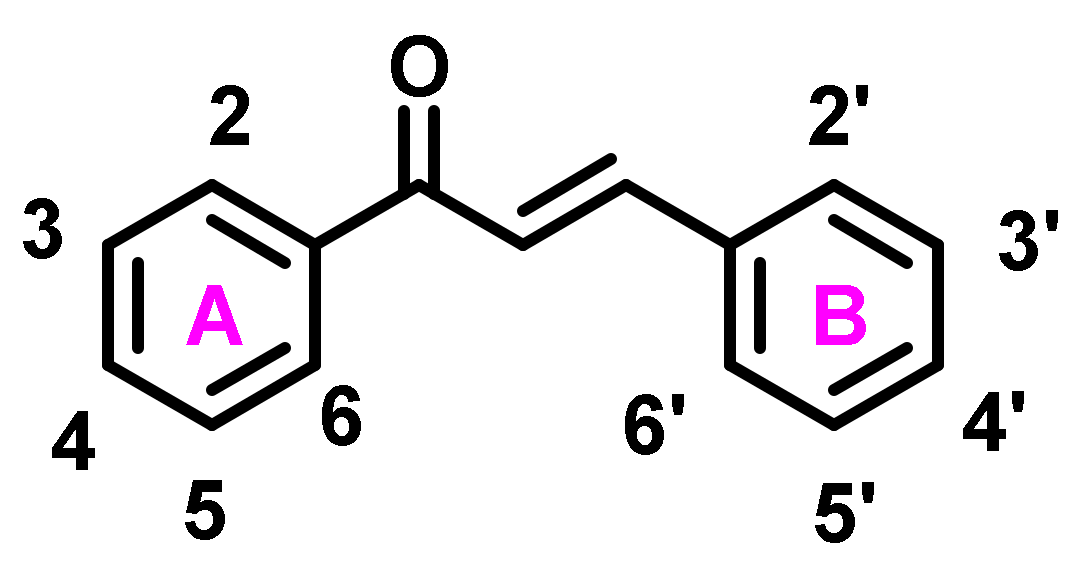
2. Structural Attachment of Various Chemical Functionalities on Chalcone Scaffold (Ring A and B)
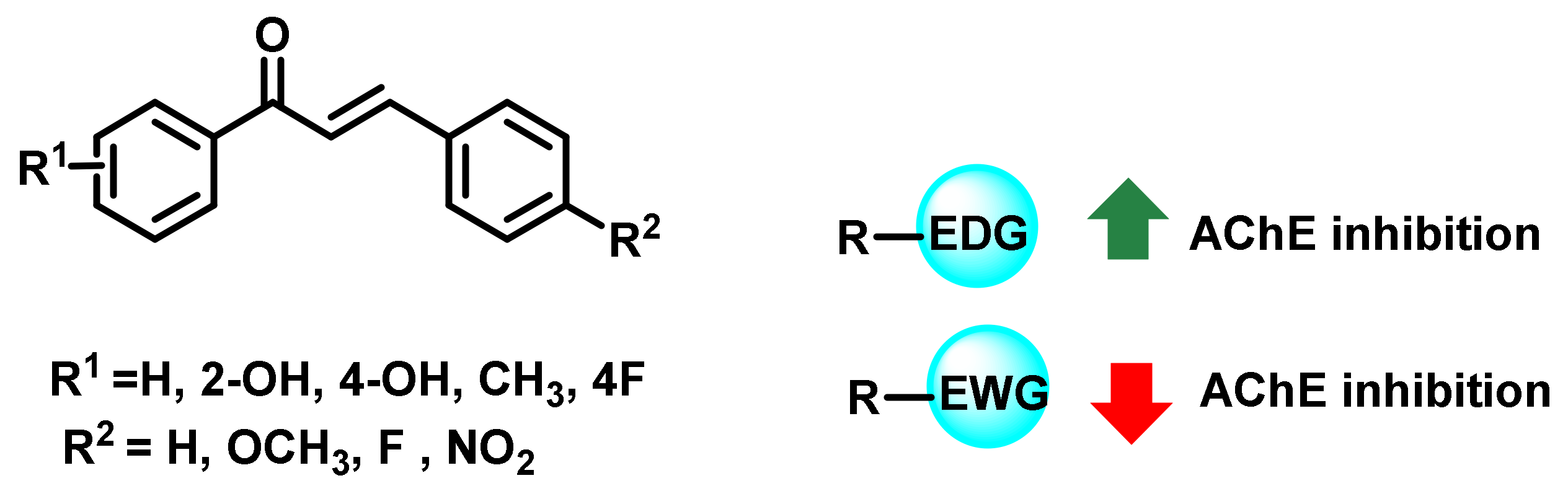
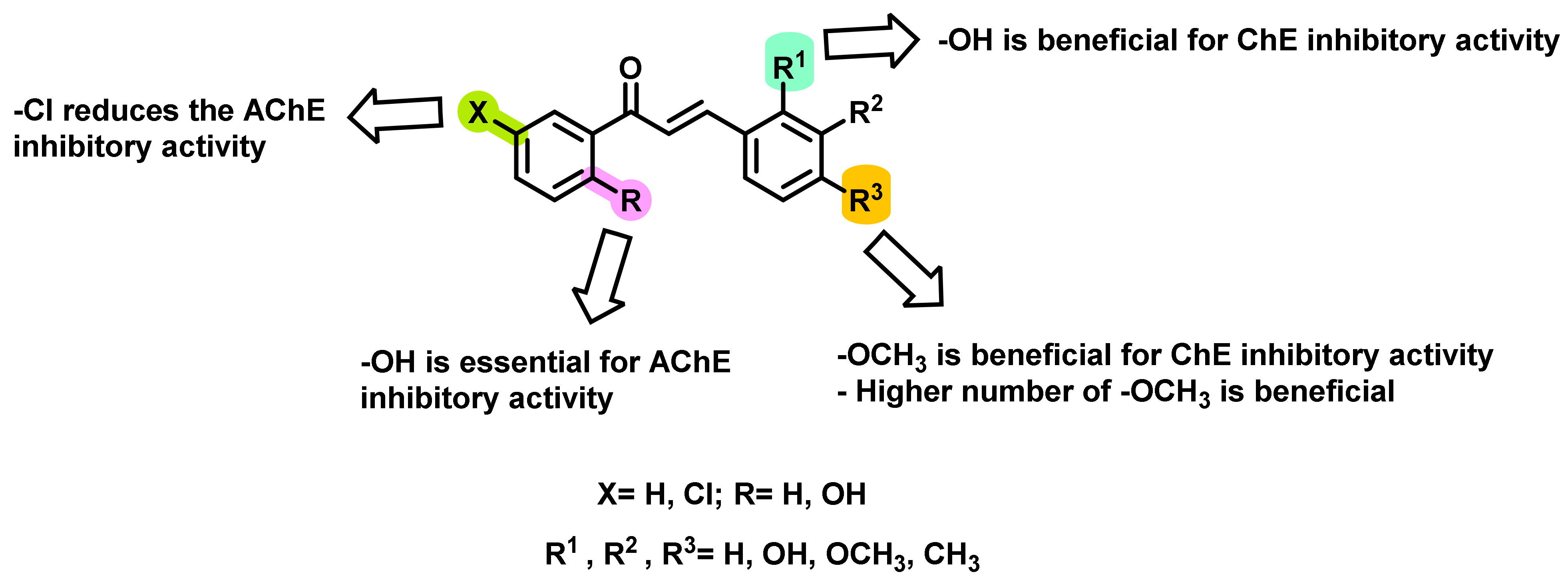
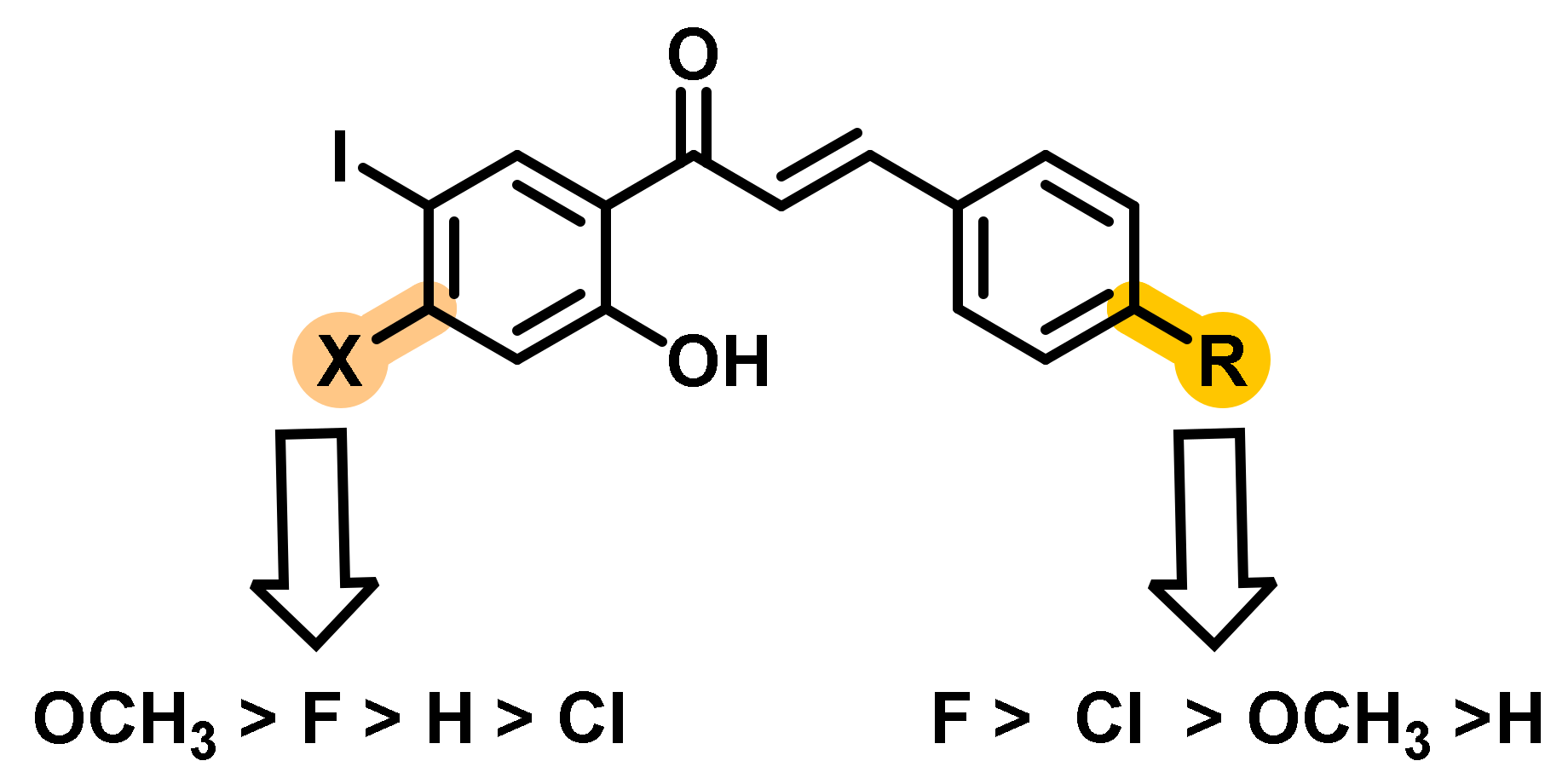
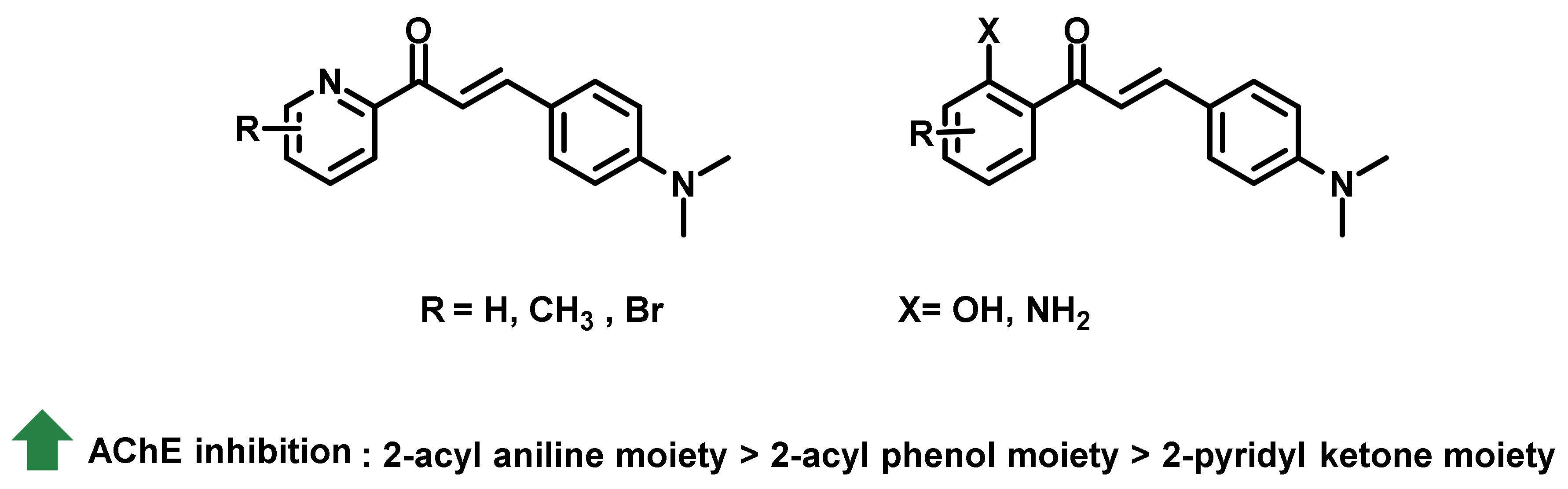
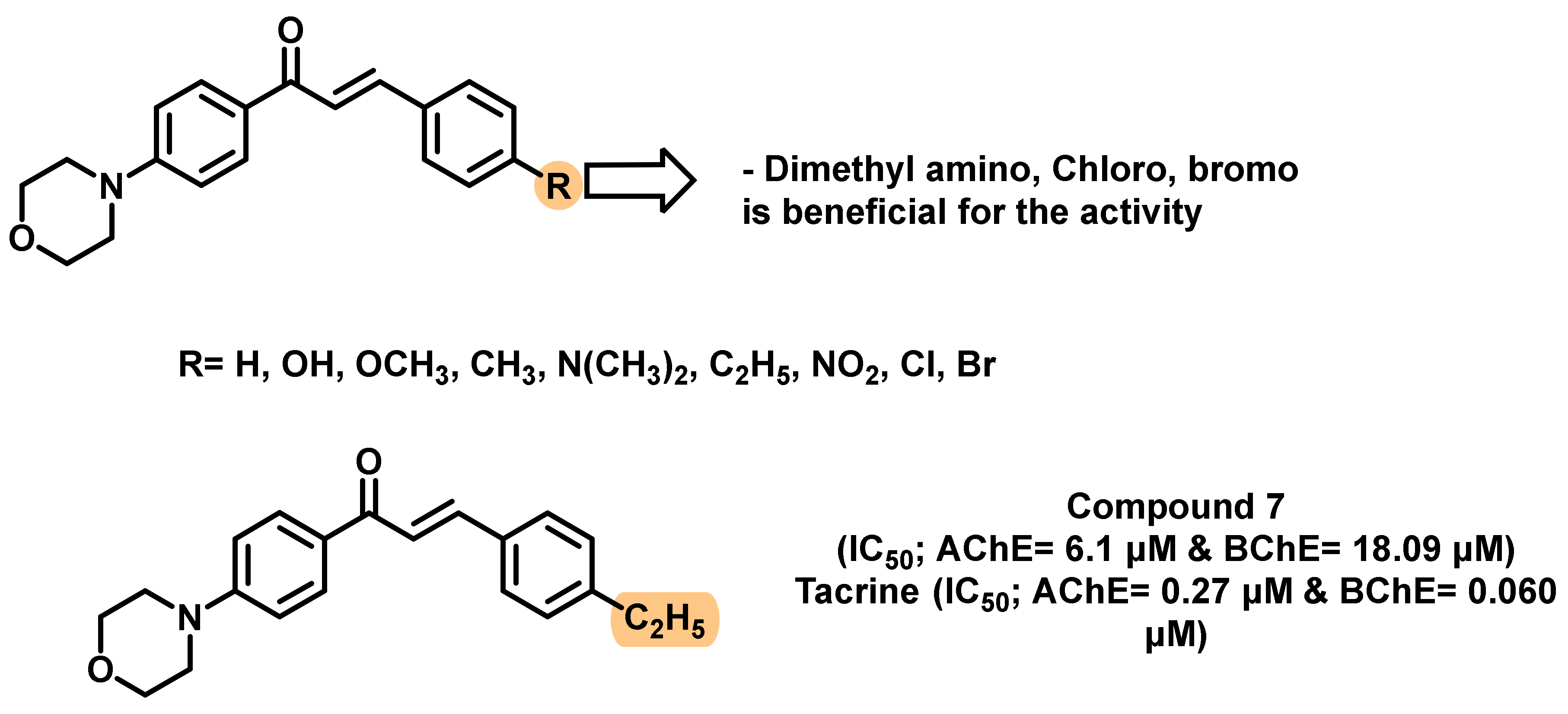
2.1. Chalcone Derivatives with Various Substituents Attachment
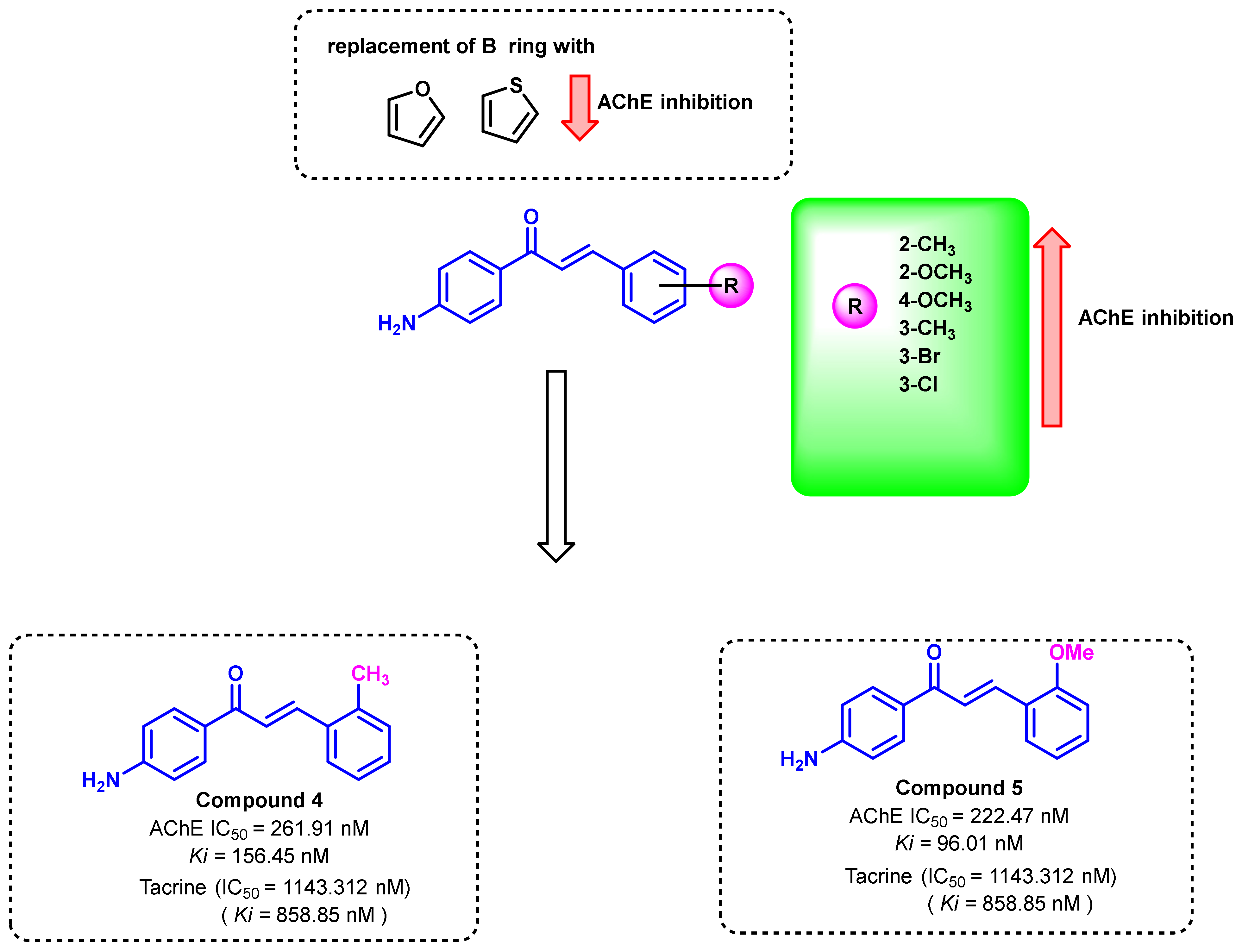
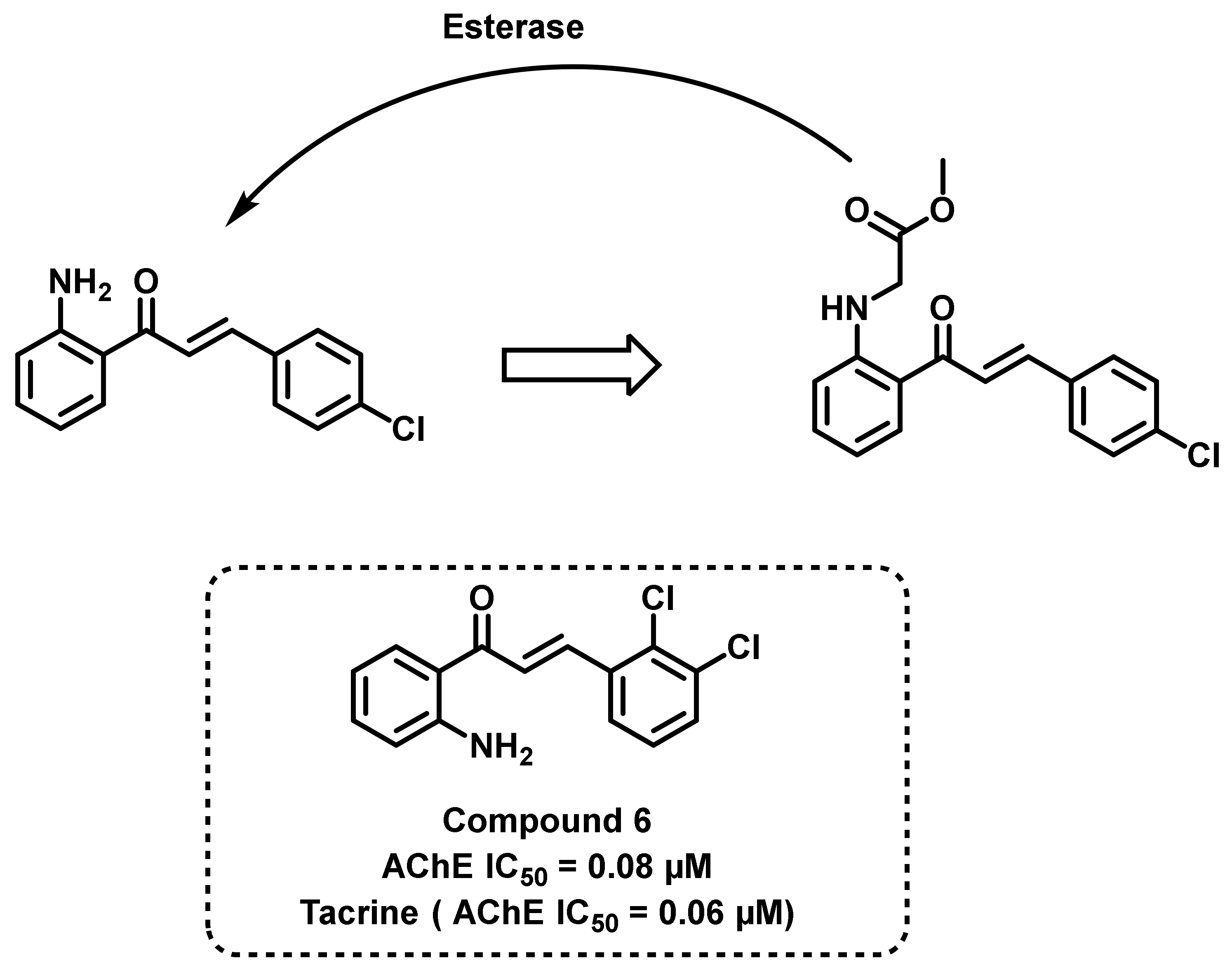
2.2. Chalcone Derivatives with Amine Substituent Modification
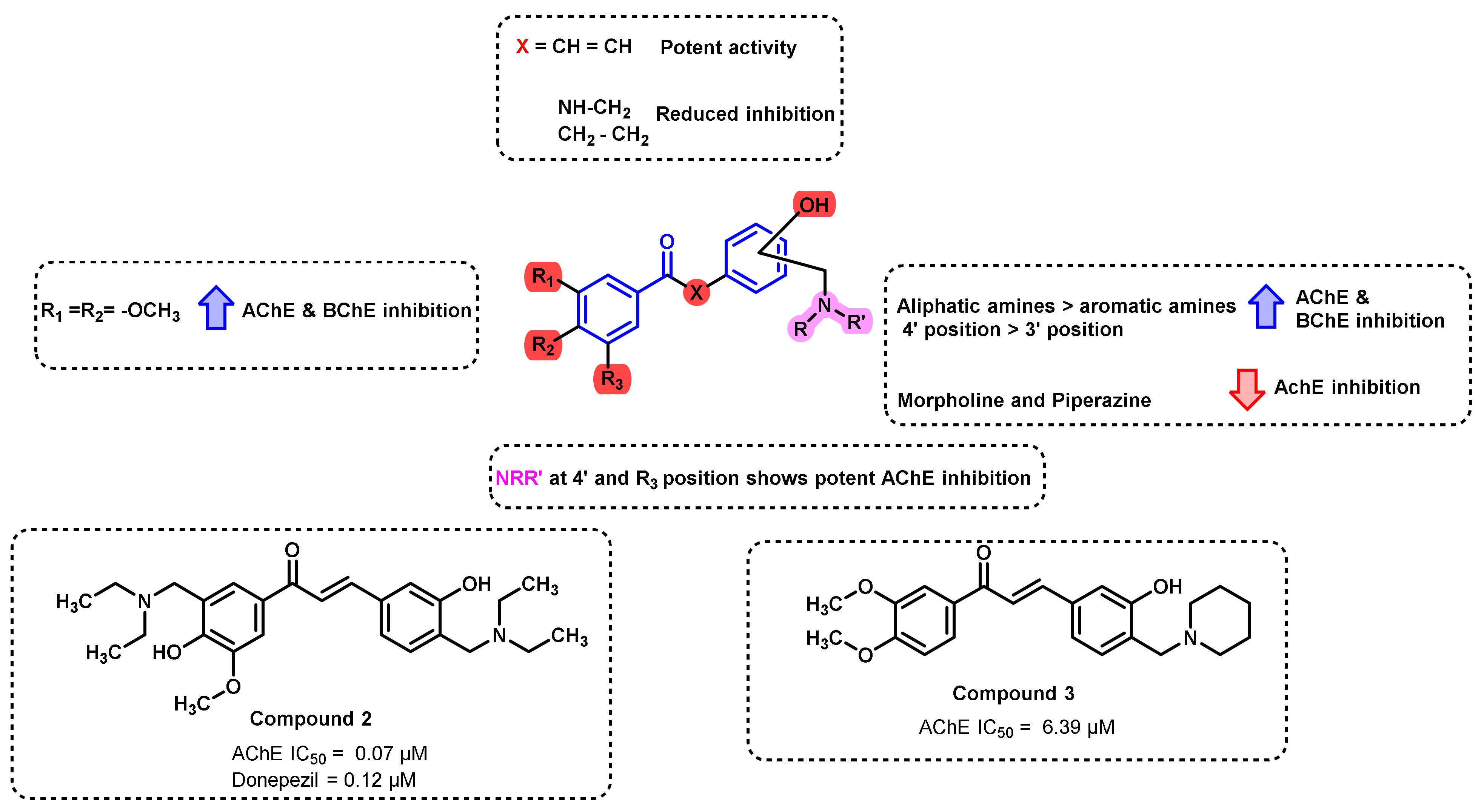
2.3. Chalcone Derivatives with Hydroxyl Substituent Modification
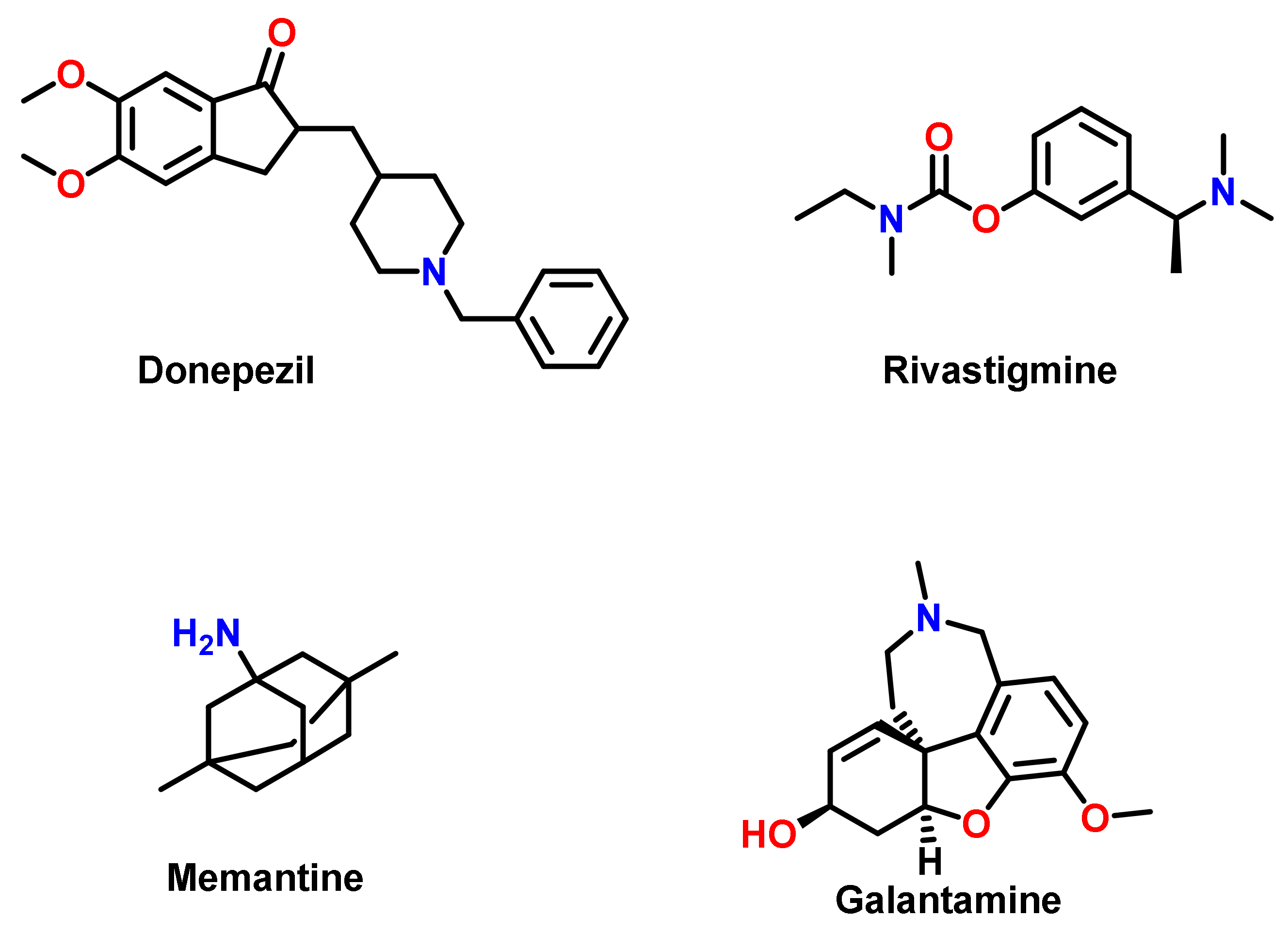
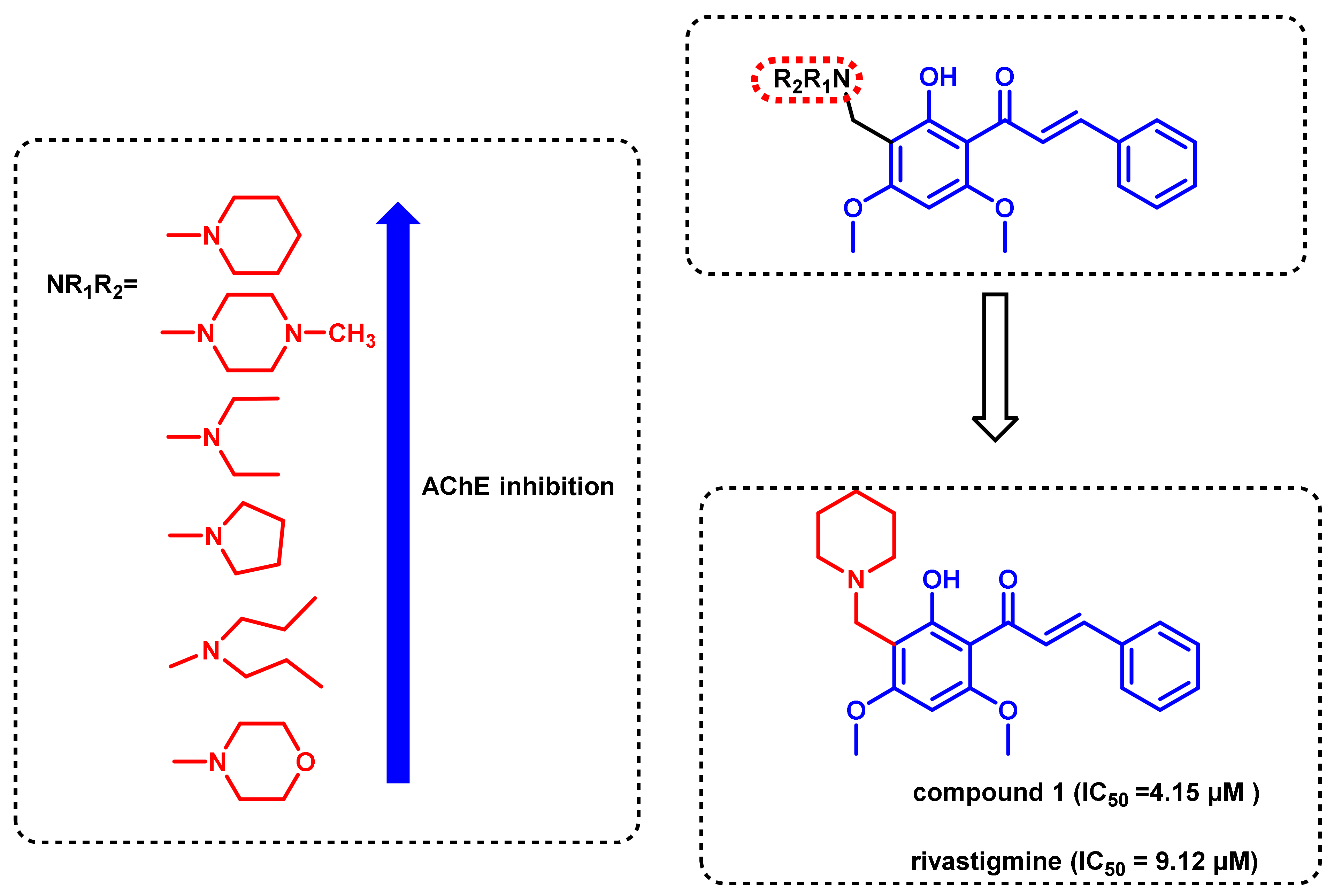
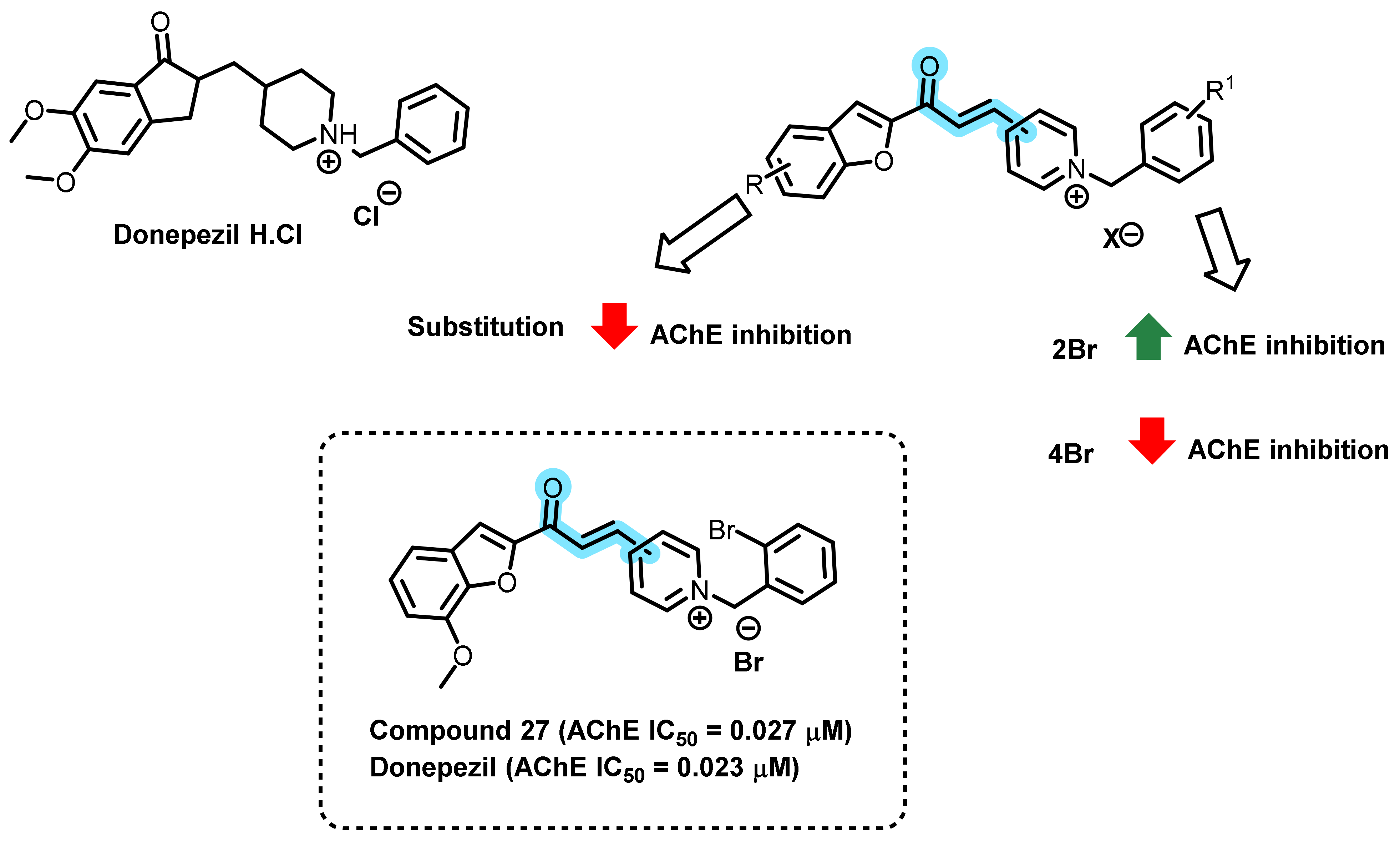
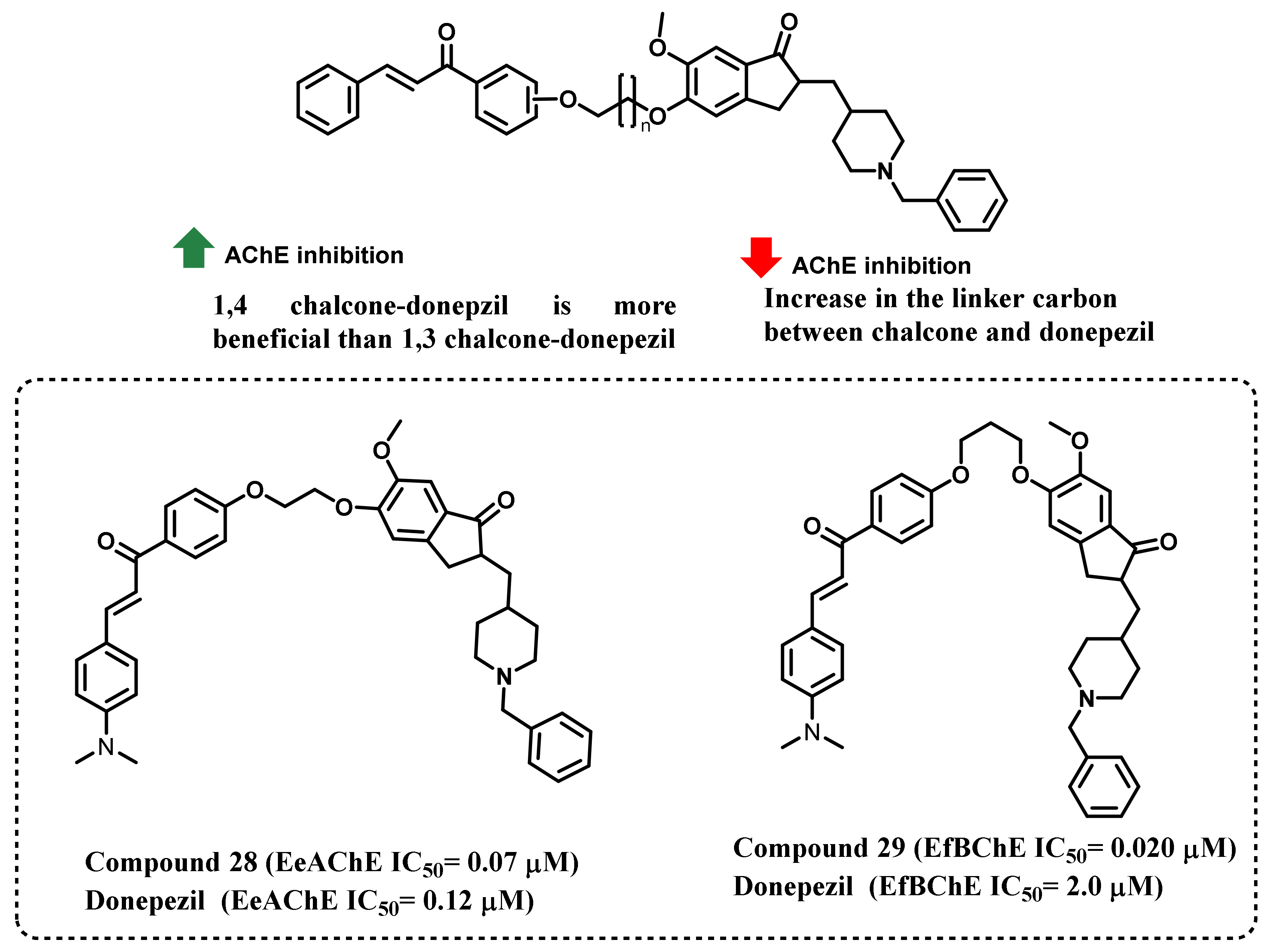
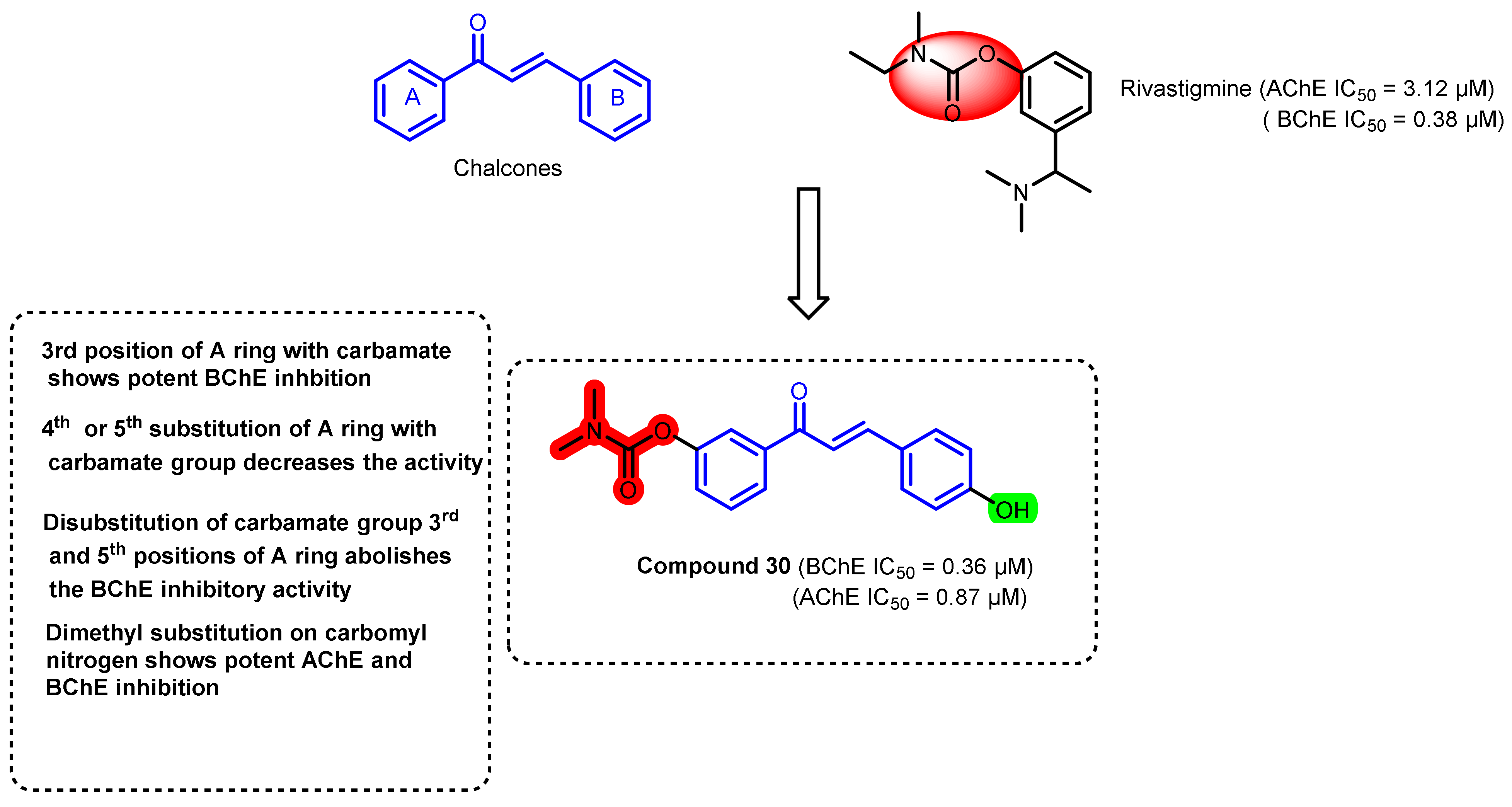
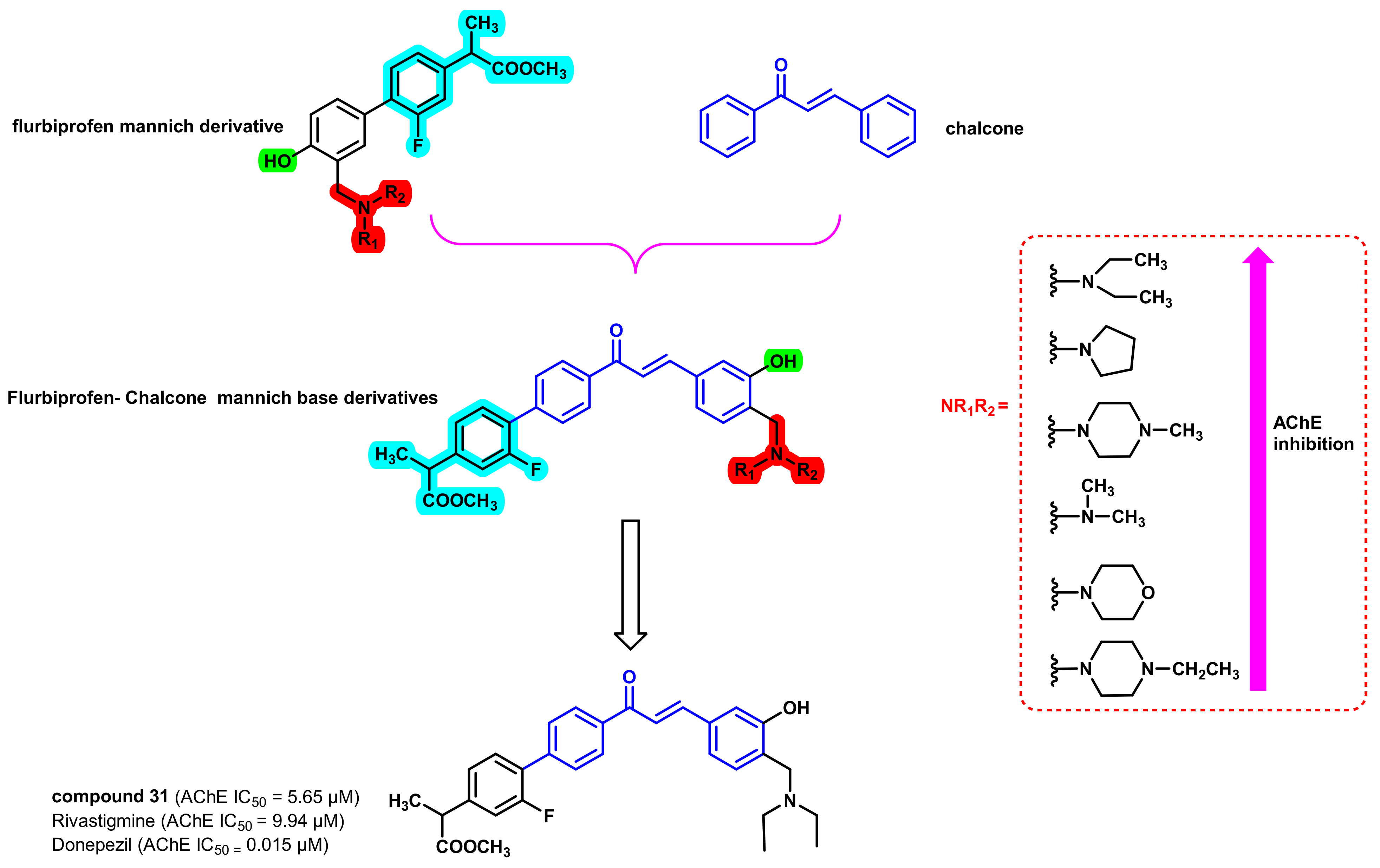
3. Chalcone-Based Hybrid with USFDA-Approved Drugs
4. Miscellaneous
4.1. Natural Chalcones
4.2. Chalcone Derivatives with Structural Modifications
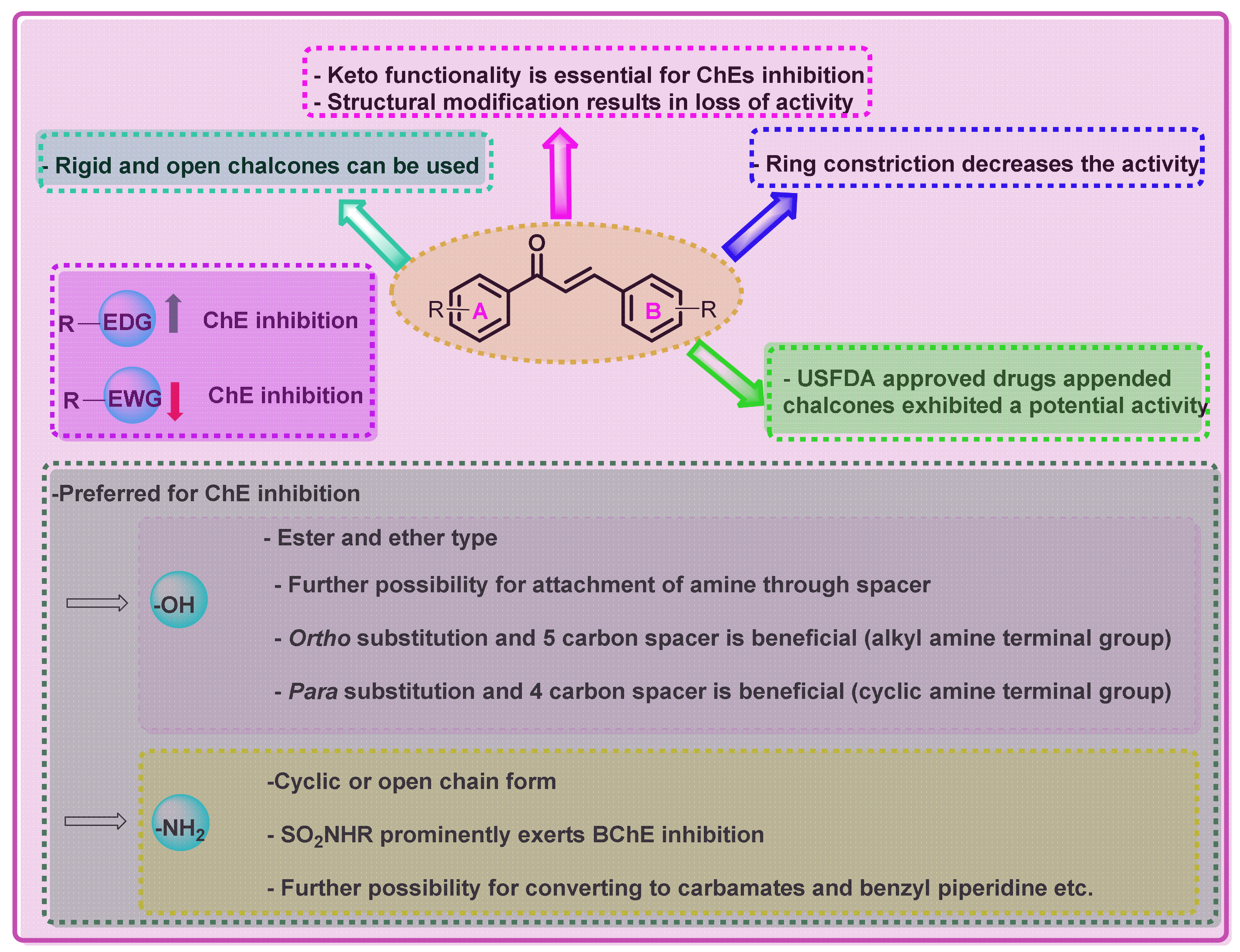
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dvir, H.; Silman, I.; Harel, M.; Rosenberry, T.L.; Sussman, J.L. Acetylcholinesterase: From 3D structure to function. Chem.-Biol. Interactions 2010, 187, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M.-M.; Guillozet, A.; Shaw, P.; Levey, A.; Duysen, E.G.; Lockridge, O. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 2002, 110, 627–639. [Google Scholar] [CrossRef]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Soreq, H.; Seidman, S. Acetylcholinesterase—New roles for an old actor. Nat. Rev. Neurosci. 2001, 2, 294–302. [Google Scholar] [CrossRef]
- Rees, T.M.; Brimijoin, S. The role of acetylcholinesterase in the pathogenesis of Alzheimer’s disease. Drugs Today 2003, 39, 75–83. [Google Scholar] [CrossRef]
- Paroni, G.; Bisceglia, P.; Seripa, D. Understanding the Amyloid Hypothesis in Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 68, 493–510. [Google Scholar] [CrossRef]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic Structure of Acetylcholinesterase from Torpedo californica: A Prototypic Acetylcholine-Binding Protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef]
- Raves, M.L.; Harel, M.; Pang, Y.-P.; Silman, I.; Kozikowski, A.P.; Sussman, J.L. Structure of acetylcholinesterase complexed with the nootropic alkaloid, (–)-huperzine A. Nat. Struct. Biol. 1997, 4, 57–63. [Google Scholar] [CrossRef]
- Sharma, A.; Mishra, A. In silico Screening of Pyridoxine Carbamates for Anti-Alzheimer’s Activities. Central Nerv. Syst. Agents Med. Chem. 2021, 21, 39–52. [Google Scholar] [CrossRef]
- Sever, B.; Altıntop, M.D.; Temel, H.E. In vitro and in silico studies on AChE inhibitory effects of a series of donepezil-like arylidene indanones. Turk. J. Biochem. 2020, 45, 359–363. [Google Scholar] [CrossRef]
- Mansha, M.; Taha, M.; Anouar, E.H.; Ullah, N. The design of fluoroquinolone-based cholinesterase inhibitors: Synthesis, biological evaluation and in silico docking studies. Arab. J. Chem. 2021, 14, 103211. [Google Scholar] [CrossRef]
- Da Silva, V.B.; de Andrade, P.; Kawano, D.F.; Morais, P.A.B.; de Almeida, J.R.; Carvalho, I.; Taft, C.A.; Silva, C.H.T.D.P.D. In silico design and search for acetylcholinesterase inhibitors in Alzheimer’s disease with a suitable pharmacokinetic profile and low toxicity. Futur. Med. Chem. 2011, 3, 947–960. [Google Scholar] [CrossRef]
- Pathak, A.; Madar, I.H.; Raithatha, K.; Gupta, J.K. In-Silico Identification of Potential Inhibitors Against AChE Using Cheminformatics Approach. MOJ Proteom. Bioinform. 2014, 1, 96–100. [Google Scholar] [CrossRef]
- Jyothi, P.; Yellamma, K. Molecular docking studies on the therapeutic targets of Alzheimer’s disease (AChE and BChE) using natural bioactive alkaloids. Int. J. Pharm. Pharm. Sci. 2016, 8, 108–112. [Google Scholar] [CrossRef][Green Version]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef]
- Huang, Y.-J.; Baldassarre, H.; Wang, B.; Lazaris, A.; Leduc, M.; Bilodeau, A.S.; Bellemare, A.; Côté, M.; Herskovits, P.; Touati, M.; et al. Recombinant human butyrylcholinesterase from milk of transgenic animals to protect against organophosphate poisoning. Proc. Natl. Acad. Sci. USA 2007, 104, 13603–13608. [Google Scholar] [CrossRef]
- Hyde, C.; Peters, J.; Bond, M.; Rogers, G.; Hoyle, M.; Anderson, R.; Jeffreys, M.; Davis, S.; Thokala, P.; Moxham, T. Evolution of the evidence on the effectiveness and cost-effectiveness of acetylcholinesterase inhibitors and memantine for Alzheimer’s disease: Systematic review and economic model. Age Ageing 2013, 42, 14–20. [Google Scholar] [CrossRef]
- Salloway, S.; Ferris, S.; Kluger, A.; Goldman, R.; Griesing, T.; Kumar, D.; Richardson, S. Efficacy of donepezil in mild cognitive impairment: A randomized placebo-controlled trial. Neurology 2004, 63, 651–657. [Google Scholar] [CrossRef]
- Arce, M.P.; Rodríguez-Franco, M.I.; González-Muñoz, G.C.; Pérez, C.; López, B.; Villarroya, M.; Lopez, M.G.; Garcia, A.G.; Conde, S. Neuroprotective and Cholinergic Properties of Multifunctional Glutamic Acid Derivatives for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2009, 52, 7249–7257. [Google Scholar] [CrossRef]
- Castro, A.; Martinez, A. Targeting Beta-Amyloid Pathogenesis Through Acetylcholinesterase Inhibitors. Curr. Pharm. Des. 2006, 12, 4377–4387. [Google Scholar] [CrossRef]
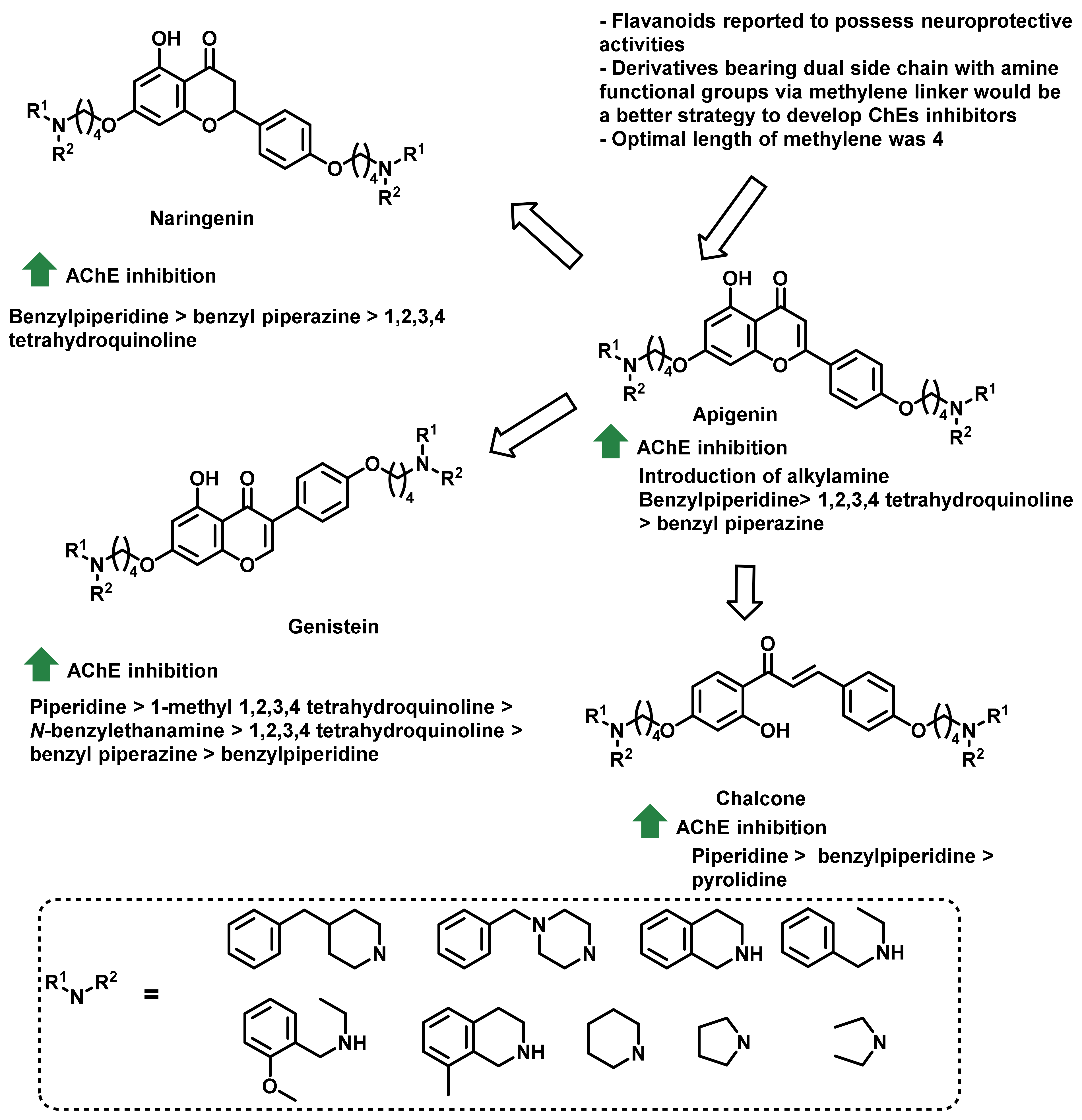
- Thomas-Barberan, F.A.; Clifford, M.N. Flavanones, chalcones and dihydrochalcones—Nature, occurrence and dietary burden. J. Sci. Food Agric. 2020, 80, 1073–1080. [Google Scholar] [CrossRef]
- Mathew, B.; Mathew, G.E.; Uçar, G.; Baysal, I.; Suresh, J.; Vilapurathu, J.K.; Prakasan, A.; Suresh, J.K.; Thomas, A. Development of fluorinated methoxylated chalcones as selective monoamine oxidase-B inhibitors: Synthesis, biochemistry and molecular docking studies. Bioorg. Chem. 2015, 62, 22–29. [Google Scholar] [CrossRef]
- Sasidharan, R.; Manju, S.L.; Uçar, G.; Baysal, I.; Mathew, B. Identification of Indole-Based Chalcones: Discovery of a Potent, Selective, and Reversible Class of MAO-B Inhibitors. Arch. Pharm. 2016, 349, 627–637. [Google Scholar] [CrossRef]
- Mathew, B.; Haridas, A.; Suresh, J.; Mathew, G.E.; Uçar, G.; Jayaprakash, V. Monoamine Oxidase Inhibitory Action of Chalcones: A Mini Review. Central Nerv. Syst. Agents Med. Chem. 2016, 16, 120–136. [Google Scholar] [CrossRef]
- Robinson, S.J.; Petzer, J.P.; Petzer, A.; Bergh, J.J.; Lourens, A.C. Selected furanochalcones as inhibitors of monoamine oxidase. Bioorg. Med. Chem. Lett. 2013, 23, 4985–4989. [Google Scholar] [CrossRef]
- De Mello, M.V.P.; Abrahim-Vieira, B.D.A.; Domingos, T.F.S.; de Jesus, J.B.; de Sousa, A.C.C.; Rodrigues, C.R.; de Souza, A.M.T. A comprehensive review of chalcone derivatives as antileishmanial agents. Eur. J. Med. Chem. 2018, 150, 920–929. [Google Scholar] [CrossRef]
- Thapa, P.; Upadhyay, S.P.; Suo, W.Z.; Singh, V.; Gurung, P.; Lee, E.S.; Sharma, R.; Sharma, M. Chalcone and its analogs: Therapeutic and diagnostic applications in Alzheimer’s disease. Bioorg. Chem. 2021, 108, 104681. [Google Scholar] [CrossRef]
- Sang, Z.; Song, Q.; Cao, Z.; Deng, Y.; Zhang, L. Design, synthesis, and evaluation of chalcone-Vitamin E-donepezil hybrids as multi-target-directed ligands for the treatment of Alzheimer’s disease. J. Enzym. Inhib. Med. Chem. 2022, 37, 69–85. [Google Scholar] [CrossRef]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef]
- Matos, M.J.; Vazquez-Rodriguez, S.; Uriarte, E.; Santana, L. Potential pharmacological uses of chalcones: A patent review (from June 2011–2014). Expert Opin. Ther. Pat. 2015, 25, 351–366. [Google Scholar] [CrossRef]
- Koyiparambath, V.P.; Rajappan, K.P.; Rangarajan, T.M.; Al-Sehemi, A.G.; Pannipara, M.; Bhaskar, V.; Nair, A.S.; Sudevan, S.T.; Kumar, S.; Mathew, B. Deciphering the detailed structure–activity relationship of coumarins as Monoamine oxidase enzyme inhibitors—An updated review. Chem. Biol. Drug Des. 2021, 98, 655–673. [Google Scholar] [CrossRef] [PubMed]
- Vishal, P.; Oh, J.; Khames, A.; Abdelgawad, M.; Nair, A.; Nath, L.; Gambacorta, N.; Ciriaco, F.; Nicolotti, O.; Kim, H.; et al. Trimethoxylated Halogenated Chalcones as Dual Inhibitors of MAO-B and BACE-1 for the Treatment of Neurodegenerative Disorders. Pharmaceutics 2021, 13, 850. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.; Ribeiro, D.; Fernandes, E.; Freitas, M. A Systematic Review on Anti-diabetic Properties of Chalcones. Curr. Med. Chem. 2020, 27, 2257–2321. [Google Scholar] [CrossRef] [PubMed]
- Karaca, H.; Kazancı, S. The metal sensing applications of chalcones: The synthesis, characterization and theoretical calculations. J. Mol. Struct. 2022, 1248, 131454. [Google Scholar] [CrossRef]
- Hanif, N.; Iswantini, D.; Hioki, Y.; Murni, A.; Kita, M.; Tanaka, J. Flavokawains, Plant-derived Chalcones, Inhibit Differentiation of Murine Pre-adipocytes. Chem. Lett. 2022, 51, 54–57. [Google Scholar] [CrossRef]
- Liu, W.; He, M.; Li, Y.; Peng, Z.; Wang, G. A review on synthetic chalcone derivatives as tubulin polymerisation inhibitors. J. Enzym. Inhib. Med. Chem. 2022, 37, 9–38. [Google Scholar] [CrossRef]
- Elzupir, A.; Ibnaouf, K. Synthesis of novel pyrrolidinyl chalcone derivatives: Spectral properties, energy band-gap tailoring, and amplified spontaneous emission. J. Mol. Struct. 2022, 1250, 131698. [Google Scholar] [CrossRef]
- Aslan, H.E.; Demir, Y.; Özaslan, M.S.; Türkan, F.; Beydemir, Ş.; Küfrevioğlu, Ö.I. The behavior of some chalcones on acetylcholinesterase and carbonic anhydrase activity. Drug Chem. Toxicol. 2019, 42, 634–640. [Google Scholar] [CrossRef]
- Hasan, A.; Khan, K.M.; Sher, M.; Maharvi, G.M.; Nawaz, S.A.; Choudhary, M.; Rahman, A.U.; Supuran, C.T. Synthesis and inhibitory potential towards acetylcholinesterase, butyrylcholinesterase and lipoxygenase of some variably substituted chalcones. J. Enzym. Inhib. Med. Chem. 2005, 20, 41–47. [Google Scholar] [CrossRef]
- Mphahlele, M.J.; Agbo, E.N.; Gildenhuys, S. Synthesis and Evaluation of the 4-Substituted 2-Hydroxy-5-Iodochalcones and Their 7-Substituted 6-Iodoflavonol Derivatives for Inhibitory Effect on Cholinesterases and β-Secretase. Int. J. Mol. Sci. 2018, 19, 4112. [Google Scholar] [CrossRef]
- Fosso, M.Y.; Rd, H.L.; Green, K.D.; Tsodikov, O.V.; Garneautsodikova, S. Effects of structural modifications on the metal binding, anti-amyloid activity, and cholinesterase inhibitory activity of chalcones. Org. Biomol. Chem. 2015, 13, 9418–9426. [Google Scholar] [CrossRef]
- Liu, H.-R.; Huang, X.-Q.; Lou, D.-H.; Liu, X.-J.; Liu, W.-K.; Wang, Q.-A. Synthesis and acetylcholinesterase inhibitory activity of Mannich base derivatives flavokawain B. Bioorg. Med. Chem. Lett. 2014, 24, 4749–4753. [Google Scholar] [CrossRef]
- Zhang, X.; Song, Q.; Cao, Z.; Li, Y.; Tian, C.; Yang, Z.; Zhang, H.; Deng, Y. Design, synthesis and evaluation of chalcone Mannich base derivatives as multifunctional agents for the potential treatment of Alzheimer’s disease. Bioorg. Chem. 2019, 87, 395–408. [Google Scholar] [CrossRef]
- Gürdere, M.B.; Budak, Y.; Kocyigit, U.M.; Taslimi, P.; Tüzün, B.; Ceylan, M. ADME properties, bioactivity and molecular docking studies of 4-amino-chalcone derivatives: New analogues for the treatment of Alzheimer, glaucoma and epileptic diseases. Silico Pharmacol. 2021, 9, 34. [Google Scholar] [CrossRef]
- Sakata, R.P.; Antoniolli, G.; Lancellotti, M.; Kawano, D.F.; Barbosa, E.G.; Almeida, W.P. Synthesis and biological evaluation of 2′-Aminochalcone: A multi-target approach to find drug candidates to treat Alzheimer’s disease. Bioorg. Chem. 2020, 103, 104201. [Google Scholar] [CrossRef]
- Sasidharan, R.; Eom, B.H.; Heo, J.H.; Park, J.E.; Abdelgawad, M.A.; Musa, A.; Gambacorta, N.; Nicolotti, O.; Manju, S.L.; Mathew, B. Morpholine-based chalcones as dual-acting monoamine oxidase-B and acetylcholinesterase inhibitors: Synthesis and biochemical investigations. J. Enz. Inhib. Med. Chem. 2021, 36, 188–197. [Google Scholar] [CrossRef]
- Mathew, B.; Oh, J.M.; Baty, R.S.; Batiha, G.E.-S.; Parambi, D.G.T.; Gambacorta, N.; Nicolotti, O.; Kim, H. Piperazine-substituted chalcones: A new class of MAO-B, AChE, and BACE-1 inhibitors for the treatment of neurological disorders. Environ. Sci. Pollut. Res. 2021, 28, 38855–38866. [Google Scholar] [CrossRef]
- Kocyigit, U.M.; Budak, Y.; Gürdere, M.B.; Ertürk, F.; Yencilek, B.; Taslimi, P.; Gülçin, I.; Ceylan, M. Synthesis of chalcone-imide derivatives and investigation of their anticancer and antimicrobial activities, carbonic anhydrase and acetylcholinesterase enzymes inhibition profiles. Arch. Physiol. Biochem. 2018, 124, 61–68. [Google Scholar] [CrossRef]
- Kang, J.E.; Cho, J.K.; Long, M.; Ryu, H.W.; Kim, J.H.; Kim, H.J.; Yuk, H.J.; Kim, D.W.; Park, K.H. Inhibitory Evaluation of Sulfonamide Chalcones on β-Secretase and Acylcholinesterase. Molecules 2013, 18, 140–153. [Google Scholar] [CrossRef]
- Liu, H.; Fan, H.; Gao, X.; Huang, X.; Liu, X.; Liu, L.; Zhou, C.; Tang, J.; Wang, Q.; Liu, W. Design, synthesis and preliminary structure–activity relationship investigation of nitrogen-containing chalcone derivatives as acetylcholinesterase and butyrylcholinesterase inhibitors: A further study based on Flavokawain B Mannich base derivatives. J. Enzym. Inhib. Med. Chem. 2016, 31, 580–589. [Google Scholar] [CrossRef]
- Liu, H.-R.; Zhou, C.; Fan, H.-Q.; Tang, J.-J.; Liu, L.-B.; Gao, X.-H.; Wang, Q.-A.; Liu, W.-K. Novel Potent and Selective Acetylcholinesterase Inhibitors as Potential Drugs for the Treatment of Alzheimer’s Disease: Synthesis, Pharmacological Evaluation, and Molecular Modeling of Amino-Alkyl-Substituted Fluoro-Chalcones Derivatives. Chem. Biol. Drug Des. 2015, 86, 517–522. [Google Scholar] [CrossRef]
- Liu, H.-R.; Liu, X.-J.; Fan, H.-Q.; Tang, J.-J.; Gao, X.-H.; Liu, W.-K. Design, synthesis and pharmacological evaluation of chalcone derivatives as acetylcholinesterase inhibitors. Bioorg. Med. Chem. 2014, 22, 6124–6133. [Google Scholar] [CrossRef]
- Rampa, A.; Montanari, S.; Pruccoli, L.; Bartolini, M.; Falchi, F.; Feoli, A.; Cavalli, A.; Belluti, F.; Gobbi, S.; Tarozzi, A.; et al. Chalcone-based carbamates for Alzheimer’s disease treatment. Futur. Med. Chem. 2017, 9, 749–764. [Google Scholar] [CrossRef]
- Zhao, F.-C.; Wu, Y.; Song, X.-J. Design and Development of a Novel Chalcone Derivative as an Anticholinesterase Inhibitor for Possible Treatment of Dementia. Med. Sci. Monit. 2017, 23, 3311–3317. [Google Scholar] [CrossRef]
- Rampa, A.; Bartolini, M.; Pruccoli, L.; Naldi, M.; Iriepa, I.; Moraleda, I.; Belluti, F.; Gobbi, S.; Tarozzi, A.; Bisi, A. Exploiting the Chalcone Scaffold to Develop Multifunctional Agents for Alzheimer’s Disease. Molecules 2018, 23, 1902. [Google Scholar] [CrossRef]
- Sang, Z.; Wang, K.; Zhang, P.; Shi, J.; Liu, W.; Tan, Z. Design, synthesis, in-silico and biological evaluation of novel chalcone derivatives as multi-function agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 180, 238–252. [Google Scholar] [CrossRef]
- Bai, P.; Wang, K.; Zhang, P.; Shi, J.; Cheng, X.; Zhang, Q.; Zheng, C.; Cheng, Y.; Yang, J.; Lu, X.; et al. Development of chalcone-O-alkylamine derivatives as multifunctional agents against Alzheimer’s disease. Eur. J. Med. Chem. 2019, 183, 111737. [Google Scholar] [CrossRef]
- Gao, X.-H.; Zhou, C.; Liu, H.-R.; Liu, L.-B.; Tang, J.-J.; Xia, X.-H. Tertiary amine derivatives of chlorochalcone as acetylcholinesterase (AChE) and buthylcholinesterase (BuChE) inhibitors: The influence of chlorine, alkyl amine side chain and α,β-unsaturated ketone group. J. Enzym. Inhib. Med. Chem. 2017, 32, 146–152. [Google Scholar] [CrossRef]
- Mathew, B.; Parambi, D.G.T.; Mathew, G.E.; Uddin, M.S.; Inasu, S.T.; Kim, H.; Marathakam, A.; Unnikrishnan, M.K.; Carradori, S. Emerging therapeutic potentials of dual-acting MAO and AChE inhibitors in Alzheimer’s and Parkinson’s diseases. Arch. Pharm. Chem. Life Sci. 2019, 352, e1900177. [Google Scholar] [CrossRef]
- Mostofi, M.; Ziarani, G.M.; Mahdavi, M.; Moradi, A.; Nadri, H.; Emami, S.; Alinezhad, H.; Foroumadi, A.; Shafiee, A. Synthesis and structure-activity relationship study of benzofuran-based chalconoids bearing benzylpyridinium moiety as potent acetylcholinesterase inhibitors. Eur. J. Med. Chem. 2015, 103, 361–369. [Google Scholar] [CrossRef]
- Chandrika, N.T.; Fosso, M.Y.; Tsodikov, O.V.; Levine, H.; Garneau-Tsodikova, S.; Iii, H.L. Combining Chalcones with Donepezil to Inhibit Both Cholinesterases and Aβ Fibril Assembly. Molecules 2020, 25, 77. [Google Scholar] [CrossRef] [PubMed]
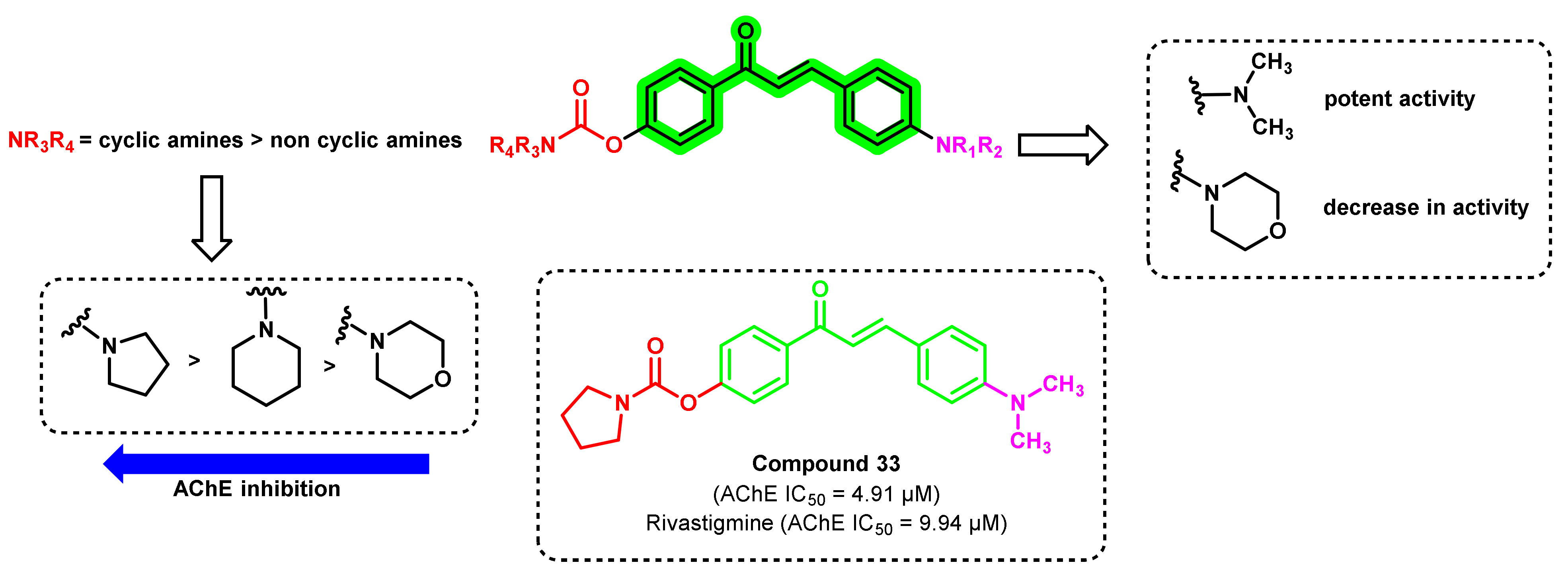
- Wang, L.; Wang, Y.; Tian, Y.; Shang, J.; Sun, X.; Chen, H.; Wang, H.; Tan, W. Design, synthesis, biological evaluation, and molecular modeling studies of chalcone-rivastigmine hybrids as cholinesterase inhibitors. Bioorg. Med. Chem. 2017, 25, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Qiang, X.; Song, Q.; Cao, Z.; Ye, C.; He, Y.; Deng, Y.; Zhang, L. Flurbiprofen-chalcone hybrid Mannich base derivatives as balanced multifunctional agents against Alzheimer’s disease: Design, synthesis and biological evaluation. Bioorg. Chem. 2020, 94, 103477. [Google Scholar] [CrossRef] [PubMed]
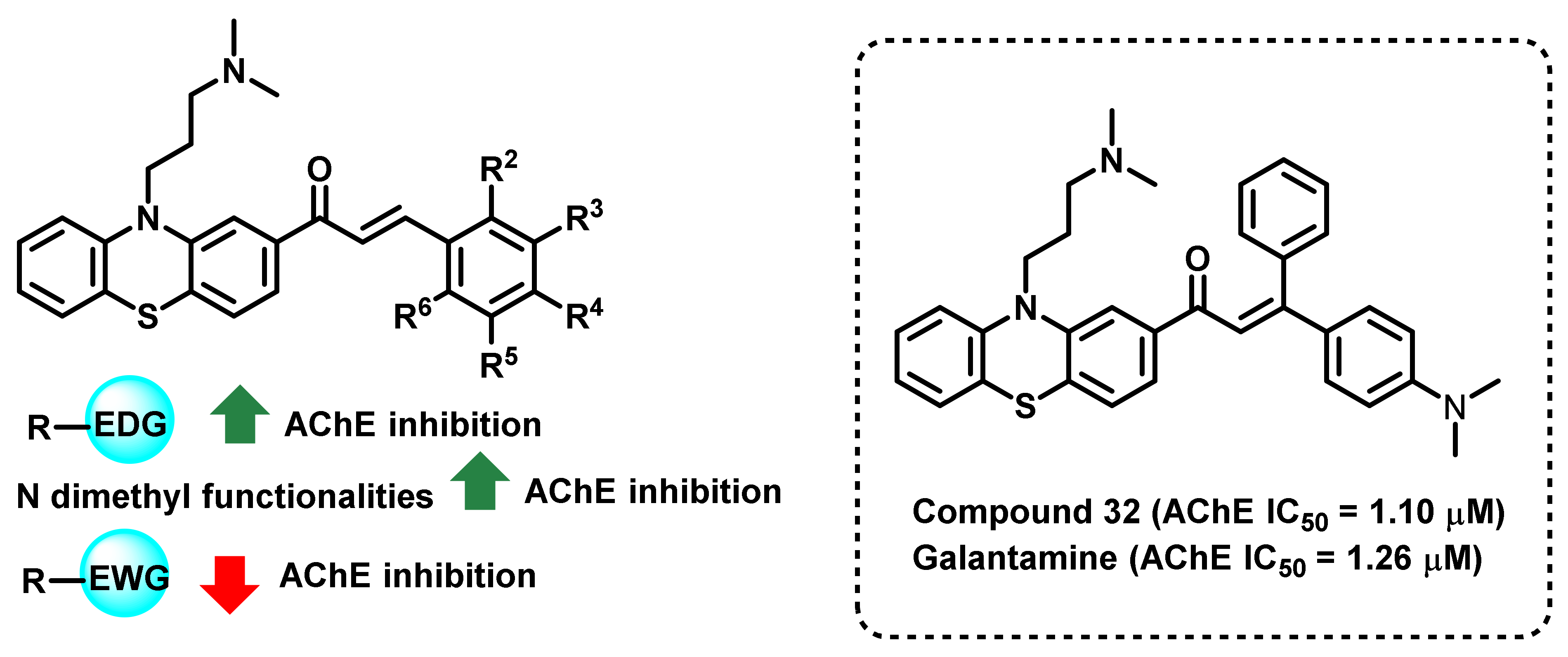
- Tran, T.-S.; Le, M.-T.; Nguyen, T.-C.; Tran, T.-H.; Tran, T.-D.; Thai, K.-M. Synthesis, In Silico and In Vitro Evaluation for Acetylcholinesterase and BACE-1 Inhibitory Activity of Some N-Substituted-4-Phenothiazine-Chalcones. Molecules 2020, 25, 3916. [Google Scholar] [CrossRef]
- Xiao, G.; Li, Y.; Qiang, X.; Xu, R.; Zheng, Y.; Cao, Z.; Luo, L.; Yang, X.; Sang, Z.; Su, F.; et al. Design, synthesis and biological evaluation of 4′-aminochalcone-rivastigmine hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2017, 25, 1030–1041. [Google Scholar] [CrossRef]
- Sang, Z.; Wang, K.; Shi, J.; Liu, W.; Cheng, X.; Zhu, G.; Wang, Y.; Zhao, Y.; Qiao, Z.; Wu, A.; et al. The development of advanced structural framework as multi-target-directed ligands for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2020, 192, 112180. [Google Scholar] [CrossRef]
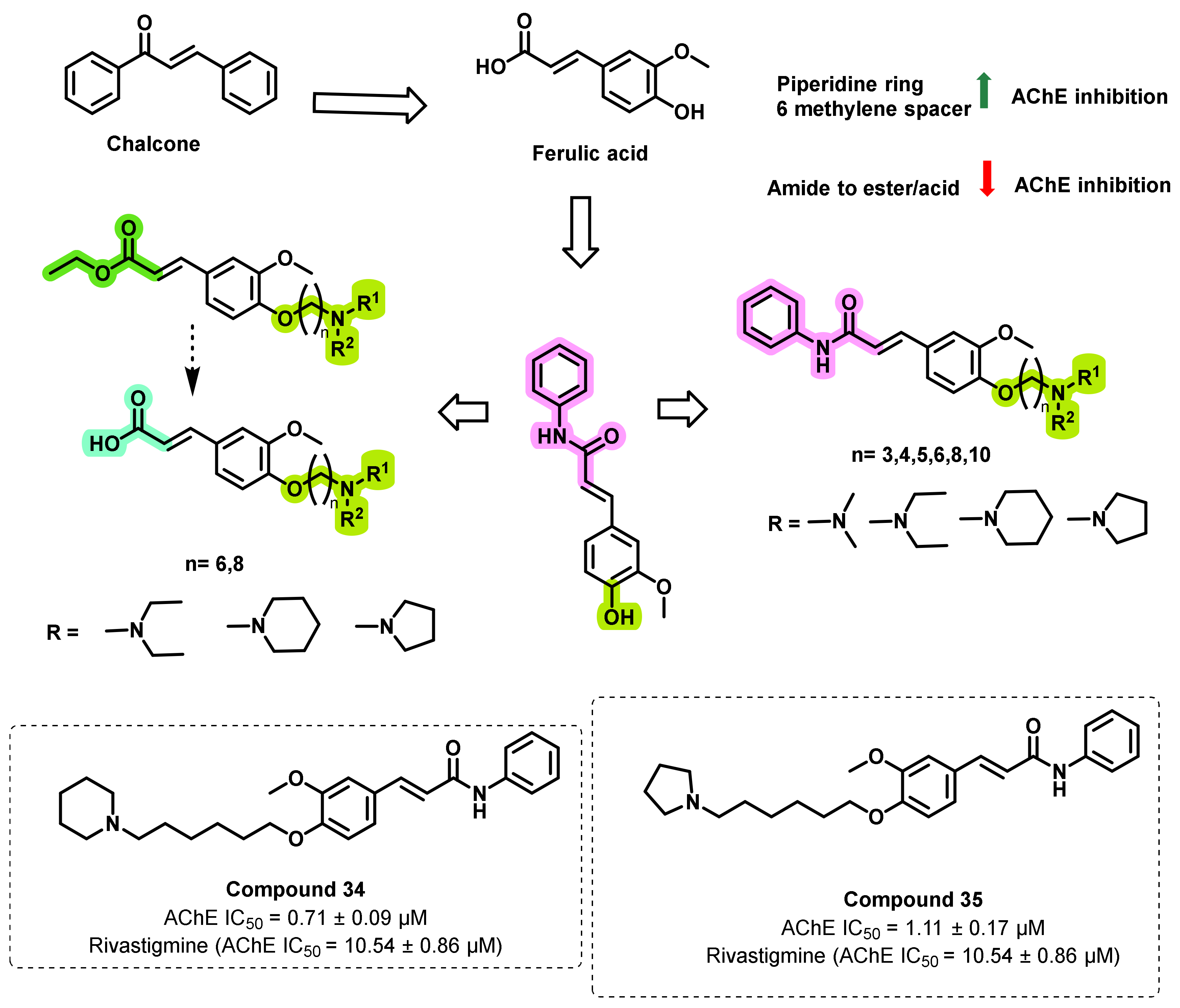
- Liu, H.; Liu, L.; Gao, X.; Liu, Y.; Xu, W.; He, W.; Jiang, H.; Tang, J.; Fan, H.; Xia, X. Novel ferulic amide derivatives with tertiary amine side chain as acetylcholinesterase and butyrylcholinesterase inhibitors: The influence of carbon spacer length, alkylamine and aromatic group. Eur. J. Med. Chem. 2017, 126, 810–822. [Google Scholar] [CrossRef]
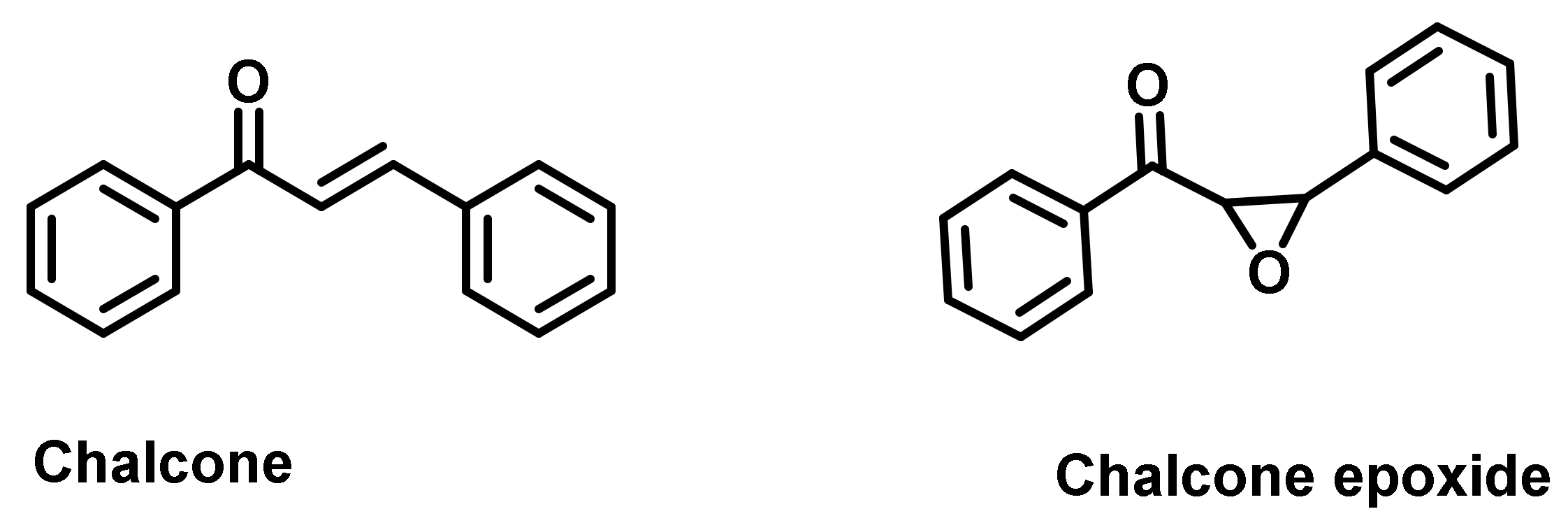
- Stellenboom, N. Comparison of the inhibitory potential towards carbonic anhydrase, acetylcholinesterase and butyrylcholinesterase of chalcone and chalcone epoxide. J. Biochem. Mol. Toxicol. 2019, 33, e22240. [Google Scholar] [CrossRef]
- Liargkova, T.; Eleftheriadis, N.; Dekker, F.; Voulgari, E.; Avgoustakis, C.; Sagnou, M.; Mavroidi, B.; Pelecanou, M.; Hadjipavlou-Litina, D. Small Multitarget Molecules Incorporating the Enone Moiety. Molecules 2019, 24, 199. [Google Scholar] [CrossRef]
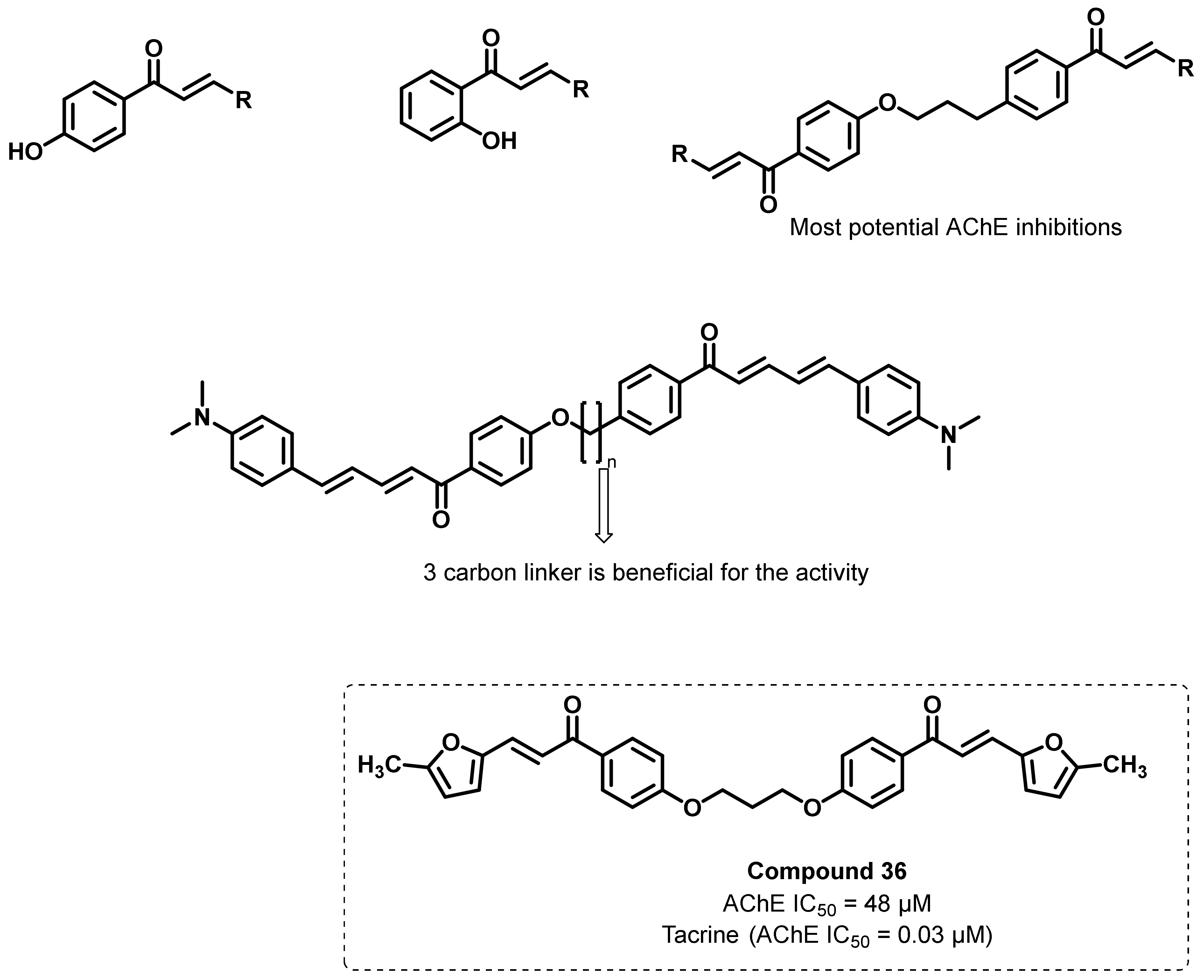
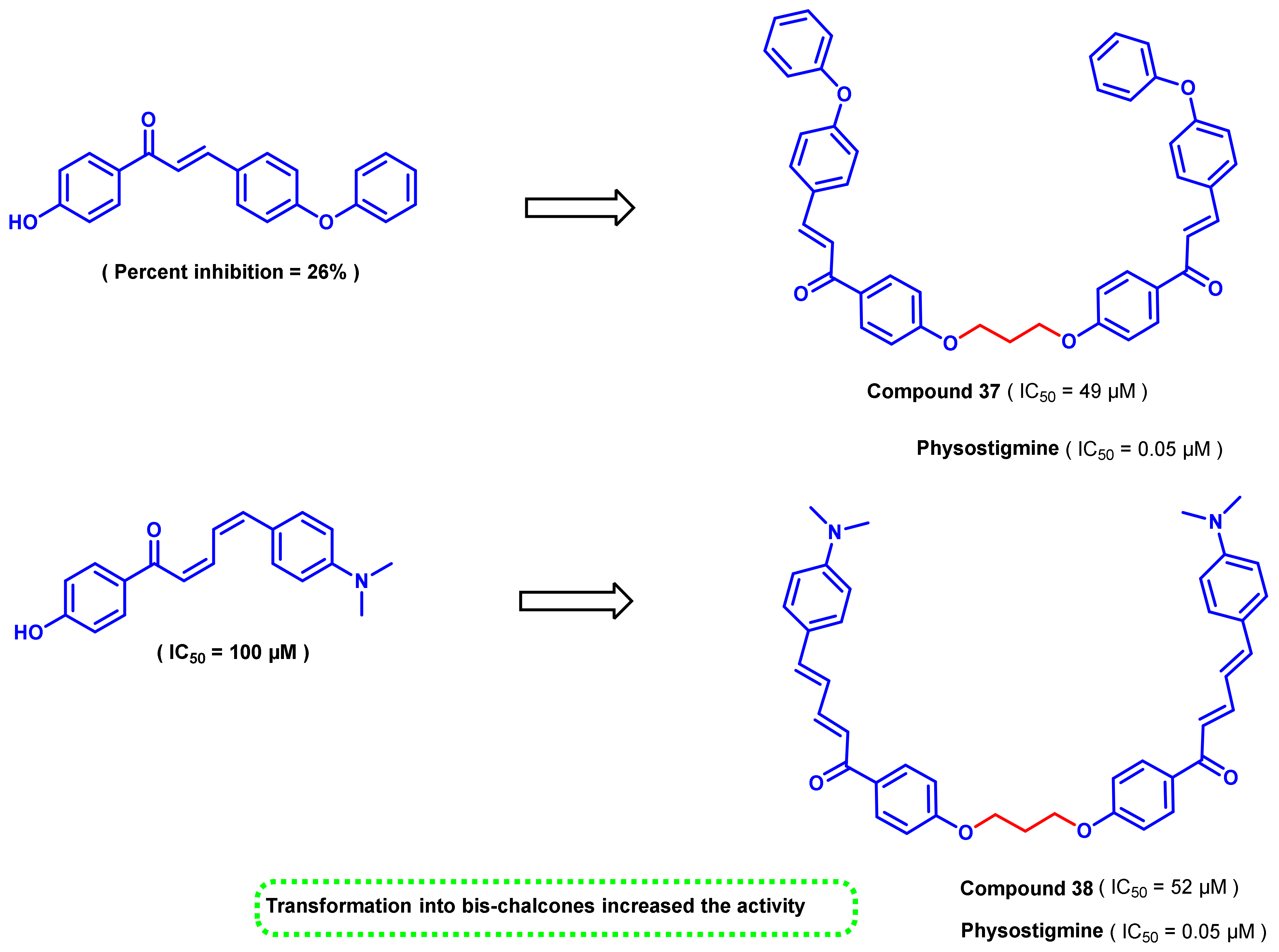
- Liargkova, T.; Hadjipavlou-Litina, D.J.; Koukoulitsa, C.; Voulgari, E.; Avgoustakis, C. Simple chalcones and bis-chalcones ethers as possible pleiotropic agents. J. Enzym. Inhib. Med. Chem. 2016, 31, 302–313. [Google Scholar] [CrossRef]
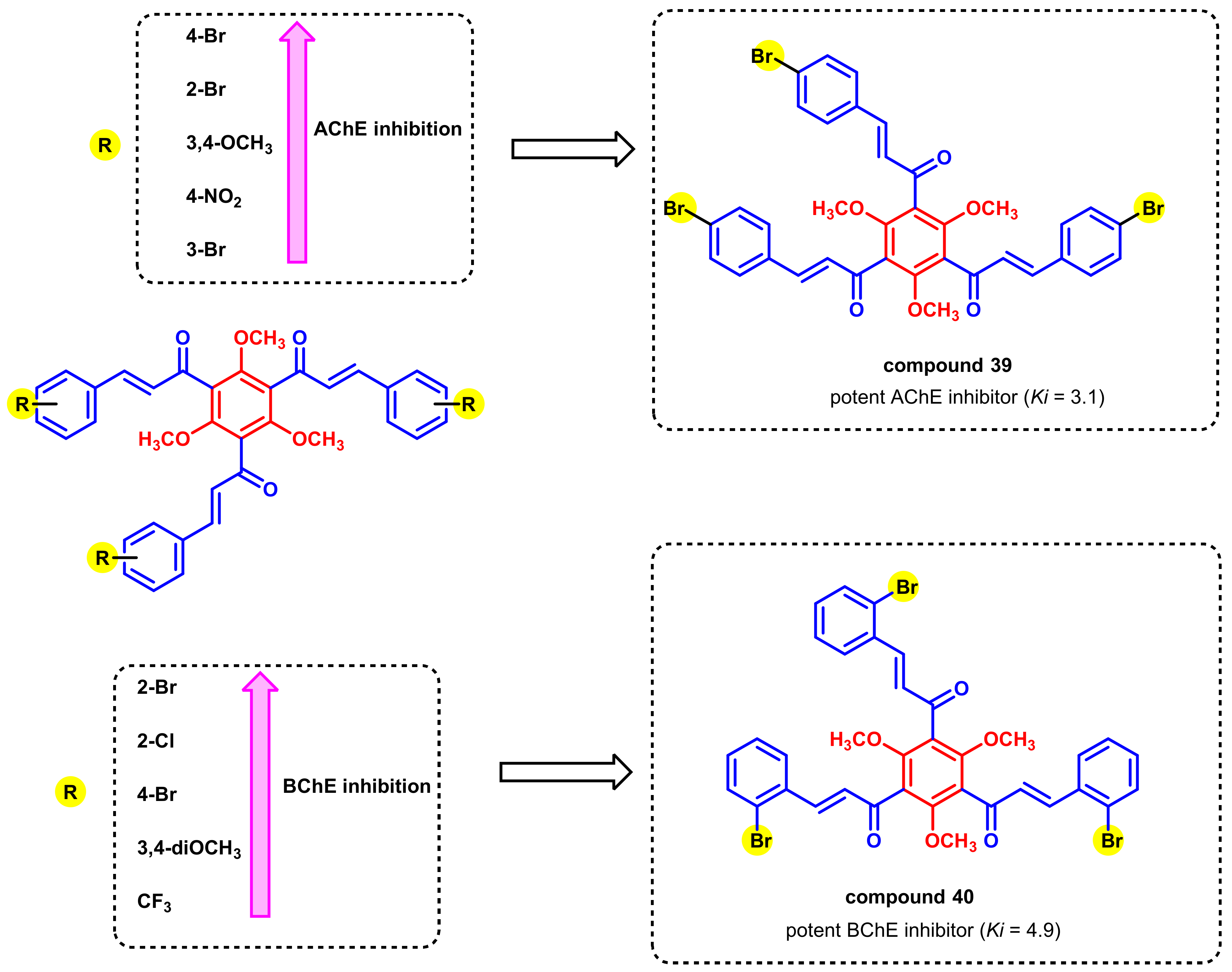
- Burmaoglu, S.; Yilmaz, A.O.; Polat, M.F.; Kaya, R.; Gulcin, I.; Algul, O. Synthesis of novel tris-chalcones and determination of their inhibition profiles against some metabolic enzymes. Arch. Physiol. Biochem. 2019, 127, 153–161. [Google Scholar] [CrossRef]
- Burmaoglu, S.; Yilmaz, A.O.; Polat, M.F.; Kaya, R.; Gulcin, I.; Algul, O. Synthesis and biological evaluation of novel tris-chalcones as potent carbonic anhydrase, acetylcholinesterase, butyrylcholinesterase and α-glycosidase inhibitors. Bioorg. Chem. 2019, 85, 191–197. [Google Scholar] [CrossRef]
- Sang, Z.; Wang, K.; Shi, J.; Liu, W.; Tan, Z. Design, synthesis, in-silico and biological evaluation of novel chalcone-O-carbamate derivatives as multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 178, 726–739. [Google Scholar] [CrossRef]
- Bag, S.; Ghosh, S.; Tulsan, R.; Sood, A.; Zhou, W.; Schifone, C.; Foster, M.; LeVine, H.; Török, B.; Török, M. Design, synthesis and biological activity of multifunctional α,β-unsaturated carbonyl scaffolds for Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2013, 23, 2614–2618. [Google Scholar] [CrossRef]
- Kang, L.; Gao, X.-H.; Liu, H.-R.; Men, X.; Wu, H.-N.; Cui, P.-W.; Oldfield, E.; Yan, J.-Y. Structure–activity relationship investigation of coumarin–chalcone hybrids with diverse side-chains as acetylcholinesterase and butyrylcholinesterase inhibitors. Mol. Divers. 2018, 22, 893–906. [Google Scholar] [CrossRef]
- Shah, M.S.; Khan, S.U.; Ejaz, S.A.; Afridi, S.; Rizvi, S.U.F.; Najam-Ul-Haq, M.; Iqbal, J. Cholinesterases inhibition and molecular modeling studies of piperidyl-thienyl and 2-pyrazoline derivatives of chalcones. Biochem. Biophys. Res. Commun. 2017, 482, 615–624. [Google Scholar] [CrossRef]
- Polo, E.; Ibarra-Arellano, N.; Prent-Peñaloza, L.; Morales-Bayuelo, A.; Henao, J.; Galdámez, A.; Gutiérrez, M. Ultrasound-assisted synthesis of novel chalcone, heterochalcone and bis-chalcone derivatives and the evaluation of their antioxidant properties and as acetylcholinesterase inhibitors. Bioorg. Chem. 2019, 90, 103034. [Google Scholar] [CrossRef]
- Shah, M.S.; Najam-Ul-Haq, M.; Shah, H.S.; Rizvi, S.U.F.; Iqbal, J. Quinoline containing chalcone derivatives as cholinesterase inhibitors and their in silico modeling studies. Comput. Biol. Chem. 2018, 76, 310–317. [Google Scholar] [CrossRef]
- Çelik, F.; Türkan, F.; Aras, A.; Atalar, M.N.; Karaman, H.S.; Ünver, Y.; Kahriman, N. Synthesis of novel 1,2,3 triazole derivatives and assessment of their potential cholinesterases, glutathione S-transferase enzymes inhibitory properties: An in vitro and in silico study. Bioorg. Chem. 2021, 107, 104606. [Google Scholar] [CrossRef]
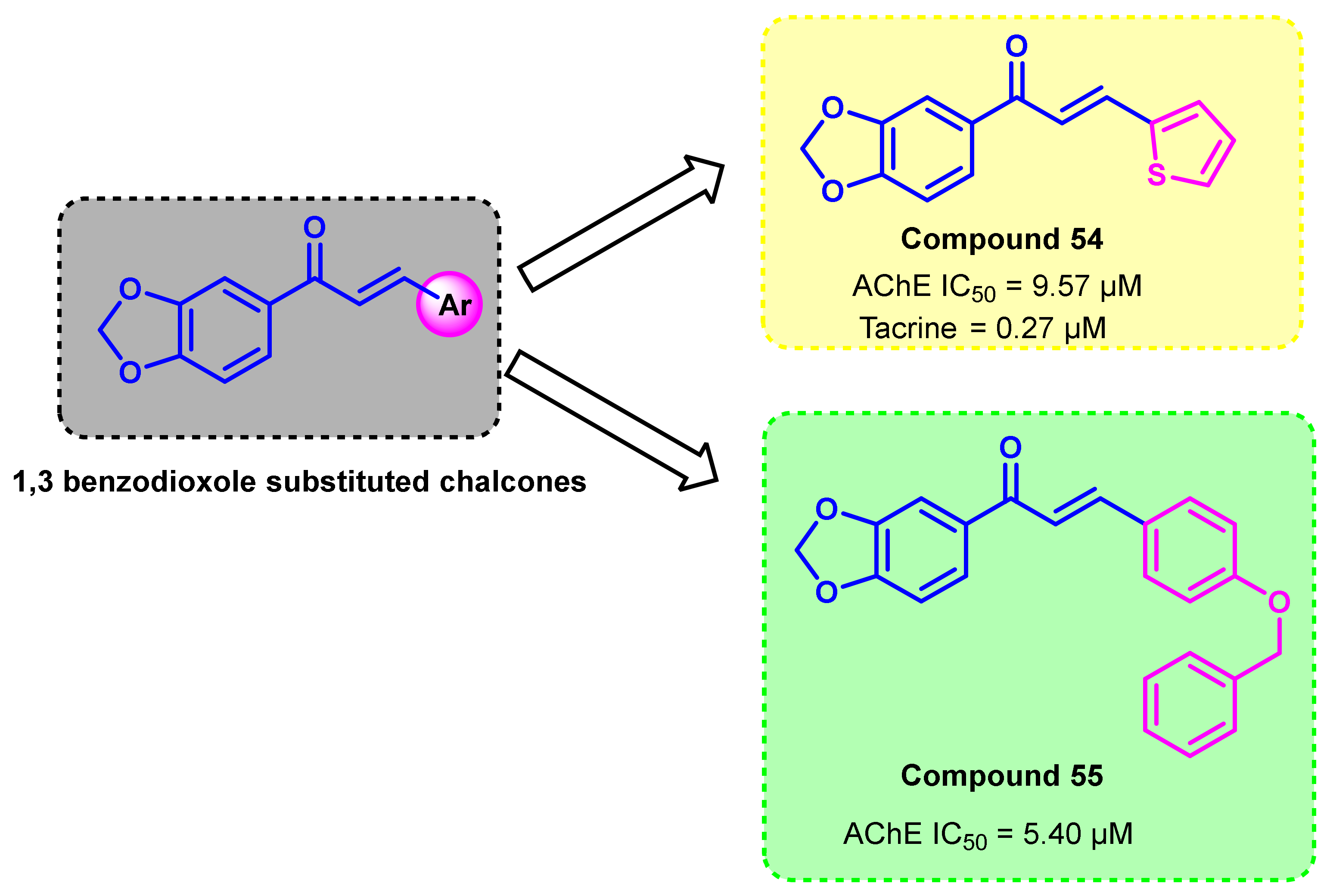
- Jeong, G.S.; Kaipakasseri, S.; Lee, S.R.; Marraiki, N.; Batiha, G.E.; Dev, S.; Palakkathondi, A.; Kavully, F.S.; Gambacorta, N.; Nicolotti, O.; et al. Selected 1,3-Benzodioxine-Containing Chalcones as Multipotent Oxidase and Acetylcholinesterase Inhibitors. ChemMedChem 2020, 15, 2257–2263. [Google Scholar] [CrossRef]
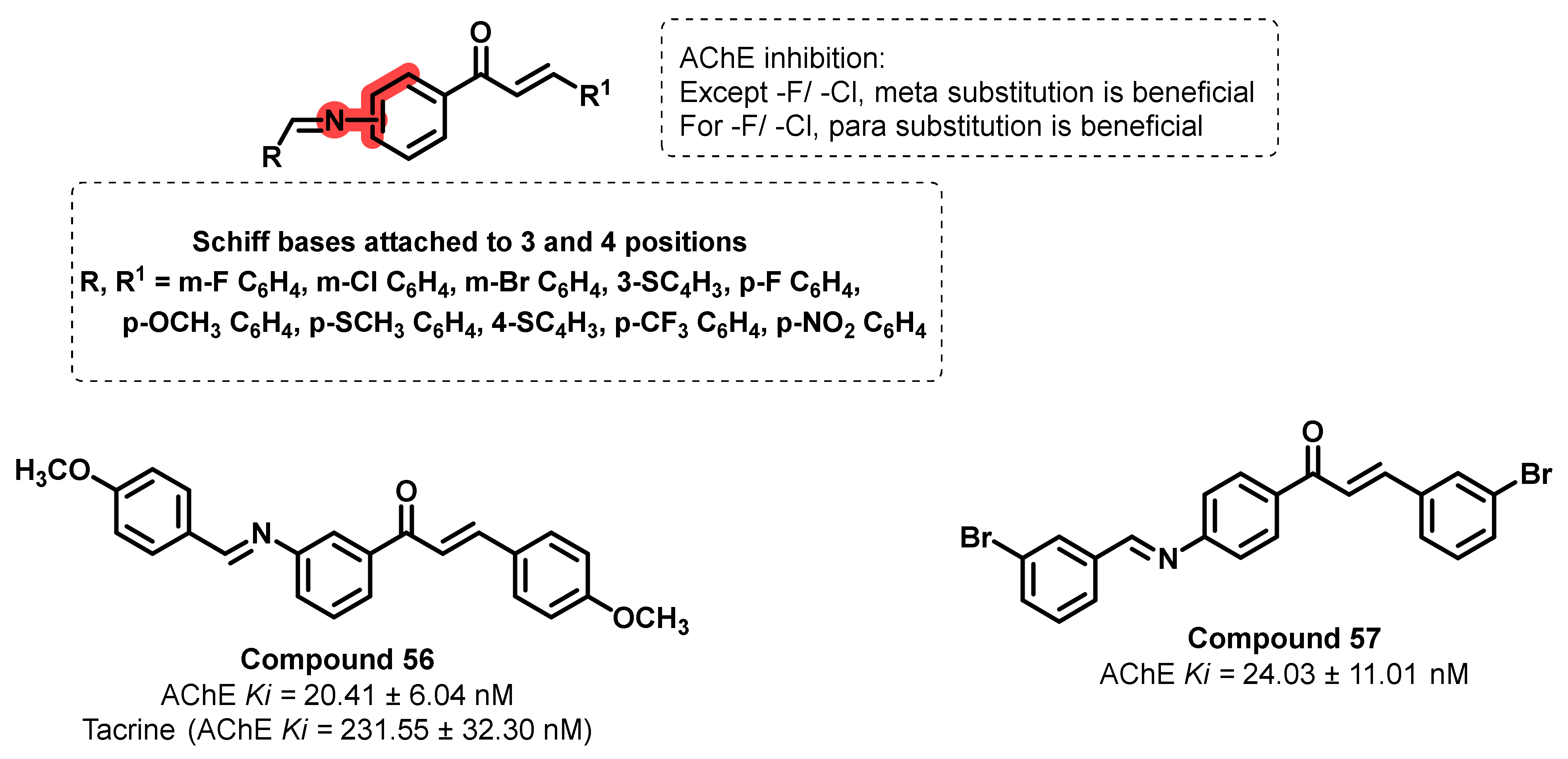
- Koçyiğit, Ü.M.; Gezegen, H.; Taslimi, P. Synthesis, characterization, and biological studies of chalcone derivatives containing Schiff bases: Synthetic derivatives for the treatment of epilepsy and Alzheimer’s disease. Arch. Pharm. 2020, 353, 2000202. [Google Scholar] [CrossRef]
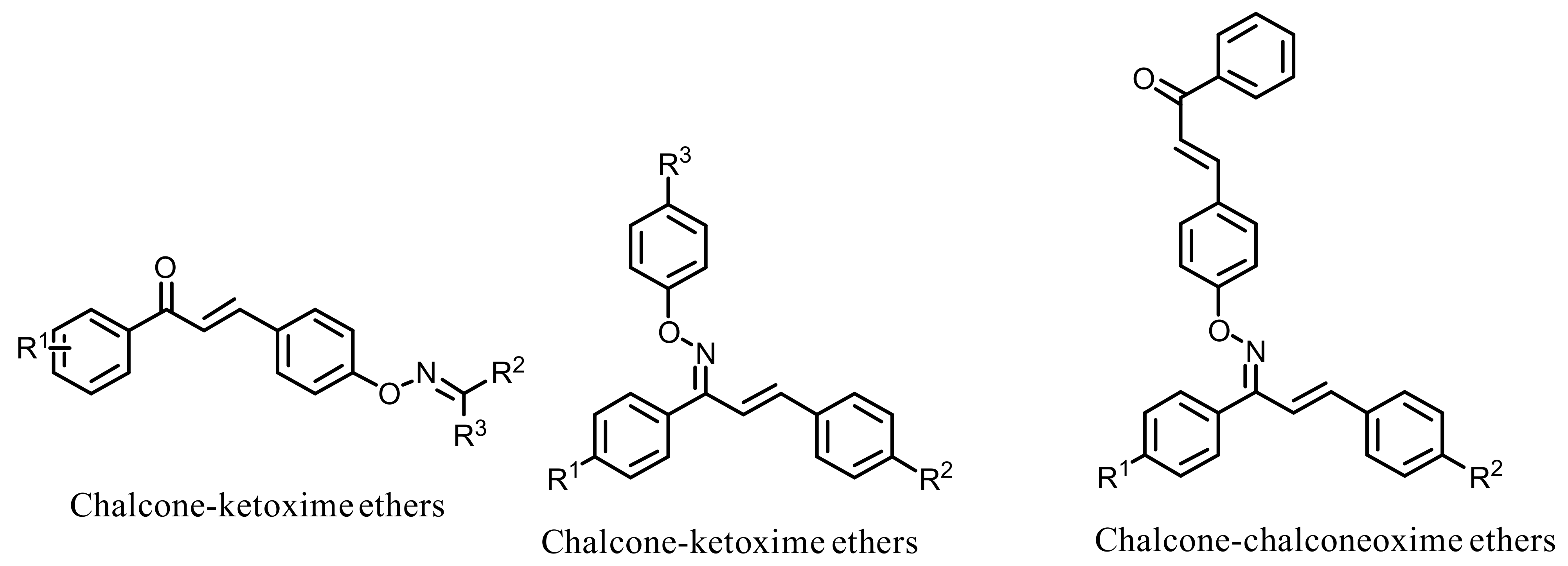
- Oh, J.M.; Rangarajan, T.M.; Chaudhary, R.; Singh, R.P.; Singh, M.; Singh, R.P.; Tondo, A.R.; Gambacorta, N.; Nicolotti, O.; Mathew, B.; et al. Novel Class of Chalcone Oxime Ethers as Potent Monoamine Oxidase-B and Acetylcholinesterase Inhibitors. Molecules 2020, 25, 2356. [Google Scholar] [CrossRef]
















































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
George, G.; Koyiparambath, V.P.; Sukumaran, S.; Nair, A.S.; Pappachan, L.K.; Al-Sehemi, A.G.; Kim, H.; Mathew, B. Structural Modifications on Chalcone Framework for Developing New Class of Cholinesterase Inhibitors. Int. J. Mol. Sci. 2022, 23, 3121. https://doi.org/10.3390/ijms23063121
George G, Koyiparambath VP, Sukumaran S, Nair AS, Pappachan LK, Al-Sehemi AG, Kim H, Mathew B. Structural Modifications on Chalcone Framework for Developing New Class of Cholinesterase Inhibitors. International Journal of Molecular Sciences. 2022; 23(6):3121. https://doi.org/10.3390/ijms23063121
Chicago/Turabian StyleGeorge, Ginson, Vishal Payyalot Koyiparambath, Sunitha Sukumaran, Aathira Sujathan Nair, Leena K. Pappachan, Abdullah G. Al-Sehemi, Hoon Kim, and Bijo Mathew. 2022. "Structural Modifications on Chalcone Framework for Developing New Class of Cholinesterase Inhibitors" International Journal of Molecular Sciences 23, no. 6: 3121. https://doi.org/10.3390/ijms23063121
APA StyleGeorge, G., Koyiparambath, V. P., Sukumaran, S., Nair, A. S., Pappachan, L. K., Al-Sehemi, A. G., Kim, H., & Mathew, B. (2022). Structural Modifications on Chalcone Framework for Developing New Class of Cholinesterase Inhibitors. International Journal of Molecular Sciences, 23(6), 3121. https://doi.org/10.3390/ijms23063121