
Multitargeting the Action of 5-HT6 Serotonin Receptor Ligands by Additional Modulation of Kinases in the Search for a New Therapy for Alzheimer’s Disease: Can It Work from a Molecular Point of View?

 ,
,  and
and 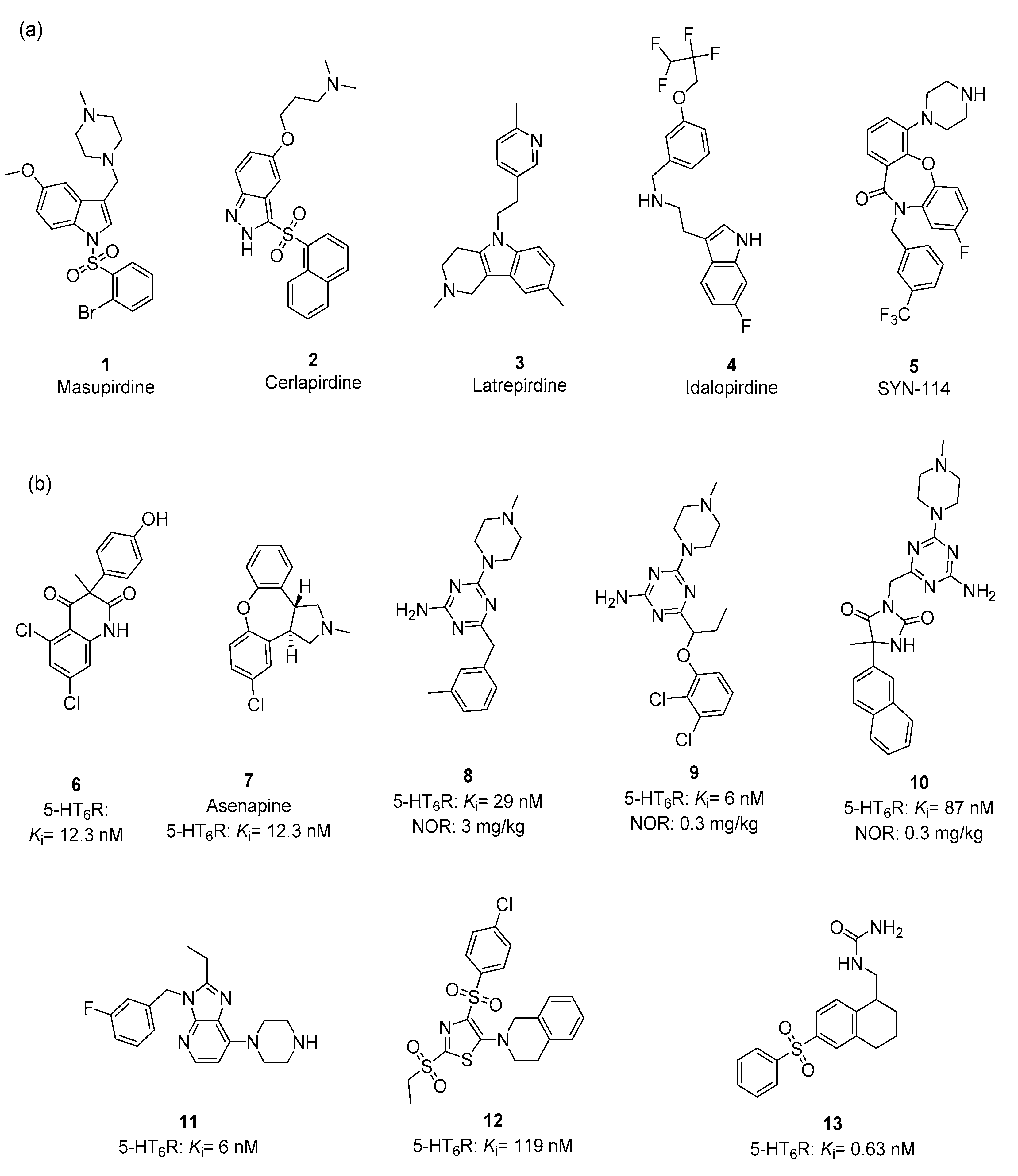{kind=link}
{kind=link}
{kind=link}
{kind=link}
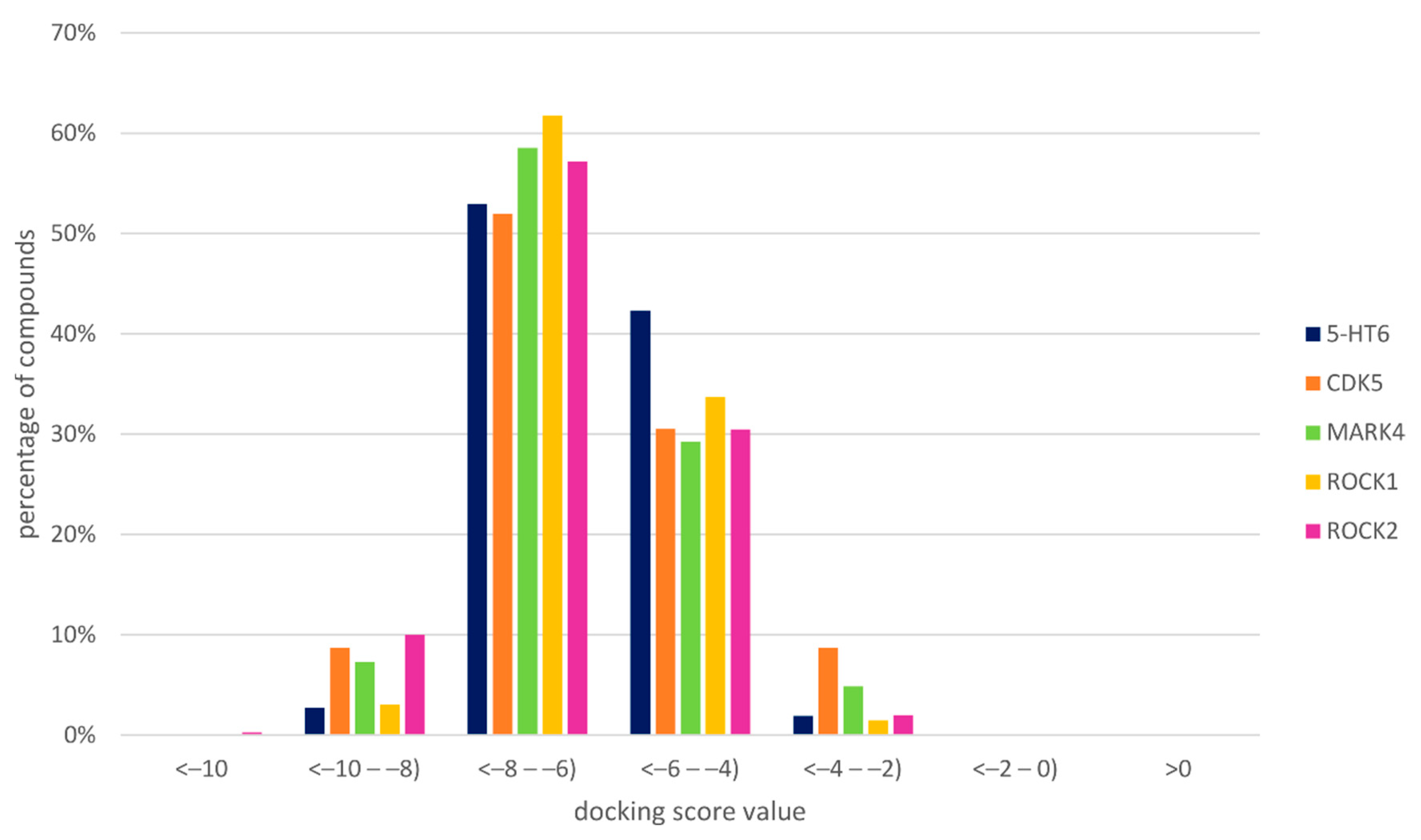{kind=link}
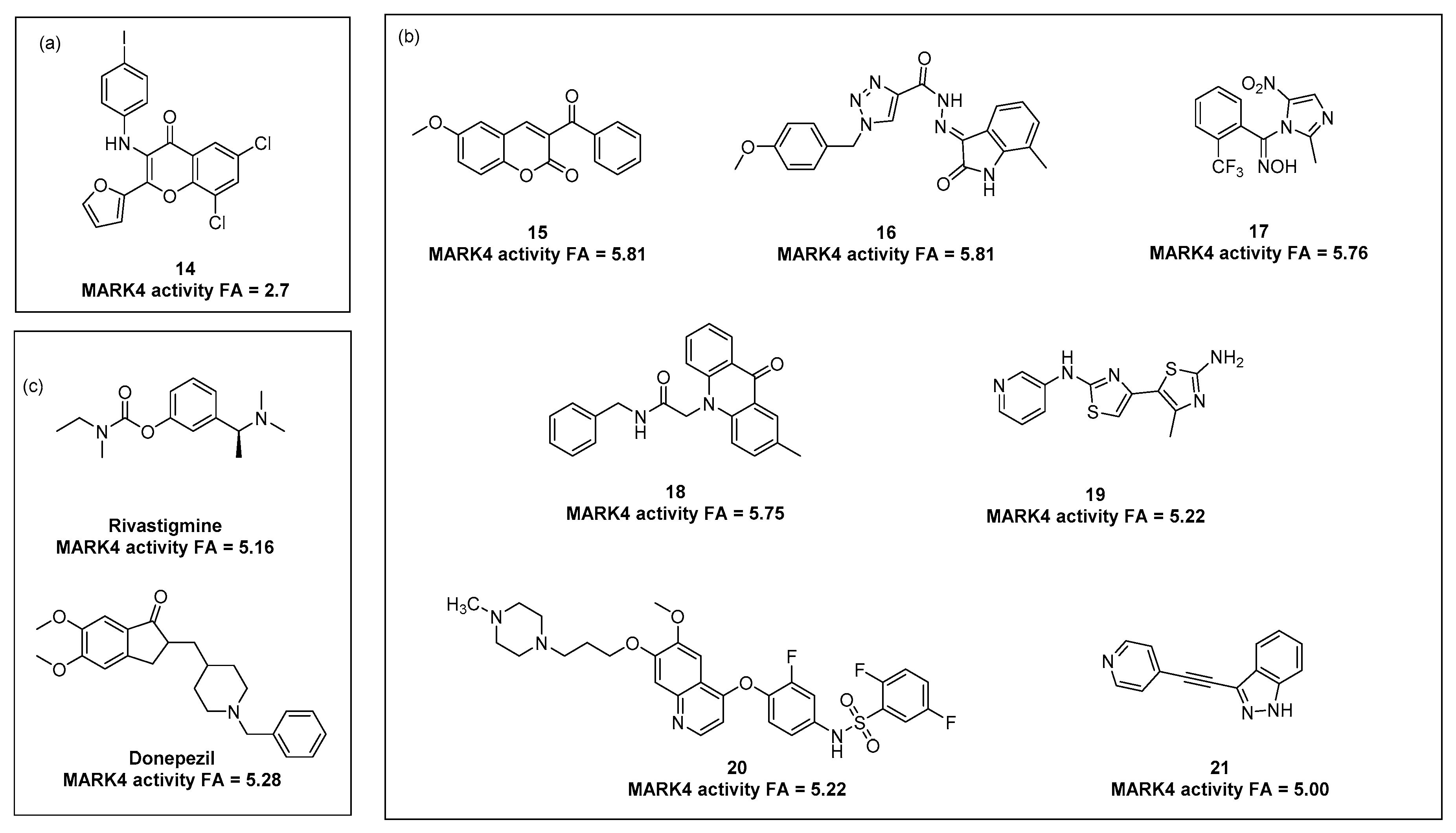{kind=link}
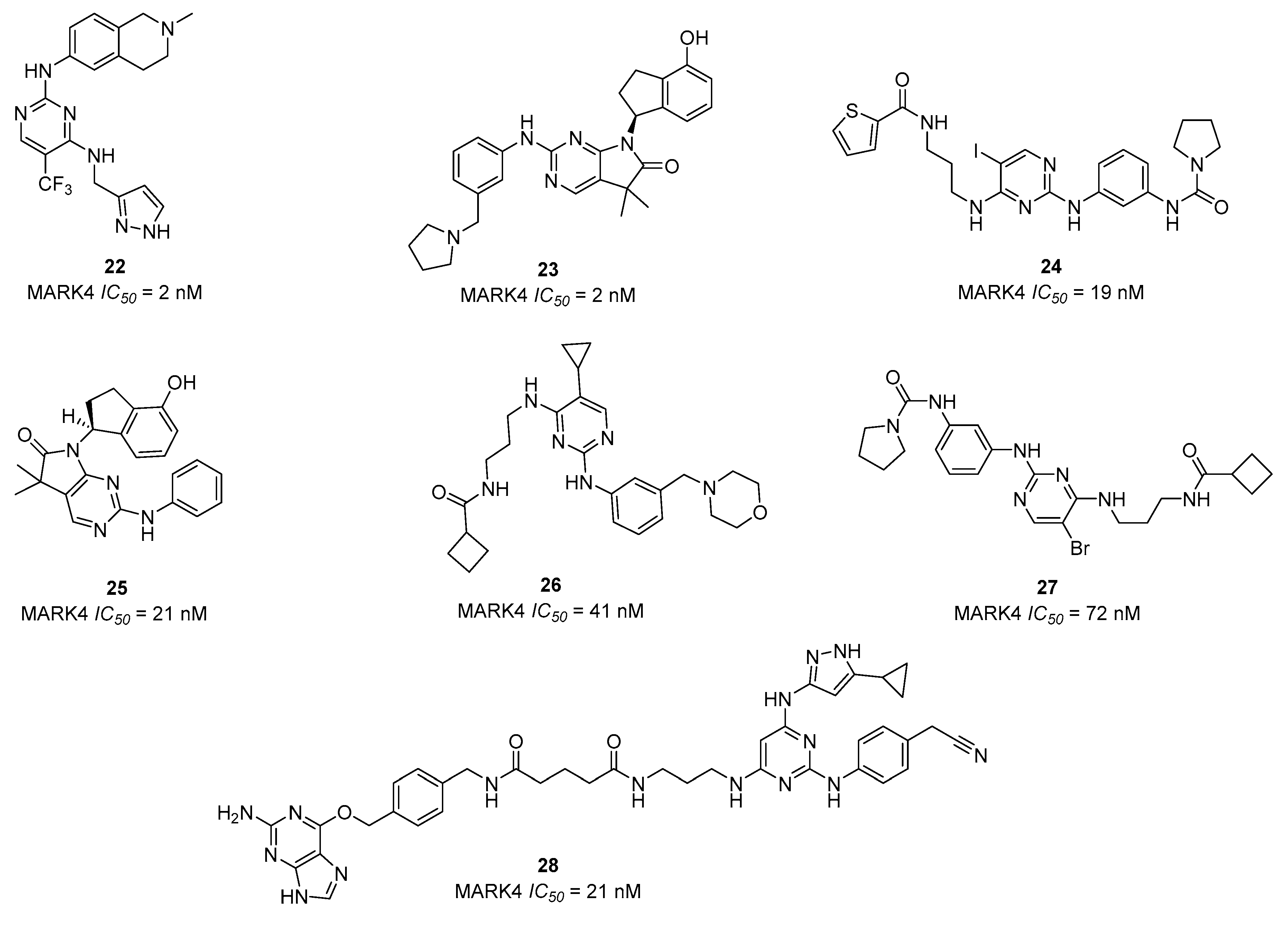{kind=link}
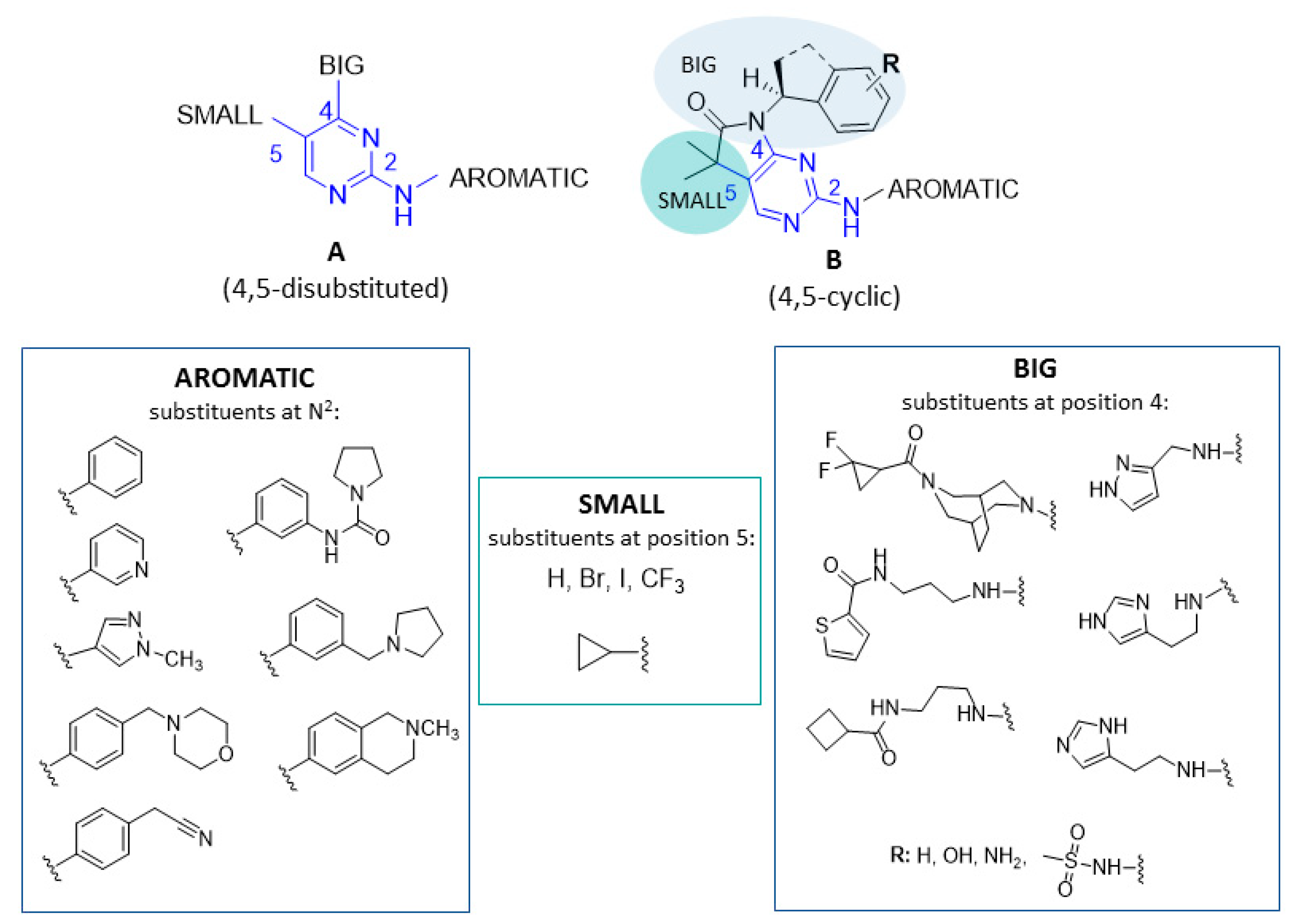{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. 5-HT6R Ligands—Pharmacophore Features
2.1. Chemical Variety of 5-HT6R Ligands
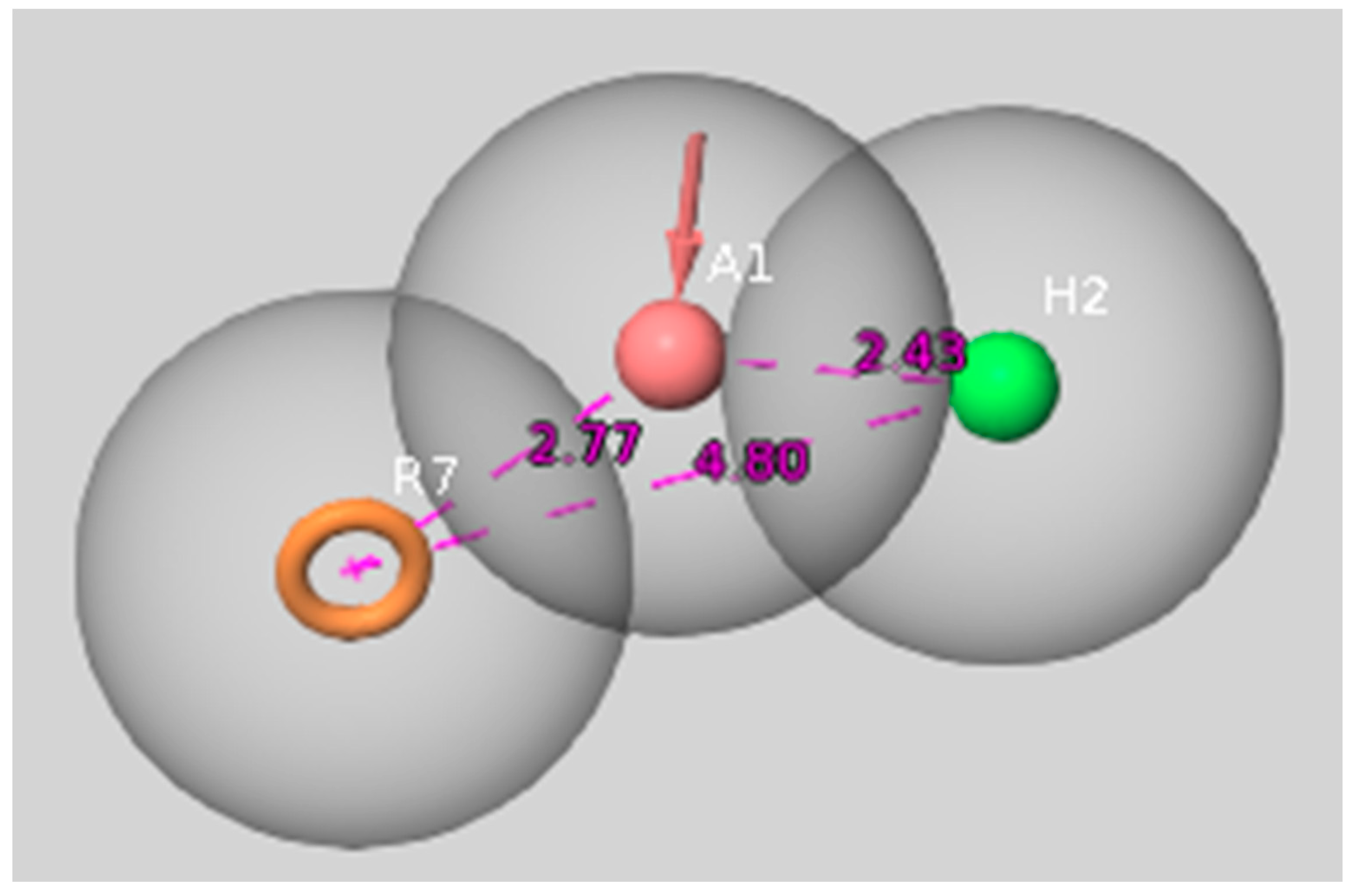
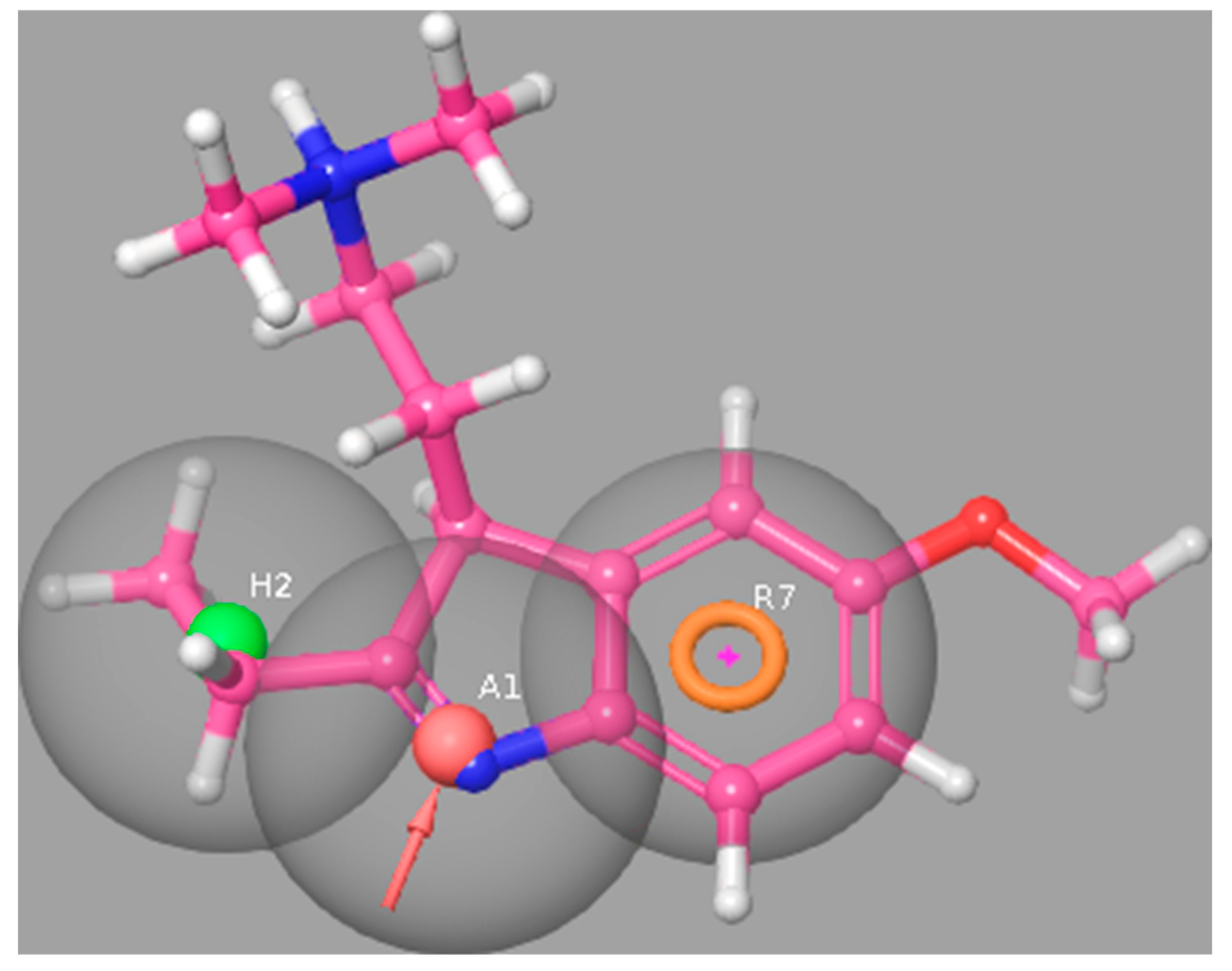
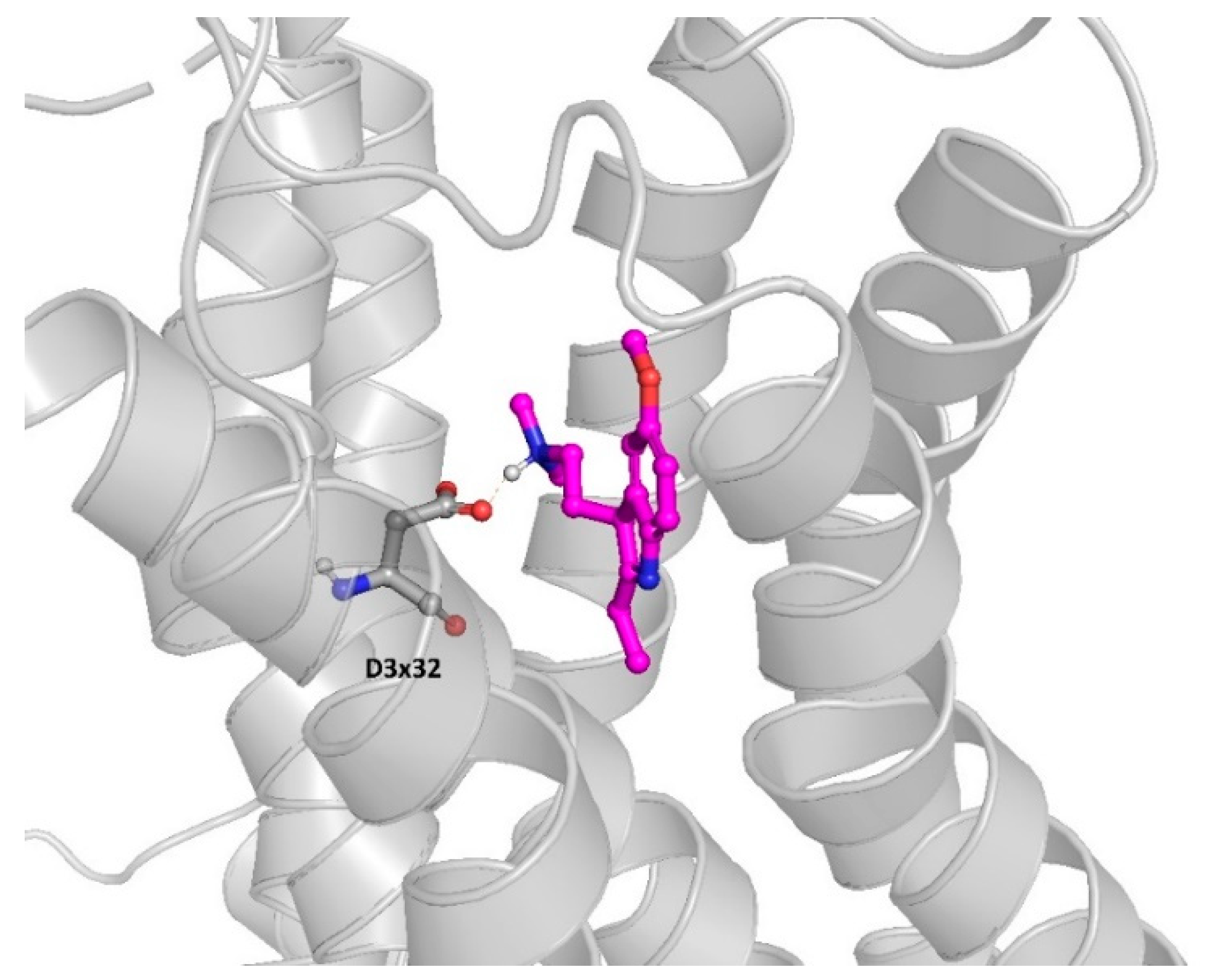
2.2. Molecular Modeling Approaches to Evaluate the Potential 5-HT6R Compound Activity
3. 5-HT6R/MARK4 as Dual Target Approach in Search for Therapeutic Solution against AD
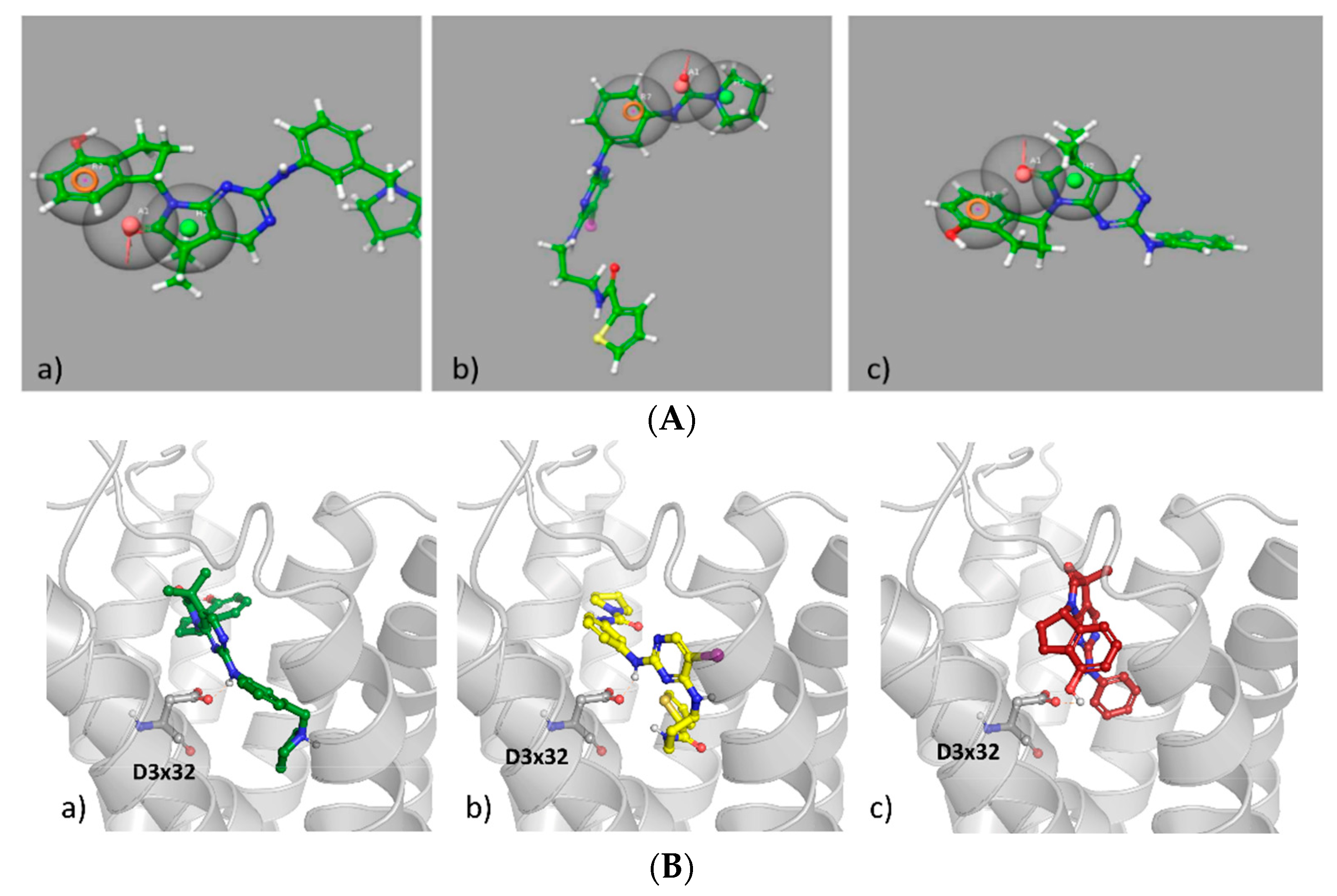
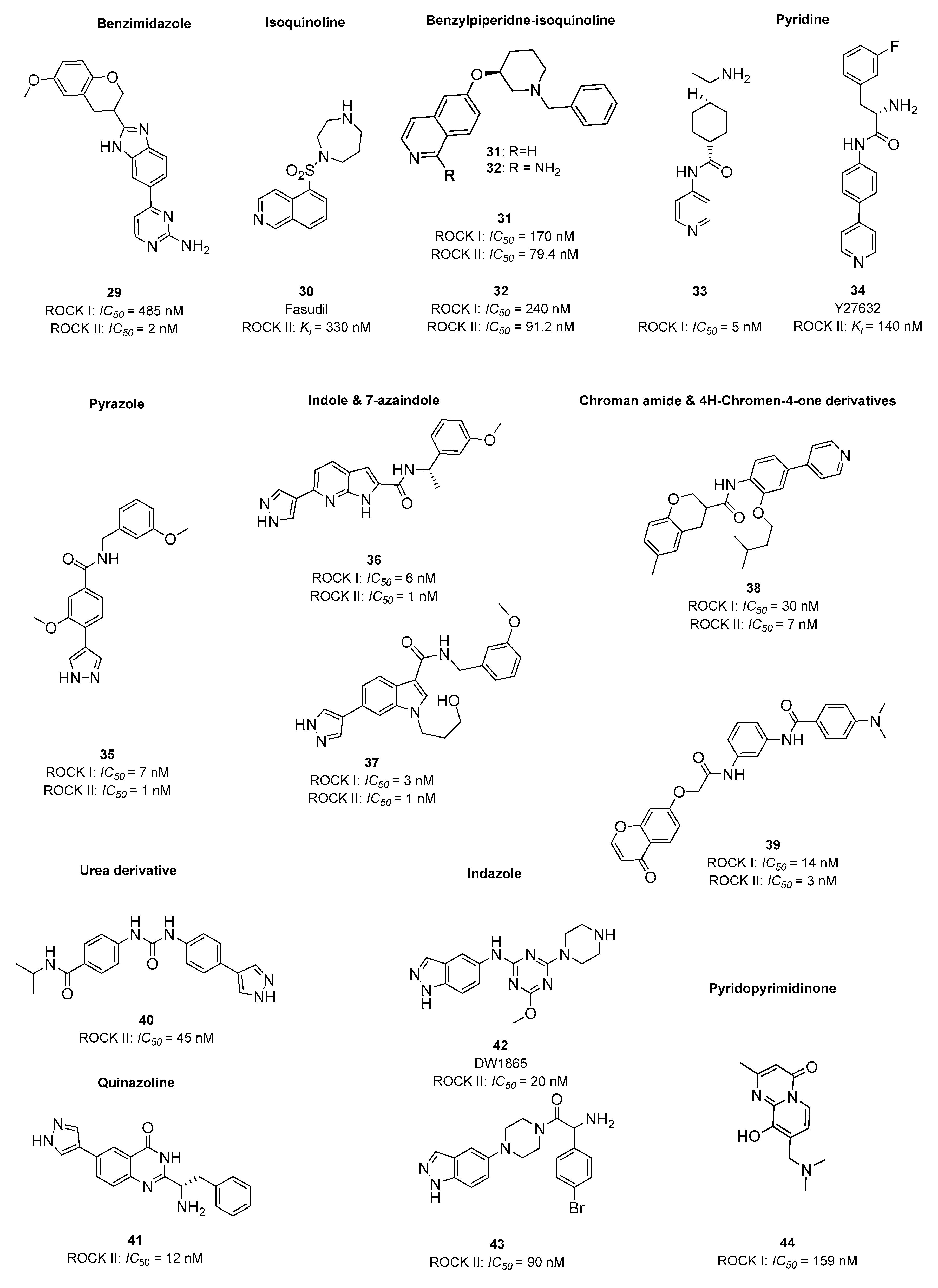
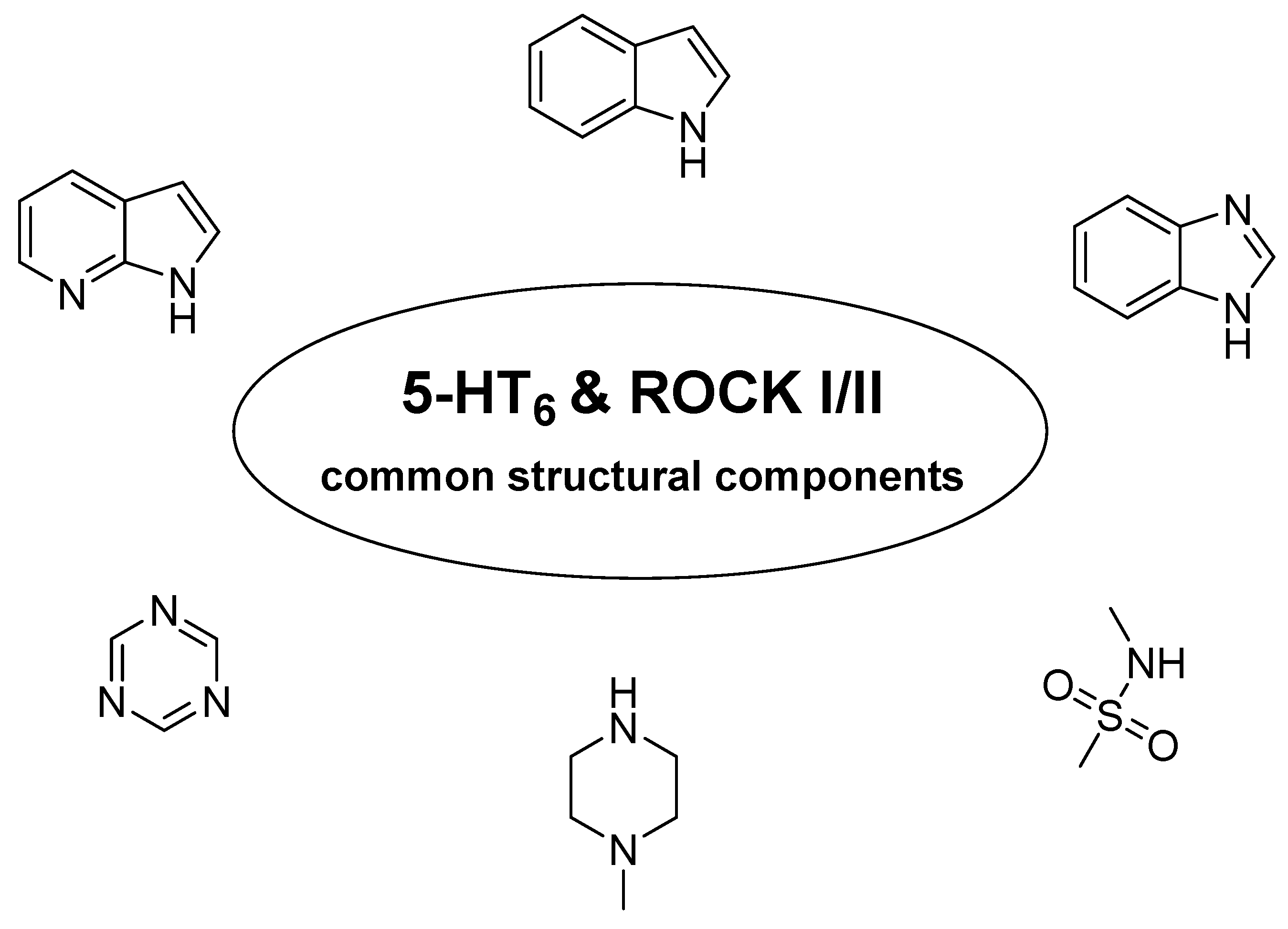
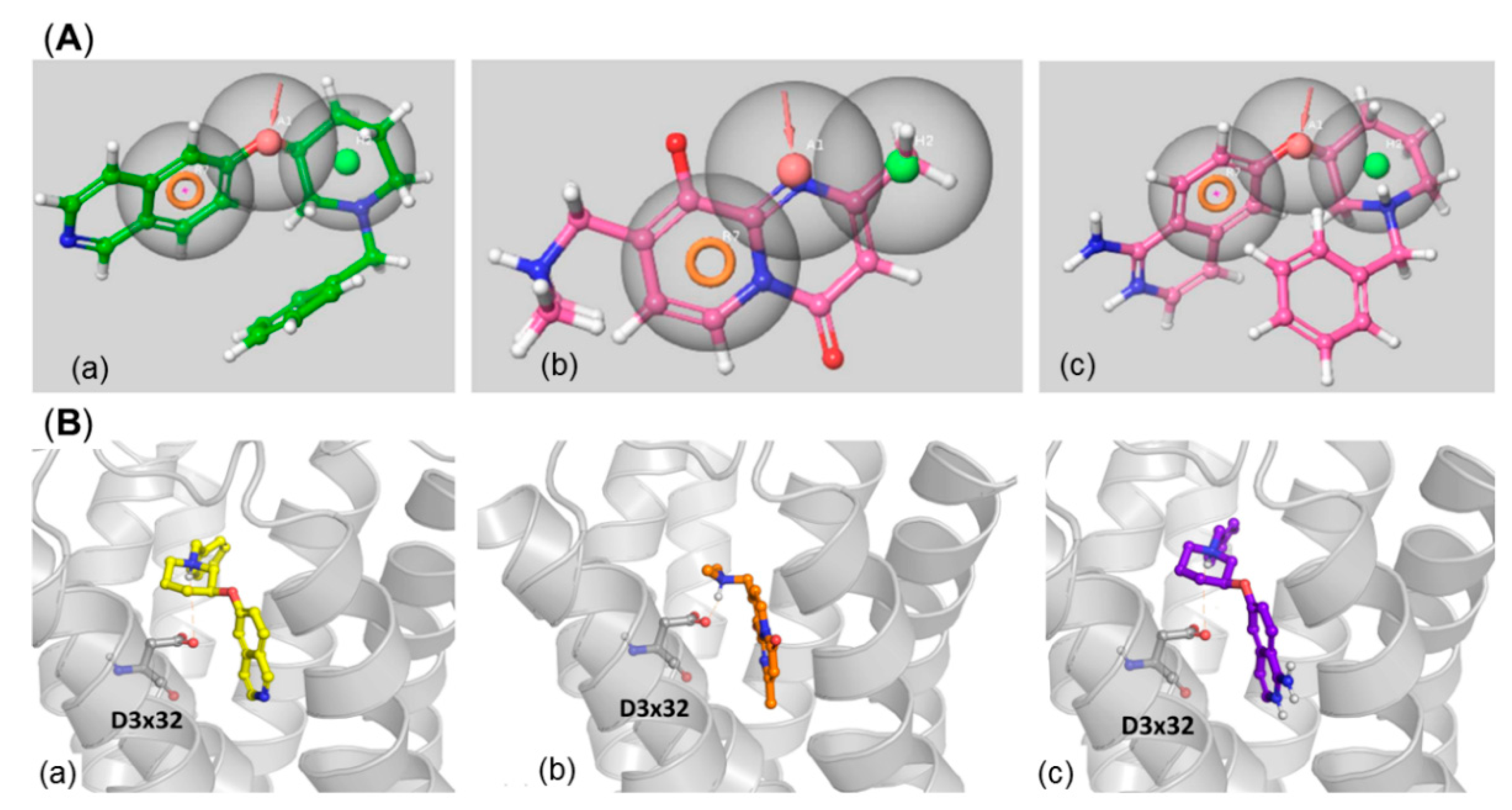
4. 5-HT6R/ROCKI/ROCKII as Multitarget Approach to AD Therapy
5. 5-HT6R/CDK5 as Possible Dual Target Approach in Search for Innovative Therapy
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blandr, J. Mild Cognitive Impairment, Neurodegeneration, and Personalized Lifestyle Medicine. Integr. Med. 2016, 15, 12–14. [Google Scholar]
- da Silva, E.R.; Rodrigues Menezes, I.R.; Brys, I. Effects of Transcranial Direct Current Stimulation on Memory of Elderly People with Mild Cognitive Impairment or Alzheimer’s Disease: A Systematic Review. J. Cent. Nerv. Syst. Dis. 2022, 14, 117957352211068. [Google Scholar] [CrossRef] [PubMed]
- García-Casares, N.; Fuentes, P.G.; Barbancho, M.Á.; López-Gigosos, R.; García-Rodríguez, A.; Gutiérrez-Bedmar, M. Alzheimer’s Disease, Mild Cognitive Impairment and Mediterranean Diet. A Systematic Review and Dose-Response Meta-Analysis. J. Clin. Med. 2021, 10, 4642. [Google Scholar] [CrossRef] [PubMed]
- Association, A. 2018 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Zvěřová, M. Clinical Aspects of Alzheimer’s Disease. Clin. Biochem. 2019, 72, 3–6. [Google Scholar] [CrossRef]
- Srivastava, S.; Ahmad, R.; Khare, S.K. Alzheimer’s Disease and Its Treatment by Different Approaches: A Review. Eur. J. Med. Chem. 2021, 216, 113320. [Google Scholar] [CrossRef]
- Pardo-Moreno, T.; González-Acedo, A.; Rivas-Domínguez, A.; García-Morales, V.; García-Cozar, F.J.; Ramos-Rodríguez, J.J.; Melguizo-Rodríguez, L. Therapeutic Approach to Alzheimer’s Disease: Current Treatments and New Perspectives. Pharmaceutics 2022, 14, 1117. [Google Scholar] [CrossRef]
- Beshir, S.A.; Aadithsoorya, A.M.; Parveen, A.; Goh, S.S.L.; Hussain, N.; Menon, V.B. Aducanumab Therapy to Treat Alzheimer’s Disease: A Narrative Review. Int. J. Alzheimer’s Dis. 2022, 2022, 9343514. [Google Scholar] [CrossRef]
- Whitehouse, P.; Gandy, S.; Saini, V.; George, D.R.; Larson, E.B.; Alexander, G.C.; Avorn, J.; Brownlee, S.; Camp, C.; Chertkow, H.; et al. Making the Case for Accelerated Withdrawal of Aducanumab. J. Alzheimer’s Dis. 2022, 87, 1003–1007. [Google Scholar] [CrossRef]
- Gunawardena, I.P.C.; Retinasamy, T.; Shaikh, M.F. Is Aducanumab for LMICs? Promises and Challenges. Brain Sci. 2021, 11, 1547. [Google Scholar] [CrossRef]
- Karila, D.; Freret, T.; Bouet, V.; Boulouard, M.; Dallemagne, P.; Rochais, C. Therapeutic Potential of 5-HT6 Receptor Agonists. J. Med. Chem. 2015, 58, 7901–7912. [Google Scholar] [CrossRef] [PubMed]
- Benhamú, B.; Martín-Fontecha, M.; Vázquez-Villa, H.; Pardo, L.; López-Rodríguez, M.L. Serotonin 5-HT6receptor Antagonists for the Treatment of Cognitive Deficiency in Alzheimer’s Disease. J. Med. Chem. 2014, 57, 7160–7181. [Google Scholar] [CrossRef] [PubMed]
- Khoury, R.; Grysman, N.; Gold, J.; Patel, K.; Grossberg, G.T. The Role of 5 HT6-Receptor Antagonists in Alzheimer’s Disease: An Update. Expert Opin. Investig. Drugs 2018, 27, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Kucwaj-Brysz, K.; Baltrukevich, H.; Czarnota, K.; Handzlik, J. Chemical Update on the Potential for Serotonin 5-HT6 and 5-HT7 Receptor Agents in the Treatment of Alzheimer’s Disease. Bioorg. Med. Chem. Lett. 2021, 49, 128275. [Google Scholar] [CrossRef]
- Ferrari, C.; Sorbi, S. The Complexity of Alzheimer’s Disease: An Evolving Puzzle. Physiol Rev. 2021, 101, 1047–1081. [Google Scholar] [CrossRef]
- Sampietro, A.; Pérez-Areales, F.J.; Martínez, P.; Arce, E.M.; Galdeano, C.; Muñoz-Torrero, D. Unveiling the Multitarget Anti-Alzheimer Drug Discovery Landscape: A Bibliometric Analysis. Pharmaceuticals 2022, 15, 545. [Google Scholar] [CrossRef]
- Zięba, A.; Stępnicki, P.; Matosiuk, D.; Kaczor, A.A. What Are the Challenges with Multi-Targeted Drug Design for Complex Diseases? Expert Opin. Drug Discov. 2022, 17, 673–683. [Google Scholar] [CrossRef]
- Kabir, A.; Muth, A. Polypharmacology: The Science of Multi-Targeting Molecules. Pharmacol. Res. 2022, 176, 106055. [Google Scholar] [CrossRef]
- Wiȩckowska, A.; Wichur, T.; Godyń, J.; Bucki, A.; Marcinkowska, M.; Siwek, A.; Wiȩckowski, K.; Zarȩba, P.; Knez, D.; Głuch-Lutwin, M.; et al. Novel Multitarget-Directed Ligands Aiming at Symptoms and Causes of Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 1195–1214. [Google Scholar] [CrossRef]
- Millan, M.J.; Dekeyne, A.; Gobert, A.; Brocco, M.; Mannoury la Cour, C.; Ortuno, J.C.; Watson, D.; Fone, K.C.F. Dual-Acting Agents for Improving Cognition and Real-World Function in Alzheimer’s Disease: Focus on 5-HT6 and D3 Receptors as Hubs. Neuropharmacology 2020, 177, 108099. [Google Scholar] [CrossRef]
- Yahiaoui, S.; Hamidouche, K.; Ballandonne, C.; Davis, A.; De Oliveira Santos, J.S.; Freret, T.; Boulouard, M.; Rochais, C.; Dallemagne, P. Design, Synthesis, and Pharmacological Evaluation of Multitarget-Directed Ligands with Both Serotonergic Subtype 4 Receptor (5-HT4R) Partial Agonist and 5-HT6R Antagonist Activities, as Potential Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2016, 121, 283–293. [Google Scholar] [CrossRef]
- López-Rodríguez, M.L.; Benhamú, B.; de la Fuente, T.; Sanz, A.; Pardo, L.; Campillo, M. A Three-Dimensional Pharmacophore Model for 5-Hydroxytryptamine 6 (5-HT6) Receptor Antagonists. J. Med. Chem. 2005, 48, 4216–4219. [Google Scholar] [CrossRef]
- Nirogi, R.; Goyal, V.K.; Bhyrapuneni, G.; Palacharla, V.R.C.; Ravulu, J.; Jetta, S.; Jasti, V. Masupirdine in Combination with Donepezil and Memantine in Patients with Moderate Alzheimer’s Disease: Subgroup Analyses of Memantine Regimen, Plasma Concentrations and Duration of Treatment. Alzheimer’s Dement. 2020, 16, e039254. [Google Scholar] [CrossRef]
- Nirogi, R.; Jayarajan, P.; Shinde, A.K.; Abraham, R.; Goyal, V.K.; Benade, V.; Ravulu, J.; Jasti, V. Masupirdine (SUVN-502): Novel Treatment Option for the Management of Behavioral and Psychological Symptoms in Patients with Alzheimer’s Disease. Alzheimer’s Dement. 2020, 16, e039303. [Google Scholar] [CrossRef]
- Study Evaluating the Safety, Tolerability, PK and PD of SAM-531 in the Subjects with Mild to Moderate Alzheimer’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT00481520 (accessed on 20 July 2022).
- Bezprozvanny, I. The Rise and Fall of Dimebon. Drug News Perspect. 2010, 23, 518. [Google Scholar] [CrossRef] [PubMed]
- Okun, I.; Tkachenko, S.; Khvat, A.; Mitkin, O.; Kazey, V.; Ivachtchenko, A. From Anti-Allergic to Anti-Alzheimer’s: Molecular Pharmacology of Dimebon™. Curr. Alzheimer Res. 2010, 7, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Eckert, S.H.; Gaca, J.; Kolesova, N.; Friedland, K.; Eckert, G.P.; Muller, W.E. Mitochondrial Pharmacology of Dimebon (Latrepirdine) Calls for a New Look at Its Possible Therapeutic Potential in Alzheimer’s Disease. Aging Dis. 2018, 9, 729. [Google Scholar] [CrossRef]
- Chau, S.; Herrmann, N.; Ruthirakuhan, M.T.; Chen, J.J.; Lanctôt, K.L. Latrepirdine for Alzheimer’s Disease. Cochrane Database Syst. Rev. 2015, 4, CD009524. [Google Scholar] [CrossRef]
- Andrews, M.; Tousi, B.; Sabbagh, M.N. 5HT6 Antagonists in the Treatment of Alzheimer’s Dementia: Current Progress. Neurol. Ther. 2018, 7, 51–58. [Google Scholar] [CrossRef]
- Herrik, K.F.; Mørk, A.; Richard, N.; Bundgaard, C.; Bastlund, J.F.; de Jong, I.E.M. The 5-HT 6 Receptor Antagonist Idalopirdine Potentiates the Effects of Acetylcholinesterase Inhibition on Neuronal Network Oscillations and Extracellular Acetylcholine Levels in the Rat Dorsal Hippocampus. Neuropharmacology 2016, 107, 351–363. [Google Scholar] [CrossRef]
- Matsunaga, S.; Fujishiro, H.; Takechi, H. Efficacy and Safety of Idalopirdine for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Int. Psychogeriatr. 2019, 31, 1627–1633. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.N., III; Kress, J.M.; Repke, D.B.; Stabler, R.S. Dibenzoxazepinone Derivatives as 5-HT6 and 5-HT2A Receptor Antagonists, Their Preparation, Pharmaceutical Compositions, and Use in Therapy. WO2006061126A2, 15 June 2006. [Google Scholar]
- Seong, C.M.; Park, W.K.; Park, C.M.; Kong, J.Y.; Park, N.S. Discovery of 3-Aryl-3-Methyl-1H-Quinoline-2,4-Diones as a New Class of Selective 5-HT6 Receptor Antagonists. Bioorg. Med. Chem. Lett. 2008, 18, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Shahid, M.; Walker, G.; Zorn, S.; Wong, E. Asenapine: A Novel Psychopharmacologic Agent with a Unique Human Receptor Signature. J. Psychopharmacol. 2009, 23, 65–73. [Google Scholar] [CrossRef]
- Łażewska, D.; Kurczab, R.; Więcek, M.; Kamińska, K.; Satała, G.; Jastrzębska-Więsek, M.; Partyka, A.; Bojarski, A.J.; Wesołowska, A.; Kieć-Kononowicz, K.; et al. The Computer-Aided Discovery of Novel Family of the 5-HT6 Serotonin Receptor Ligands among Derivatives of 4-Benzyl-1,3,5-Triazine. Eur. J. Med. Chem. 2017, 135, 117–124. [Google Scholar] [CrossRef]
- Latacz, G.; Lubelska, A.; Jastrzębska-Więsek, M.; Partyka, A.; Marć, M.A.; Satała, G.; Wilczyńska, D.; Kotańska, M.; Więcek, M.; Kamińska, K.; et al. The 1,3,5-Triazine Derivatives as Innovative Chemical Family of 5-HT6 Serotonin Receptor Agents with Therapeutic Perspectives for Cognitive Impairment. Int. J. Mol. Sci. 2019, 20, 3420. [Google Scholar] [CrossRef] [PubMed]
- Sudoł, S.; Cios, A.; Jastrzębska-Więsek, M.; Honkisz-Orzechowska, E.; Mordyl, B.; Wilczyńska-Zawal, N.; Satała, G.; Kucwaj-Brysz, K.; Partyka, A.; Latacz, G.; et al. The Phenoxyalkyltriazine Antagonists for 5-HT6 Receptor with Promising Procognitive and Pharmacokinetic Properties In Vivo in Search for a Novel Therapeutic Approach to Dementia Diseases. Int. J. Mol. Sci. 2021, 22, 10773. [Google Scholar] [CrossRef]
- Sudoł, S.; Kucwaj-Brysz, K.; Kurczab, R.; Wilczyńska, N.; Jastrzębska-Więsek, M.; Satała, G.; Latacz, G.; Głuch-Lutwin, M.; Mordyl, B.; Żesławska, E.; et al. Chlorine Substituents and Linker Topology as Factors of 5-HT6R Activity for Novel Highly Active 1,3,5-Triazine Derivatives with Procognitive Properties in Vivo. Eur. J. Med. Chem. 2020, 203, 112529. [Google Scholar] [CrossRef] [PubMed]
- Lubelska, A.; Latacz, G.; Jastrzębska-Więsek, M.; Kotańska, M.; Kurczab, R.; Partyka, A.; Marć, M.A.; Wilczyńska, D.; Doroz-Płonka, A.; Łażewska, D.; et al. Are the Hydantoin-1,3,5-Triazine 5-HT6R Ligands a Hope to a Find New Procognitive and Anti-Obesity Drug? Considerations Based on Primary In Vivo Assays and ADME-Tox Profile In Vitro. Molecules 2019, 24, 4472. [Google Scholar] [CrossRef]
- Kurczab, R.; Ali, W.; Łażewska, D.; Kotańska, M.; Jastrzębska-Więsek, M.; Satała, G.; Więcek, M.; Lubelska, A.; Latacz, G.; Partyka, A.; et al. Computer-Aided Studies for Novel Arylhydantoin 1,3,5-Triazine Derivatives as 5-HT6 Serotonin Receptor Ligands with Antidepressive-Like, Anxiolytic and Antiobesity Action In Vivo. Molecules 2018, 23, 2529. [Google Scholar] [CrossRef]
- Vanda, D.; Soural, M.; Canale, V.; Chaumont-Dubel, S.; Satała, G.; Kos, T.; Funk, P.; Fülöpová, V.; Lemrová, B.; Koczurkiewicz, P.; et al. Novel Non-Sulfonamide 5-HT 6 Receptor Partial Inverse Agonist in a Group of Imidazo[4,5- b ]Pyridines with Cognition Enhancing Properties. Eur. J. Med. Chem. 2018, 144, 716–729. [Google Scholar] [CrossRef]
- Smusz, S.; Kurczab, R.; Satała, G.; Bojarski, A.J. Fingerprint-Based Consensus Virtual Screening towards Structurally New 5-HT6R Ligands. Bioorg. Med. Chem. Lett. 2015, 25, 1827–1830. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.N.; Stabler, R.S.; Repke, D.B.; Kress, J.M.; Walker, K.A.; Martin, R.S.; Brothers, J.M.; Ilnicka, M.; Lee, S.W.; Mirzadegan, T. Highly Potent, Non-Basic 5-HT6 Ligands. Site Mutagenesis Evidence for a Second Binding Mode at 5-HT6 for Antagonism. Bioorg. Med. Chem. Lett. 2010, 20, 3436–3440. [Google Scholar] [CrossRef] [PubMed]
- Ivanenkov, Y.A.; Majouga, A.G.; Veselov, M.S.; Chufarova, N.V.; Baranovsky, S.S.; Filkov, G.I. Computational Approaches to the Design of Novel 5-HT6 R Ligands. Rev. Neurosci. 2014, 25, 451–467. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Xu, P.; Shen, D.; Simon, I.A.; Mao, C.; Tan, Y.; Zhang, H.; Harpsøe, K.; Li, H.; Zhang, Y.; et al. GPCRs Steer Gi and Gs Selectivity via TM5-TM6 Switches as Revealed by Structures of Serotonin Receptors. Mol. Cell 2022, 82, 2681–2695.e6. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A Large-Scale Bioactivity Database for Drug Discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef]
- Bender, A.; Mussa, H.Y.; Glen, R.C.; Reiling, S. Similarity Searching of Chemical Databases Using Atom Environment Descriptors (MOLPRINT 2D): Evaluation of Performance. J. Chem. Inf. Comput. Sci. 2004, 44, 1708–1718. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A New Engine for Pharmacophore Perception, 3D QSAR Model Development, and 3D Database Screening: 1. Methodology and Preliminary Results. J. Comput.–Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Pándy-Szekeres, G.; Esguerra, M.; Hauser, A.S.; Caroli, J.; Munk, C.; Pilger, S.; Keserű, G.M.; Kooistra, A.J.; Gloriam, D.E. The G protein database, GproteinDb. Nucleic Acids Res. 2022, 50, D518–D525. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR Structure Models and Ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef]
- Potemkin, V.; Potemkin, A.; Grishina, M. Internet Resources for Drug Discovery and Design. Curr. Top Med. Chem. 2018, 18, 1955–1975. [Google Scholar] [CrossRef] [PubMed]
- Begley, D.J. Delivery of Therapeutic Agents to the Central Nervous System: The Problems and the Possibilities. Pharmacol. Ther. 2004, 104, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Misra, A.; Ganesh, S.; Shahiwala, A.; Shah, S.P. Drug delivery to the central nervous system: A review. J. Pharm. Pharm. Sci. 2003, 6, 252–273. [Google Scholar]
- Sun, W.; Lee, S.; Huang, X.; Liu, S.; Inayathullah, M.; Kim, K.M.; Tang, H.; Ashford, J.W.; Rajadas, J. Attenuation of Synaptic Toxicity and MARK4/PAR1-Mediated Tau Phosphorylation by Methylene Blue for Alzheimer’s Disease Treatment. Sci. Rep. 2016, 6, 34784. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, A.; Anwar, S.; Mohammad, T.; Alajmi, M.F.; Hussain, A.; Rehman, M.T.; Hasan, G.M.; Islam, A.; Hassan, M.I. MARK4 Inhibited by AChE Inhibitors, Donepezil and Rivastigmine Tartrate: Insights into Alzheimer’s Disease Therapy. Biomolecules 2020, 10, 789. [Google Scholar] [CrossRef] [PubMed]
- Lund, H.; Gustafsson, E.; Svensson, A.; Nilsson, M.; Berg, M.; Sunnemark, D.; von Euler, G. MARK4 and MARK3 Associate with Early Tau Phosphorylation in Alzheimer’s Disease Granulovacuolar Degeneration Bodies. Acta Neuropathol. Commun. 2014, 2, 22. [Google Scholar] [CrossRef]
- Anand, K.; Abdul, N.S.; Ghazi, T.; Ramesh, M.; Gupta, G.; Tambuwala, M.M.; Dureja, H.; Singh, S.K.; Chellappan, D.K.; Dua, K.; et al. Induction of Caspase-Mediated Apoptosis in HepG2 Liver Carcinoma Cells Using Mutagen–Antioxidant Conjugated Self-Assembled Novel Carbazole Nanoparticles and In Silico Modeling Studies. ACS Omega 2021, 6, 265–277. [Google Scholar] [CrossRef]
- Shamsi, A.; DasGupta, D.; Alhumaydhi, F.A.; Khan, M.S.; Alsagaby, S.A.; al Abdulmonem, W.; Hassan, M.I.; Yadav, D.K. Inhibition of MARK4 by Serotonin as an Attractive Therapeutic Approach to Combat Alzheimer’s Disease and Neuroinflammation. RSC Med. Chem. 2022, 13, 737–745. [Google Scholar] [CrossRef]
- Parveen, I.; Khan, P.; Ali, S.; Hassan, M.I.; Ahmed, N. Synthesis, Molecular Docking and Inhibition Studies of Novel 3-N-Aryl Substituted-2-Heteroarylchromones Targeting Microtubule Affinity Regulating Kinase 4 Inhibitors. Eur. J. Med. Chem. 2018, 159, 166–177. [Google Scholar] [CrossRef]
- Shen, X.; Liu, X.; Wan, S.; Fan, X.; He, H.; Wei, R.; Pu, W.; Peng, Y.; Wang, C. Discovery of Coumarin as Microtubule Affinity-Regulating Kinase 4 Inhibitor That Sensitize Hepatocellular Carcinoma to Paclitaxel. Front. Chem. 2019, 7, 366. [Google Scholar] [CrossRef]
- Aneja, B.; Khan, N.S.; Khan, P.; Queen, A.; Hussain, A.; Rehman, M.T.; Alajmi, M.F.; El-Seedi, H.R.; Ali, S.; Hassan, M.I.; et al. Design and Development of Isatin-Triazole Hydrazones as Potential Inhibitors of Microtubule Affinity-Regulating Kinase 4 for the Therapeutic Management of Cell Proliferation and Metastasis. Eur. J. Med. Chem. 2019, 163, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Peerzada, M.N.; Khan, P.; Khan, N.S.; Avecilla, F.; Siddiqui, S.M.; Hassan, M.I.; Azam, A. Design and Development of Small-Molecule Arylaldoxime/5-Nitroimidazole Hybrids as Potent Inhibitors of MARK4: A Promising Approach for Target-Based Cancer Therapy. ACS Omega 2020, 5, 22759–22771. [Google Scholar] [CrossRef] [PubMed]
- Voura, M.; Khan, P.; Thysiadis, S.; Katsamakas, S.; Queen, A.; Hasan, G.M.; Ali, S.; Sarli, V.; Hassan, M.I. Probing the Inhibition of Microtubule Affinity Regulating Kinase 4 by N-Substituted Acridones. Sci. Rep. 2019, 9, 1676. [Google Scholar] [CrossRef] [PubMed]
- Ngoei, K.R.W.; Ng, D.C.H.; Gooley, P.R.; Fairlie, D.P.; Stoermer, M.J.; Bogoyevitch, M.A. Identification and Characterization of Bi-Thiazole-2,2′-Diamines as Kinase Inhibitory Scaffolds. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2013, 1834, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, I.; Torka, R.; Garamvölgyi, R.; Baska, F.; Gyulavári, P.; Boros, S.; Illyés, E.; Choidas, A.; Ullrich, A.; Őrfi, L. Discovery of N-[4-(Quinolin-4-Yloxy)Phenyl]Benzenesulfonamides as Novel AXL Kinase Inhibitors. J. Med. Chem. 2018, 61, 6277–6292. [Google Scholar] [CrossRef]
- Barile, E.; De, S.K.; Carlson, C.B.; Chen, V.; Knutzen, C.; Riel-Mehan, M.; Yang, L.; Dahl, R.; Chiang, G.; Pellecchia, M. Design, Synthesis, and Structure−Activity Relationships of 3-Ethynyl-1H-Indazoles as Inhibitors of the Phosphatidylinositol 3-Kinase Signaling Pathway. J. Med. Chem. 2010, 53, 8368–8375. [Google Scholar] [CrossRef]
- Li, F.; Liu, Z.; Sun, H.; Li, C.; Wang, W.; Ye, L.; Yan, C.; Tian, J.; Wang, H. PCC0208017, a Novel Small-Molecule Inhibitor of MARK3/MARK4, Suppresses Glioma Progression In Vitro and In Vivo. Acta Pharm. Sin. B 2020, 10, 289–300. [Google Scholar] [CrossRef]
- Faisal, M.; Kim, J.H.; Yoo, K.H.; Roh, E.J.; Hong, S.S.; Lee, S.H. Development and Therapeutic Potential of NUAKs Inhibitors. J. Med. Chem. 2021, 64, 2–25. [Google Scholar] [CrossRef]
- Katz, J.D.; Haidle, A.; Childers, K.K.; Zabierek, A.A.; Jewell, J.P.; Hou, Y.; Altman, M.D.; Szewczak, A.; Chen, D.; Harsch, A.; et al. Structure Guided Design of a Series of Selective Pyrrolopyrimidinone MARK Inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 114–120. [Google Scholar] [CrossRef]
- Drewry, D.H.; Annor-Gyamfi, J.K.; Wells, C.I.; Pickett, J.E.; Dederer, V.; Preuss, F.; Mathea, S.; Axtman, A.D. Identification of Pyrimidine-Based Lead Compounds for Understudied Kinases Implicated in Driving Neurodegeneration. J. Med. Chem. 2022, 65, 1313–1328. [Google Scholar] [CrossRef]
- Gower, C.M.; Thomas, J.R.; Harrington, E.; Murphy, J.; Chang, M.E.K.; Cornella-Taracido, I.; Jain, R.K.; Schirle, M.; Maly, D.J. Conversion of a Single Polypharmacological Agent into Selective Bivalent Inhibitors of Intracellular Kinase Activity. ACS Chem. Biol. 2016, 11, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Fensome, A.; Ambler, C.M.; Arnold, E.; Banker, M.E.; Brown, M.F.; Chrencik, J.; Clark, J.D.; Dowty, M.E.; Efremov, I.V.; Flick, A.; et al. Dual Inhibition of TYK2 and JAK1 for the Treatment of Autoimmune Diseases: Discovery of ((S)-2,2-Difluorocyclopropyl)((1R,5S)-3-(2-((1-Methyl-1H-Pyrazol-4-Yl)Amino)Pyrimidin-4-Yl)-3,8-Diazabicyclo[3.2.1]Octan-8-Yl)Methanone (PF-06700841). J. Med. Chem. 2018, 61, 8597–8612. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.; Manser, E.; Tan, L.; Lim, L. A Novel Serine/Threonine Kinase Binding the Ras-Related RhoA GTPase Which Translocates the Kinase to Peripheral Membranes. J. Biol. Chem. 1995, 270, 29051–29054. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Roy, K. ROCK-2-Selective Targeting and Its Therapeutic Outcomes. Drug Discov. Today 2020, 20, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, T.; Maekawa, M.; Fujisawa, K.; Okawa, K.; Iwamatsu, A.; Fujita, A.; Watanabe, N.; Saito, Y.; Kakizuka, A.; Morii, N.; et al. The Small GTP-Binding Protein Rho Binds to and Activates a 160 KDa Ser/Thr Protein Kinase Homologous to Myotonic Dystrophy Kinase. EMBO J. 1996, 15, 1885–1893. [Google Scholar] [CrossRef]
- Nakagawa, O.; Fujisawa, K.; Ishizaki, T.; Saito, Y.; Nakao, K. ROCK-I and ROCK-II, Two Isoforms of Rho-Associated Coiled-Coil Forming Protein Serine / Threonine Kinase in Mice. FEBS Lett. 1996, 392, 189–193. [Google Scholar] [CrossRef]
- Matsui, T.; Amano, M.; Yamamoto, T.; Chihara, K.; Nakafuku, M.; Ito, M.; Nakano, T.; Okawa, K.; Iwamatsu, A.; Kaibuchi, K. Rho-Associated Kinase, a Novel Serine/Threonine Kinase, as a Putative Target for the Small GTP Binding Protein Rho. EMBO J. 1996, 15, 2208–2216. [Google Scholar] [CrossRef]
- Mueller, B.K.; Mack, H.; Teusch, N. Rho Kinase, A Promising Neurological Disorders. Nat. Rev. Drug Discov. 2005, 4, 387–398. [Google Scholar] [CrossRef]
- Shahbazi, R.; Baradaran, B.; Khordadmehr, M.; Safaei, S.; Baghbanzadeh, A.; Jigari, F.; Ezzati, H. Targeting ROCK Signaling in Health, Malignant and Non-Malignant Diseases. Immunol. Lett. 2020, 219, 15–26. [Google Scholar] [CrossRef]
- Gu, Q.F.; Yu, J.Z.; Wu, H.; Li, Y.H.; Liu, C.Y.; Feng, L.; Zhang, G.X.; Xiao, B.G.; Ma, C.G. Therapeutic Effect of Rho Kinase Inhibitor FSD–C10 in a Mouse Model of Alzheimer’s Disease. Exp. Ther. Med. 2018, 16, 3929–3938. [Google Scholar] [CrossRef]
- Chen, J.; Sun, Z.; Jin, M.; Tu, Y.; Wang, S.; Yang, X.; Chen, Q.; Zhang, X.; Han, Y.; Pi, R. Inhibition of AGEs/RAGE/Rho/ROCK Pathway Suppresses Non-Specific Neuroinflammation by Regulating BV2 Microglial M1/M2 Polarization through the NF-ΚB Pathway. J. Neuroimmunol. 2017, 305, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.C.; Tatenhorst, L.; Roser, A.E.; Saal, K.A.; Tönges, L.; Lingor, P. ROCK Inhibition in Models of Neurodegeneration and Its Potential for Clinical Translation. Pharmacol. Ther. 2018, 189, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.L.; Wang, Y.Y.; Huang, Z.T.; Zou, Q.; Pu, Y.S.; Yu, C.; Cai, Z. Role of RhoA/ROCK Signaling in Alzheimer’s Disease. Behav. Brain Res. 2021, 414, 113481. [Google Scholar] [CrossRef]
- Zhou, H.; Li, Y. Rho Kinase Inhibitors: Potential Treatments for Diabetes and Diabetic Complications. Curr. Pharm. Des. 2012, 18, 2964–2973. [Google Scholar] [CrossRef]
- Morgan-fisher, M.; Wewer, U.M.; Yoneda, A. Regulation of ROCK Activity in Cancer. J. Histochem. Cytochem. 2013, 61, 185–198. [Google Scholar] [CrossRef]
- Berrino, E.; Supuran, C.T. Expert Opinion on Therapeutic Patents Rho-Kinase Inhibitors in the Management of Glaucoma. Expert Opin. Ther. Patients 2019, 29, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Urol, N.R. Understanding and Targeting the Rho Kinase Pathway in Erectile Dysfunction. Nat. Rev. Urol. 2015, 11, 622–628. [Google Scholar] [CrossRef]
- Ohnaka, K.; Shimoda, S.; Nawata, H.; Shimokawa, H.; Kaibuchi, K.; Iwamoto, Y.; Takayanagi, R. Pitavastatin Enhanced BMP-2 and Osteocalcin Expression by Inhibition of Rho-Associated Kinase in Human Osteoblasts. Biochem. Biophys. Res. Commun. 2001, 342, 337–342. [Google Scholar] [CrossRef]
- Satoh, K.; Fukumoto, Y.; Shimokawa, H. Rho-Kinase: Important New Therapeutic Target in Cardiovascular Diseases. Am. J. Physiol.-Heart Circ. Physiol. 2022, 301, H287–H296. [Google Scholar] [CrossRef]
- Amigoni, F.; Legnaghi, E.; Pevarello, P. Kinase Inhibitors for CNS Diseases: An Lysis of the Recent Patent Literature. Pharm. Pat. Anal 2012, 1, 177–192. [Google Scholar] [CrossRef]
- Julian, L.; Olson, M.F. Rho-Associated Coiled-Coil Containing Kinases (ROCK) Structure, Regulation, and Functions. Small GTPases 2014, 5, e29846. [Google Scholar] [CrossRef] [PubMed]
- Sessions, E.H.; Yin, Y.; Bannister, T.D.; Weiser, A.; Griffin, E.; Pocas, J.; Cameron, M.D.; Ruiz, C.; Lin, L.; Schürer, S.C.; et al. Benzimidazole- and Benzoxazole-Based Inhibitors of Rho Kinase. Bioorg. Med. Chem. Lett. 2008, 18, 6390–6393. [Google Scholar] [CrossRef]
- Ray, P.; Wright, J.; Adam, J.; Bennett, J.; Boucharens, S.; Black, D.; Cook, A.; Brown, A.R.; Epemolu, O.; Fletcher, D.; et al. Fragment-Based Discovery of 6-Substituted Isoquinolin-1-Amine Based ROCK-I Inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 97–101. [Google Scholar] [CrossRef]
- Activation, R.; Uehata, M.; Ishizaki, T.; Satoh, H. Calcium Sensitization of Smooth Muscle Mediated by a Rho-Associated Protein Kinase in Hypertension. Nature 1997, 389, 6654. [Google Scholar]
- Feng, Y.; Lograsso, P.V.; Defert, O.; Li, R. Rho Kinase (ROCK) Inhibitors and Their Therapeutic Potential. J. Med. Chem. 2016, 59, 2269–2300. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Sessions, E.H.; Pocas, J.R.; Grant, W.; Schröter, T.; Lin, L.; Ruiz, C.; Cameron, M.D.; Schürer, S.; Lograsso, P.; et al. Discovery and Optimization of Indoles and 7-Azaindoles as Rho Kinase (ROCK) Inhibitors (Part-I). Bioorg. Med. Chem. Lett. 2011, 21, 7107–7112. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Wang, C.; Glunz, P.W.; Li, J.; Cheadle, N.L.; Chen, A.Y.; Chen, X.Q.; Myers, J.E.; Guarino, V.R.; Rose, A.; et al. Discovery of a phenylpyrazole amide ROCK inhibitor as a tool molecule for in vivo studies. Bioorg. Med. Chem. Lett. 2020, 30, 127495. [Google Scholar] [CrossRef]
- Sessions, E.H.; Chowdhury, S.; Yin, Y.; Pocas, J.R.; Grant, W.; Schröter, T.; Lin, L.; Ruiz, C.; Cameron, M.D.; Lograsso, P.; et al. Bioorganic & Medicinal Chemistry Letters Discovery and Optimization of Indole and 7-Azaindoles as Rho Kinase (ROCK) Inhibitors (Part-II). Bioorg. Med. Chem. Lett. 2011, 21, 7113–7118. [Google Scholar] [CrossRef]
- Pan, J.; Yin, Y.; Zhao, L.; Feng, Y. Discovery of (S)-6-Methoxy-Chroman-3-Carboxylic Acid (4-Pyridin-4-Yl-Phenyl)-Amide as Potent and Isoform Selective ROCK2 Inhibitors. Bioorg. Med. Chem. 2019, 27, 1382–1390. [Google Scholar] [CrossRef]
- Yin, Y.; Zheng, K.; Eid, N.; Howard, S.; Jeong, J.; Yi, F.; Guo, J.; Park, C.M.; Bibian, M.; Wu, W.; et al. Bis-Aryl Urea Derivatives as Potent and Selective LIM Kinase (Limk) Inhibitors. J. Med. Chem. 2016, 58, 1846–1861. [Google Scholar] [CrossRef]
- Chowdhury, S.; Ting, Y.; Fang, X.; Grant, W.; Pocas, J.; Cameron, M.D.; Ruiz, C.; Lin, L.; Park, H.; Schröter, T.; et al. Amino Acid Derived Quinazolines as Rock / PKA Inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Sehon, C.A.; Wang, G.Z.; Viet, A.Q.; Goodman, K.B.; Dowdell, S.E.; Elkins, P.A.; Semus, S.F.; Evans, C.; Jolivette, L.J.; Kirkpatrick, R.B.; et al. Potent, Selective and Orally Bioavailable Dihydropyrimidine Inhibitors of Rho Kinase (ROCK1) as Potential Therapeutic Agents for Cardiovascular Diseases. J. Med. Chem. 2008, 51, 6631–6634. [Google Scholar] [CrossRef] [PubMed]
- Goodman, K.B.; Cui, H.; Dowdell, S.E.; Gaitanopoulos, D.E.; Ivy, R.L.; Sehon, C.A.; Stavenger, R.A.; Wang, G.Z.; Viet, A.Q.; Xu, W.; et al. Development of Dihydropyridone Indazole Amides as Selective Rho-Kinase Inhibitors. J. Med. Chem. 2007, 50, 6–9. [Google Scholar] [CrossRef]
- Oh, K.; Koo, B.; Ho, C.; Won, H.; Sook, N.; Hyun, J.; Soo, J.; Ho, B. Cardiovascular Effects of a Novel Selective Rho Kinase Inhibitor, 2- (1H-Indazole-5-Yl) Amino-4-Methoxy-6-Piperazino Triazine (DW1865). Eur. J. Pharmacol. 2013, 702, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Dayal, N.; Mikek, C.G.; Hernandez, D.; Naclerio, G.A.; Fei, E.; Chu, Y.; Carter-cooper, B.A.; Lapidus, R.G.; Sintim, H.O. Potently Inhibiting Cancer Cell Migration with Novel 3H-Pyrazolo[4,3-f]Quinoline Boronic Acid ROCK Inhibitors. Eur. J. Med. Chem. 2019, 180, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Cameron, M.D.; Frackowiak, B.; Griffin, E.; Lin, L.; Ruiz, C.; Schröter, T.; LoGrasso, P. Structure-Activity Relationships, and Drug Metabolism and Pharmacokinetic Properties for Indazole Piperazine and Indazole Piperidine Inhibitors of ROCK-II. Bioorg. Med. Chem. Lett. 2007, 17, 2355–2360. [Google Scholar] [CrossRef] [PubMed]
- Takayasu, M.; Suzuki, Y.; Shibuya, M.; Asano, T.; Kanamori, M.; Okada, T.; Kageyama, N.; Hidaka, H. The Effects of HA Compound Calcium Antagonists on Delayed Cerebral Vasospasm in Dogs. J. Neurosurg. 1986, 65, 80–85. [Google Scholar] [CrossRef]
- Liao, J.K.; Seto, M.; Noma, K. Rho Kinase (ROCK) Inhibitors. J. Cardiovasc. Pharmacol. 2007, 50, 17–24. [Google Scholar] [CrossRef]
- Hamano, T.; Shirafuji, N.; Yen, S.; Yoshida, H.; Nicholas, M.; Hayashi, K.; Ikawa, M.; Yamamura, O.; Fujita, Y.; Nakamoto, Y.; et al. Rho-Kinase ROCK Inhibitors Reduce Oligomeric Tau Protein. Neurobiol. Aging 2021, 89, 41–54. [Google Scholar] [CrossRef]
- Wei, W.; Wang, Y.; Zhang, J.; Gu, Q.; Liu, X.; Song, L.; Chai, Z.; Guo, M.; Yu, J.; Ma, C. Fasudil Ameliorates Cognitive Deficits, Oxidative Stress and Neuronal Apoptosis via Inhibiting ROCK/MAPK and Activating Nrf2 Signalling Pathways in APP/PS1 Mice. Folia Neuropathol. 2021, 59, 32–49. [Google Scholar] [CrossRef]
- Yan, H.; Yan, Y.; Gao, Y.; Zhang, N.; Kumar, G.; Fang, Q.; Li, Z.; Li, J.; Zhang, Y.; Song, L.; et al. Transcriptome Analysis of Fasudil Treatment in the APPswe/PSEN1dE9 Transgenic (APP/PS1) Mice Model of Alzheimer’s Disease. Sci. Rep. 2022, 12, 6625. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.F.; Zhang, H.Y.; Zhang, P.J.; Liu, X.Q.; Song, L.J.; Wei, W.Y.; Wang, Y.Y.; Mu, B.T.; Chai, Z.; Yu, J.Z.; et al. Fasudil Reduces β-Amyloid Levels and Neuronal Apoptosis in APP/PS1 Transgenic Mice via Inhibition of the Nogo-A/NgR/RhoA Signaling Axis. J. Integr. Neurosci. 2020, 19, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Logé, C.; Wallez, V.; Scalbert, E.; Cario-Tourmaniantz, C.; Loirand, G.; Pacaud, P.; Lesieur, D. Rho-Kinase Inhibitors: Pharmacomodulations on the Lead Compound Y-32885. J. Enzym. Inhib. Med. Chem. 2002, 17, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Boland, S.; Bourin, A.; Alen, J.; Geraets, J.; Schroeders, P.; Castermans, K.; Kindt, N.; Boumans, N.; Panitti, L.; Fransen, S.; et al. Design, Synthesis, and Biological Evaluation of Novel, Highly Active Soft Rock Inhibitors. J. Med. Chem. 2015, 58, 4309–4324. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Miwa, Y.; Kasa, M.; Kitano, K.; Amano, M.; Kaibuchi, K.; Hakoshima, T. Structural Basis for Induced-Fit Binding of Rho-Kinase to the Inhibitor Y-27632. J. Biochem. 2006, 140, 305–311. [Google Scholar] [CrossRef]
- Lew, J.; Beaudette, K.; Litwin, C.M.E.; Wang, J.H. Purification and Characterization of a Novel Proline-Directed Protein Kinase from Bovine Brain. J. Biol. Chem. 1992, 267, 13383–13390. [Google Scholar] [CrossRef]
- Gao, G.B.; Sun, Y.; Fang, R.D.; Wang, Y.; Wang, Y.; He, Q.Y. Post-Translational Modifications of CDK5 and Their Biological Roles in Cancer. Mol. Biomed. 2021, 2, 22. [Google Scholar] [CrossRef]
- Zhang, P.; Yu, P.C.; Tsang, A.H.K.; Chen, Y.; Fu, A.K.Y.; Fu, W.Y.; Chung, K.K.; Ip, N.Y. S-Nitrosylation of Cyclin-Dependent Kinase 5 (Cdk5) Regulates Its Kinase Activity and Dendrite Growth during Neuronal Development. J. Neurosci. 2010, 30, 14366–14370. [Google Scholar] [CrossRef]
- Lee, J.; Ko, Y.U.; Chung, Y.; Yun, N.; Kim, M.; Kim, K.; Oh, Y.J. The Acetylation of Cyclin-Dependent Kinase 5 at Lysine 33 Regulates Kinase Activity and Neurite Length in Hippocampal Neurons. Sci. Rep. 2018, 8, 13676. [Google Scholar] [CrossRef]
- Sridhar, J.; Akula, N.; Pattabiraman, N. Selectivity and Potency of Cyclin-Dependent Kinase Inhibitors. AAPS J. 2006, 8, 204–221. [Google Scholar] [CrossRef][Green Version]
- Shupp, A.; Casimiro, M.C.; Pestell, R.G. Biological Functions of CDK5 and Potential CDK5 Targeted Clinical Treatments. Oncotarget 2017, 8, 17373–17382. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK Inhibitors in Cancer Therapy, an Overview of Recent Development. Am. J. Cancer. Res. 2021, 11, 1913–1935. [Google Scholar] [PubMed]
- Lenjisa, J.L.; Tadesse, S.; Khair, N.Z.; Kumarasiri, M.; Yu, M.; Albrecht, H.; Milne, R.; Wang, S. CDK5 in Oncology: Recent Advances and Future Prospects. Future Med. Chem. 2017, 9, 1939–1962. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Cutler, S.J.; Doerksen, R.J.; Khan, I.A.; Williamson, J.S. Discovery of Thienoquinolone Derivatives as Selective and ATP Non-Competitive CDK5/P25 Inhibitors by Structure-Based Virtual Screening. Bioorg. Med. Chem. 2014, 22, 6409–6421. [Google Scholar] [CrossRef] [PubMed]
- Weishaupt, J.H.; Neusch, C.; Bähr, M. Cyclin-Dependent Kinase 5 (CDK5) and Neuronal Cell Death. Cell Tissue Res. 2003, 312, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, N.; Gupta, R.; Kumar, P. Pharmacological Relevance of CDK Inhibitors in Alzheimer’s Disease. Neurochem. Int. 2021, 148, 105115. [Google Scholar] [CrossRef]
- Mushtaq, G.; Greig, N.H.; Anwar, F.; Al-Abbasi, F.A.; Zamzami, M.A.; Al-Talhi, H.A.; Kamal, M.A. Neuroprotective Mechanisms Mediated by CDK5 Inhibition HHS Public Access. Curr. Phram. Des. 2016, 22, 527–534. [Google Scholar] [CrossRef]
- Available online: https://www.ebi.ac.uk/chembl/ (accessed on 20 July 2022).
- Helal, C.J.; Sanner, M.A.; Cooper, C.B.; Gant, T.; Adam, M.; Lucas, J.C.; Kang, Z.; Kupchinsky, S.; Ahlijanian, M.K.; Tate, B.; et al. Discovery and SAR of 2-Aminothiazole Inhibitors of Cyclin-Dependent Kinase 5/P25 as a Potential Treatment for Alzheimer’s Disease. Bioorg. Med. Chem. Lett. 2004, 14, 5521–5525. [Google Scholar] [CrossRef]
- Sonawane, Y.A.; Taylor, M.A.; Napoleon, J.V.; Rana, S.; Contreras, J.I.; Natarajan, A. Cyclin Dependent Kinase 9 Inhibitors for Cancer Therapy. J. Med. Chem. 2016, 59, 8667–8684. [Google Scholar] [CrossRef]
- Havlíček, L.; Hanuš, J.; Veselý, J.; Leclerc, S.; Meijer, L.; Shaw, G.; Strnad, M. Cytokinin-Derived Cyclin-Dependent Kinase Inhibitors: Synthesis and Cdc2 Inhibitory Activity of Olomoucine and Related Compounds. J. Med. Chem. 1997, 40, 408–412. [Google Scholar] [CrossRef]
- Vymětalová, L.; Havlíček, L.; Šturc, A.; Skrášková, Z.; Jorda, R.; Pospíšil, T.; Strnad, M.; Kryštof, V. 5-Substituted 3-Isopropyl-7-[4-(2-Pyridyl)Benzyl]Amino-1(2)H-Pyrazolo[4,3-d]Pyrimidines with Anti-Proliferative Activity as Potent and Selective Inhibitors of Cyclin-Dependent Kinases. Eur. J. Med. Chem. 2016, 110, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Schonbrunn, E.; Betzi, S.; Alam, R.; Martin, M.P.; Becker, A.; Han, H.; Francis, R.; Chakrasali, R.; Jakkaraj, S.; Kazi, A.; et al. Development of Highly Potent and Selective Diaminothiazole Inhibitors of Cyclin-Dependent Kinases. J. Med. Chem. 2013, 56, 3768–3782. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martínez, C.; Gelbert, L.M.; Lallena, M.J.; de Dios, A. Cyclin Dependent Kinase (CDK) Inhibitors as Anticancer Drugs. Bioorg. Med. Chem. Lett. 2015, 25, 3420–3435. [Google Scholar] [CrossRef]
- Li, X.; Huang, P.; Cui, J.J.; Zhang, J.; Tang, C. Novel Pyrrolyllactone and Pyrrolyllactam Indolinones as Potent Cyclin-Dependent Kinase 2 Inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 1939–1942. [Google Scholar] [CrossRef]
- William, A.D.; Lee, A.C.; Goh, K.C.; Poulsen, A.; Teo, E.L.; Nagaraj, H.; Lee, C.P.; Wang, H.; Williams, M.; Sun, E.T.; et al. Discovery of Kinase Spectrum Selective Macrocycle (16E)-14-Methyl20-Oxa-5,7,14,26-Tetraazatetracyclo[19.3.1.1(2,6).1(8,12)]Heptacosa1(25),2(26),3,5,8(27),9,11,16,21,23-Decaene (SB1317/TG02), a Potent Inhibitor of Cyclin Dependent Kinases (CDKs), Janus Kinase 2 (JAK2), and Fms-like Tyrosine Kinase-3 (FLT3) for the Treatment of Cancer. J. Med. Chem. 2012, 55, 169–196. [Google Scholar]
- Dong, K.; Wang, X.; Yang, X.; Zhu, X. Binding Mechanism of CDK5 with Roscovitine Derivatives Based on Molecular Dynamics Simulations and MM/PBSA Methods. J. Mol. Graph. Model. 2016, 68, 57–67. [Google Scholar] [CrossRef]
- Mapelli, M.; Massimiliano, L.; Crovace, C.; Seeliger, M.A.; Tsai, L.H.; Meijer, L.; Musacchio, A. Mechanism of CDK5/P25 Binding by CDK Inhibitors. J. Med. Chem. 2005, 48, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Zeb, A.; Son, M.; Yoon, S.; Kim, J.H.; Park, S.J.; Lee, K.W. Computational Simulations Identified Two Candidate Inhibitors of Cdk5/P25 to Abrogate Tau-Associated Neurological Disorders. Comput. Struct. Biotechnol. J. 2019, 17, 579–590. [Google Scholar] [CrossRef]
- National Library of Medicine (U.S.). Available online: https://Clinicaltrials.Gov/Ct2/Show/NCT01676753 (accessed on 15 July 2022).
- Pires, D.; Blundell, T.; Ascher, D. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czarnota-Łydka, K.; Kucwaj-Brysz, K.; Pyka, P.; Haberek, W.; Podlewska, S.; Handzlik, J. Multitargeting the Action of 5-HT6 Serotonin Receptor Ligands by Additional Modulation of Kinases in the Search for a New Therapy for Alzheimer’s Disease: Can It Work from a Molecular Point of View? Int. J. Mol. Sci. 2022, 23, 8768. https://doi.org/10.3390/ijms23158768
Czarnota-Łydka K, Kucwaj-Brysz K, Pyka P, Haberek W, Podlewska S, Handzlik J. Multitargeting the Action of 5-HT6 Serotonin Receptor Ligands by Additional Modulation of Kinases in the Search for a New Therapy for Alzheimer’s Disease: Can It Work from a Molecular Point of View? International Journal of Molecular Sciences. 2022; 23(15):8768. https://doi.org/10.3390/ijms23158768
Chicago/Turabian StyleCzarnota-Łydka, Kinga, Katarzyna Kucwaj-Brysz, Patryk Pyka, Wawrzyniec Haberek, Sabina Podlewska, and Jadwiga Handzlik. 2022. "Multitargeting the Action of 5-HT6 Serotonin Receptor Ligands by Additional Modulation of Kinases in the Search for a New Therapy for Alzheimer’s Disease: Can It Work from a Molecular Point of View?" International Journal of Molecular Sciences 23, no. 15: 8768. https://doi.org/10.3390/ijms23158768
APA StyleCzarnota-Łydka, K., Kucwaj-Brysz, K., Pyka, P., Haberek, W., Podlewska, S., & Handzlik, J. (2022). Multitargeting the Action of 5-HT6 Serotonin Receptor Ligands by Additional Modulation of Kinases in the Search for a New Therapy for Alzheimer’s Disease: Can It Work from a Molecular Point of View? International Journal of Molecular Sciences, 23(15), 8768. https://doi.org/10.3390/ijms23158768