
c-Myc Protein Level Affected by Unsymmetrical Bisacridines Influences Apoptosis and Senescence Induced in HCT116 Colorectal and H460 Lung Cancer Cells


Abstract
1. Introduction
2. Results
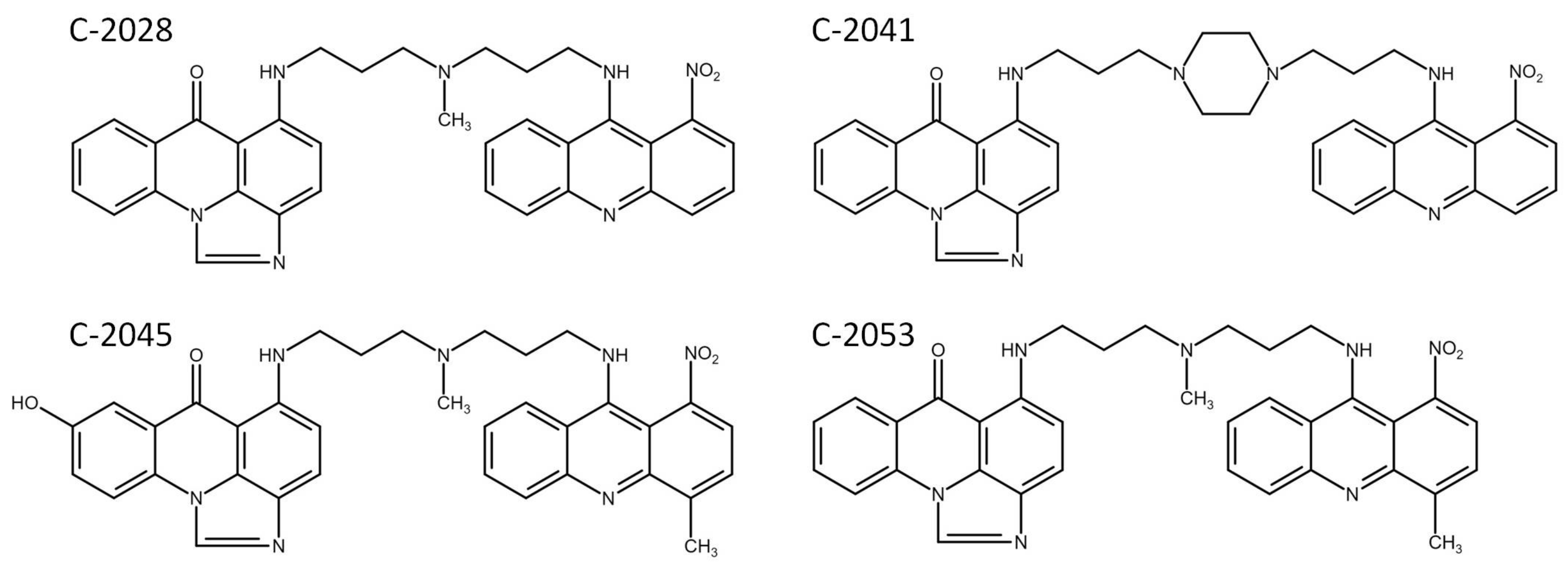
2.1. Cytotoxic Effects of UA Compounds against Colon and Lung Cancer and Normal Cells
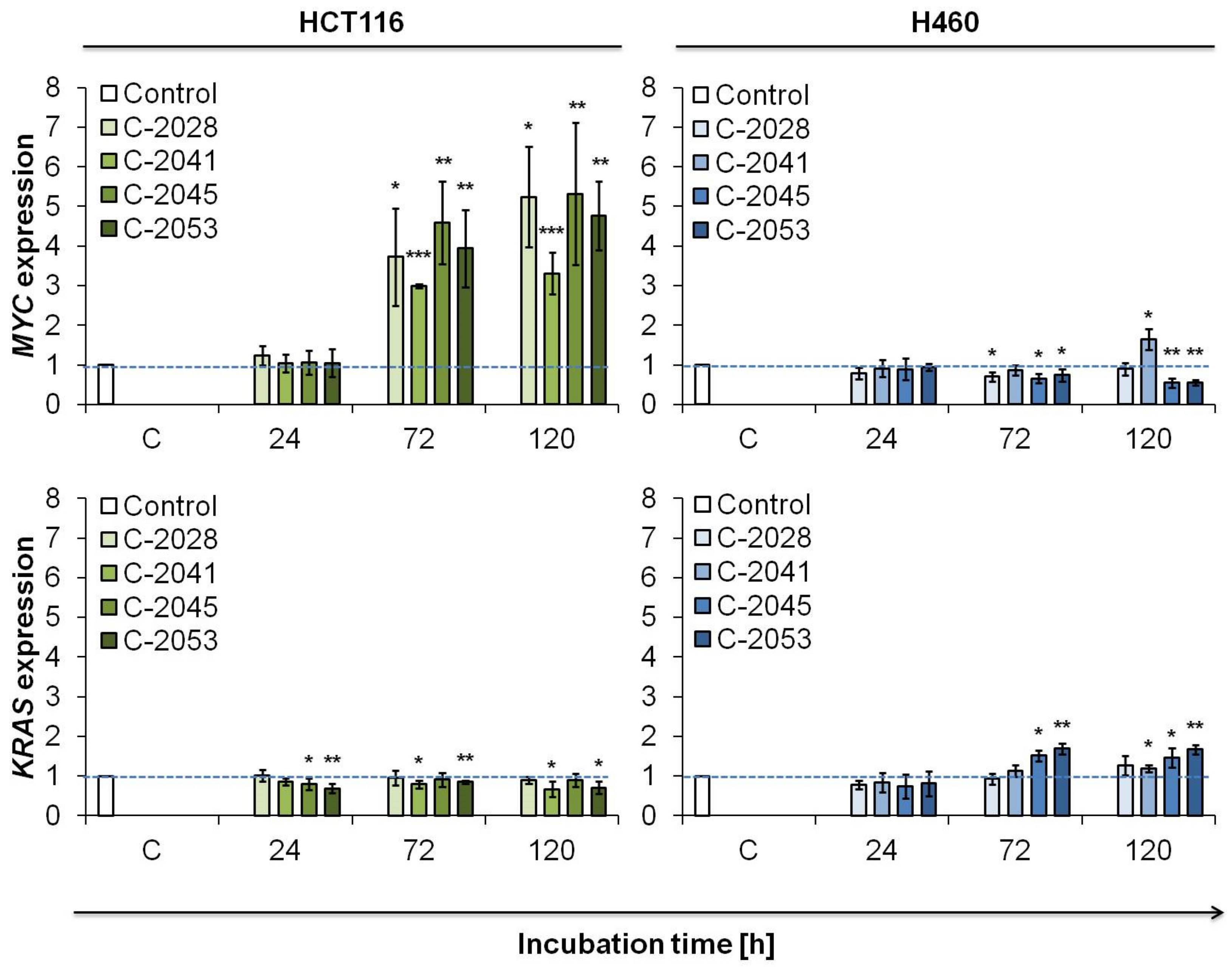
2.2. c-Myc and K-Ras Expression in Cancer and Normal Cells
2.3. Western Blotting Analysis of c-Myc and K-Ras Level in Cancer and Normal Cells
2.4. Cell Cycle Distribution of Colon and Lung Cancer and Normal Cells Treated with UAs
2.5. Morphological Changes of Nuclei Triggered by UAs in Cancer and Normal Cells
2.6. UAs Induction of Apoptosis in Cancer Cells
2.7. UAs Induction of Accelerated Senescence in Cancer Cell Lines
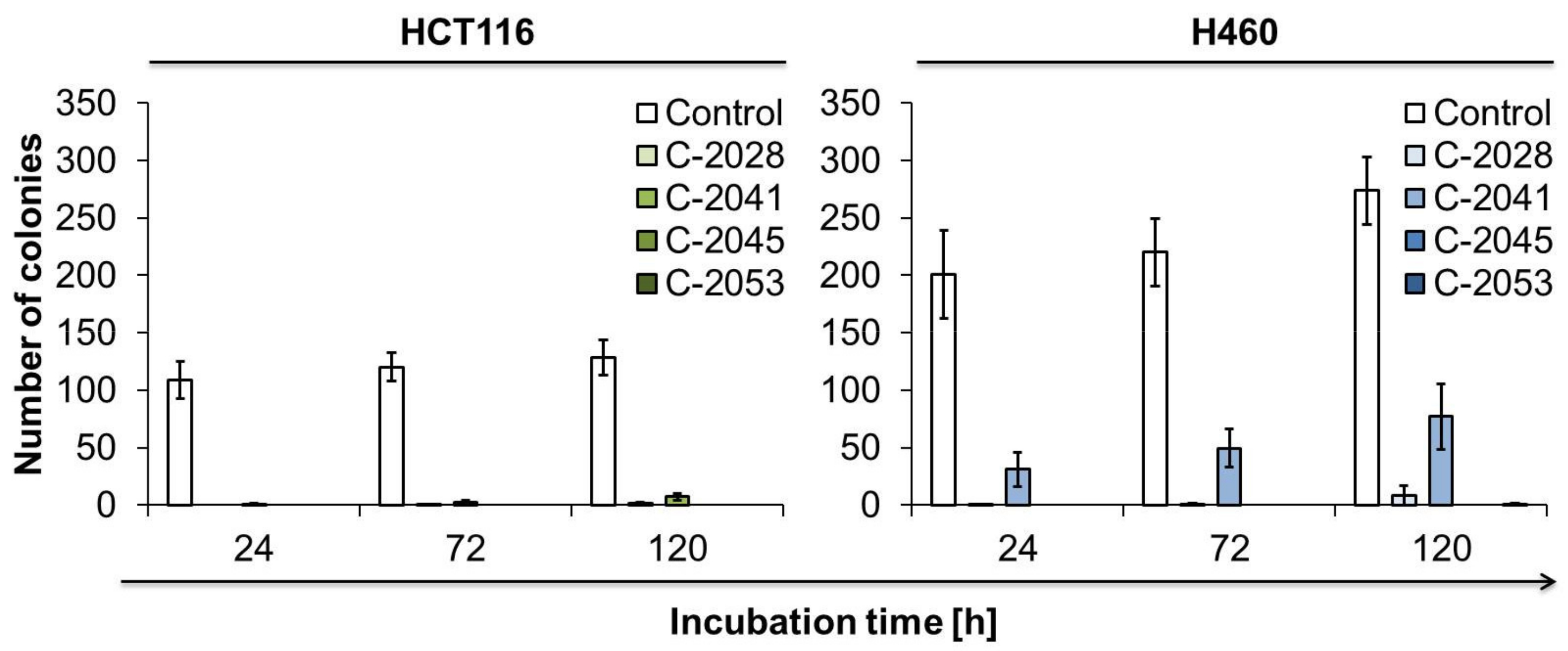
2.8. The Ability of Cancer Cells to Return to Proliferation after UAs Treatment
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Lines and Culture
4.3. Cell Growth Inhibition Assay
4.4. mRNA Isolation and Real-Time Quantitative Polymerase Chain Reaction (RT-PCR)
4.5. Western Blotting
4.6. Cell Cycle Analysis
4.7. Assessment of Cell Morphology
4.8. Annexin V/PI Dual Staining
4.9. Caspase-3 Activity Measurement
4.10. PARP Cleavage Analysis
4.11. Mitochondrial Transmembrane Potential Measurements
4.12. Accelerated Senescence Observations
4.13. Colony-Forming Assay
4.14. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Konopa, J.; Horowska, B.; Paluszkiewicz, E.; Borowa-Mazgaj, B.; Augustin, E.; Skwarska, A.; Mazerska, Z. Asymmetric Bis-Acridines with Antitumor Activity and Use Thereof. European Patent 3,070,078, 4 October 2017. [Google Scholar]
- Konopa, J.; Horowska, B.; Paluszkiewicz, E.; Borowa-Mazgaj, B.; Augustin, E.; Skwarska, A.; Mazerska, Z. Asymmetric Bis-Acridines with Antitumour Activity and Their Uses. U.S. Patent 10,202,349, 12 February 2019. [Google Scholar]
- Paluszkiewicz, E.; Horowska, B.; Borowa-Mazgaj, B.; Peszyńska-Sularz, G.; Paradziej-Łukowicz, J.; Augustin, E.; Konopa, J.; Mazerska, Z. Design, synthesis and high antitumor potential of new unsymmetrical bisacridine derivatives towards human solid tumors, specifically pancreatic cancers and their unique ability to stabilize DNA G-quadruplexes. Eur. J. Med. Chem. 2020, 204, 112599. [Google Scholar] [CrossRef] [PubMed]
- Yang, D. G-Quadruplex DNA and RNA. In G-Quadruplex Nucleic Acids. Methods in Molecular Biology; Yang, D., Lin, C., Eds.; Springer Nature; Humana: New York, NY, USA, 2019; Volume 2035, pp. 1–24. [Google Scholar] [CrossRef]
- Evan, G.I.; Littlewood, T.D. The role of c-Myc in cell growth. Curr. Opin. Genet. Dev. 1993, 3, 44–49. [Google Scholar] [CrossRef]
- Henriksson, M.; Luscher, B. Proteins of the Myc network: Essential regulators of cell growth and differentiation. Adv. Cancer Res. 1996, 68, 109–182. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. The MYC oncogene as a cancer drug target. Curr. Cancer Drug Targets 2003, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef] [PubMed]
- Prochownik, E.V. c-Myc as a therapeutic target in cancer. Expert Rev. Anticancer Ther. 2002, 4, 289–302. [Google Scholar] [CrossRef]
- Hoffman, B.; Liebermann, D.A. Apoptotic signaling by c-Myc. Oncogene 2008, 27, 6462–6472. [Google Scholar] [CrossRef]
- Wu, C.H.; Van Riggelen, J.; Yetil, A.; Fan, A.C.; Bachireddy, P.; Felsher, D.W. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc. Nat. Acad. Sci. USA 2007, 104, 13028–13033. [Google Scholar] [CrossRef]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef]
- Kalkat, M.; De Melo, J.; Hickman, K.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.; Penn, L. MYC deregulation in primary human cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef]
- Calabrese, D.R.; Chen, X.; Leon, E.C.; Gaikwad, S.M.; Phyo, Z.; Hewitt, W.M.; Alden, S.; Hilimire, T.A.; He, F.; Michalowski, A.M.; et al. Chemical and structural studies provide a mechanistic basis for recognition of the MYC G-quadruplex. Nat. Commun. 2018, 12, 4229. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef]
- Ou, T.; Lu, Y.J.; Zhang, C.; Huang, Z.S.; Wang, X.D.; Tan, J.H.; Chen, Y.; Ma, D.L.; Wong, K.Y.; Tang, J.C.; et al. Stabilization of G-quadruplex DNA and downregulation of oncogene c-myc by quindoline derivatives. J. Med. Chem. 2007, 50, 1465–1474. [Google Scholar] [CrossRef]
- Kang, H.J.; Park, H.J. Novel molecular mechanism for actinomycin D activity as an oncogenic promoter G-quadruplex binder. Biochemistry 2009, 48, 7392–7398. [Google Scholar] [CrossRef]
- Krens, L.L.; Baas, J.M.; Gelderblom, H.; Guchelaar, H.J. Therapeutic modulation of k-ras signaling in colorectal cancer. Drug Discov. Today 2010, 15, 502–516. [Google Scholar] [CrossRef]
- Ryan, M.B.; Corcoran, R.B. Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef]
- Kim, W.; Lee, S.; Kim, H.S.; Song, M.; Cha, Y.H.; Kim, Y.H.; Shin, J.; Lee, E.S.; Joo, Y.; Song, J.J.; et al. Targeting mutant KRAS with CRISPR-Cas9 controls tumor growth. Genome Res. 2018, 28, 374–382. [Google Scholar] [CrossRef]
- Shin-ya, K.; Wierzba, K.; Matsuo, K.; Ohtani, T.; Yamada, Y.; Furihata, K.; Hayakawa, Y.; Seto, H. Telomestatin, a novel telomerase inhibitor from Streptomyces anulatus. J. Am. Chem. Soc. 2001, 123, 1262–1263. [Google Scholar] [CrossRef]
- Dutta, D.; Debnath, M.; Müller, D.; Paul, R.; Das, T.; Bessi, I.; Schwalbe, H.; Dash, J. Cell penetrating thiazole peptides inhibit c-MYC expression via site-specific targeting of c-MYC G-quadruplex. Nucleic Acids Res. 2018, 46, 5355–5365. [Google Scholar] [CrossRef]
- Amato, J.; Madanayake, T.W.; Iaccarino, N.; Novellino, E.; Randazzo, A.; Hurley, L.H.; Pagano, B. HMGB1 binds to the KRAS promoter G-quadruplex: A new player in oncogene trancription regulation? Chem. Commun. 2018, 54, 6442–9445. [Google Scholar] [CrossRef]
- Agarwala, P.; Pandey, S.; Maiti, S. Role of G-quadruplex located at 5′ end of mRNAs. Biochim. Biophys. Acta 2014, 1840, 3503–3510. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Kulesza, J.; Pawłowska, M.; Augustin, E. The Influence of Antitumor Unsymmetrical Bisacridines on 3D Cancer Spheroids Growth and Viability. Molecules 2021, 26, 6262. [Google Scholar] [CrossRef] [PubMed]
- Pilch, J.; Matysiak-Brynda, E.; Kowalczyk, A.; Bujak, P.; Mazerska, Z.; Nowicka, A.M.; Augustin, E. New Unsymmetrical Bisacridine Derivatives Noncovalently Attached to Quaternary Quantum Dots Improve Cancer Therapy by Enhancing Cytotoxicity toward Cancer Cells and Protecting Normal Cells. ACS Appl. Mater. Interfaces 2020, 12, 17276–17289. [Google Scholar] [CrossRef] [PubMed]
- Pilch, J.; Kowalik, P.; Bujak, P.; Nowicka, A.M.; Augustin, E. Quantum Dots as a Good Carriers of Unsymmetrical Bisacridines for Modulating Cellular Uptake and the Biological Response in Lung and Colon Cancer Cells. Nanomaterials 2021, 11, 462. [Google Scholar] [CrossRef] [PubMed]
- Pilch, J.; Kowalik, P.; Kowalczyk, A.; Bujak, P.; Kasprzak, A.; Paluszkiewicz, E.; Augustin, E.; Nowicka, A.M. Foliate-Targeting Quantum Dots-p-Cyclodextrin Nanocarrier for Efficient Delivery of Unsymmetrical Bisacridines to Lung and Prostate Cancer Cells. Int. J. Mol. Sci. 2022, 23, 1261. [Google Scholar] [CrossRef] [PubMed]
- Vétillard, A.; Jonchère, B.; Moreau, M.; Toutain, B.; Henry, C.; Fontanel, S.; Bernard, A.C.; Campone, M.; Guette, C.; Coqueret, O. Akt inhibition improves irinotecan treatment and prevents cell emergence by switching the senescence response to apoptosis. Oncotarget 2015, 6, 43342–43362. [Google Scholar] [CrossRef]
- Zastre, J.; Anantha, M.; Ramsay, E.; Bally, M. Irinotecan-cisplatin interactions assessed in cell-based screening assays: Cytotoxicity, drug accumulation and DNA adduct formation in an NSCLC cell line. Cancer Chemother. Pharmacol. 2007, 60, 91–102. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, J.; Zhang, L.; Zhao, L.; Fan, S.; Yang, Z.; Gao, F.; Kong, Y.; Xiao, G.G.; Wang, Q. Expression of P-gp, MRP, LRP, GST-π and TopoIIα and intrinsic resistance in human lung cancer cell lines. Oncol. Rep. 2011, 26, 1081–1089. [Google Scholar] [CrossRef]
- Huang, H.; Aladelokun, O.; Ideta, T.; Giardina, C.; Ellis, L.M.; Rosenberg, D.W. Inhibition of PGE2/EP4 receptor signaling enhances oxaliplatin efficacy in resistant colon cancer cells through modulation of oxidative stress. Sci. Rep. 2019, 9, 4954. [Google Scholar] [CrossRef]
- Francisco, A.P.; Paulo, A. Oncogene expression modulation in cancer cell lines by DNA G-quadruplex-interactive small molecules. Curr. Med. Chem. 2016, 24, 4873–4904. [Google Scholar] [CrossRef]
- Seimiya, T.; Otsuka, M.; Iwata, T.; Tanaka, E.; Sekiba, K.; Shibata, C.; Moriyama, M.; Nakagawa, R.; Maruyama, R.; Koike, K. Aberrant expression of a novel circular RNA in pancreatic cancer. J. Hum. Genet. 2021, 66, 181–191. [Google Scholar] [CrossRef]
- Carvalho, J.; Mergny, J.L.; Salgado, G.F.; Queiroz, J.A.; Cruz, C. G-quadruplex, Friend or Foe: The Role of the G-quartet in Anticancer Strategies. Trends Mol. Med. 2020, 26, 848–861. [Google Scholar] [CrossRef]
- Morris, M.J.; Basu, S. An unusually stable G-quadruplex within the 5′-UTR of the MT3 matrix metalloproteinase mRNA represses translation in eukaryotic cells. Biochemistry 2009, 48, 5313–5319. [Google Scholar] [CrossRef]
- Shahid, R.; Bugaut, A.; Balasubramanian, S. The BCL-2 5′ untranslated region contains an RNA G-quadruplex-forming motif that modulates protein expression. Biochemistry 2010, 49, 8300–8306. [Google Scholar] [CrossRef]
- Herdy, B.; Mayer, C.; Varshney, D.; Marsico, G.; Murat, P.; Taylor, C.; D’Santos, C.; Tannahill, D.; Balasubramanian, S. Analysis of NRAS RNA G-quadruplex binding proteins reveals DDX3X as a novel interactor of cellular G-quadruplex containing transcripts. Nucleic Acids Res. 2019, 46, 11592–11604. [Google Scholar] [CrossRef]
- Friday, B.B.; Adjei, A.A. K-ras as a target for cancer therapy. Biochim. Biophys. Acta 2005, 1756, 127–144. [Google Scholar] [CrossRef]
- Xie, C.; Li, Y.; Li, L.L.; Fan, X.X.; Wang, Y.W.; Wei, C.L.; Liu, L.; Leung, E.L.H.; Yao, X.J. Identification of a new potent inhibitor targeting KRAS in non-small cell lung cancer cells. Front. Pharmacol. 2017, 8, 823. [Google Scholar] [CrossRef]
- Pant, S.; Hubbard, J.; Martinelli, E.; Bekaii-Saab, T. Clinical update on K-Ras targeted therapy in gastrointestinal cancers. Crit Rev. Oncol. Hematol. 2018, 130, 78–91. [Google Scholar] [CrossRef]
- Brito, H.; Martins, A.C.; Lavrado, J.; Mendes, E.; Francisco, A.P.; Santos, S.A.; Ohnmacht, S.A.; Kim, N.S.; Rodrigues, C.M.; Moreira, R.; et al. Targeting KRAS oncogene in colon cancer cells with 7-carboxylate Indolo[3,2-b]quinoline tri-Alkylamine derivatives. PLoS ONE 2015, 10, e0126891. [Google Scholar] [CrossRef] [PubMed]
- Chanvorachote, P.; Sriratanasak, N.; Nonpanya, N. C-myc contributes to malignancy of lung Cancer: A potential anticancer drug target. Anticancer Res. 2020, 40, 609–618. [Google Scholar] [CrossRef]
- Cogoi, S.; Xodo, L.E. G4 DNA in ras genes and its potential in cancer therapy. Biochim. Biophys. Acta 2016, 1859, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Lavrado, J.; Brito, H.; Borralho, P.M.; Ohnmacht, S.A.; Kim, N.S.; Leitão, C.; Pisco, S.; Gunaratnam, M.; Rodrigues, C.M.; Moreira, R.; et al. KRAS oncogene repression in colon cancer cell lines by G-quadruplex binding indolo[3,2-c]quinolines. Sci. Rep. 2015, 5, 9696. [Google Scholar] [CrossRef] [PubMed]
- Boddupally, P.V.; Hahn, S.; Beman, C.; De, B.; Brooks, T.A.; Gokhale, V.; Hurley, L.H. Anticancer activity and cellular repression of c-MYC by the G-quadruplex-stabilizing 11-piperazinylquindoline is not dependent on direct targeting of the G-quadruplex in the c-MYC promoter. J. Med. Chem. 2012, 55, 6076–6086. [Google Scholar] [CrossRef] [PubMed]
- Ou, T.M.; Lin, J.; Lu, Y.J.; Hou, J.Q.; Tan, J.H.; Chen, S.H.; Li, Z.; Li, Y.P.; Li, D.; Gu, L.Q.; et al. Inhibition of cell proliferation by quindoline derivative (SYUIQ-05) through its preferential interaction with c-myc promoter G-quadruplex. J. Med. Chem. 2011, 54, 5671–5679. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Hu, J.; He, B.; Abril, Y.L.N.; Stupinski, J.; Weiser, K.; Carbonaro, M.; Chiang, Y.L.; Southard, T.; Giannakakou, P.; et al. A SIRT2-selective inhibitor promotes c-Myc oncoprotein degradation and exhibits broad anticancer activity. Cancer Cell 2016, 29, 297–310. [Google Scholar] [CrossRef]
- Skwarska, A.; Augustin, E.; Beffinger, M.; Wojtczyk, A.; Konicz, S.; Laskowska, K.; Polewska, J. Targeting of FLT3-ITD kinase contributes to high selectivity of imidazoacridinone C-1311 against FLT3-activated leukemia cells. Biochem. Pharmacol. 2015, 95, 238–252. [Google Scholar] [CrossRef]
- Augustin, E.; Moś-Rompa, A.; Nowak-Ziatyk, D.; Konopa, J. Antitumor 1-nitroacridine derivative C-1748, induces apoptosis, necrosis or senescence in human colon carcinoma HCT8 and HT29 cells. Biochem. Pharmacol. 2009, 79, 1231–1241. [Google Scholar] [CrossRef][Green Version]
- Capizzi, R.L.; Roman, L.A.; Tjulandin, S.; Smirnova, I.; Manikhas, A.; Paterson, J.S.; Major, A.; Lundberg, A.S.; Fumoleau, P. Phase II trial of C1311, a novel inhibitor of topoisomerase II in advanced breast cancer. J. Clin. Oncol. 2008, 26, 1055. [Google Scholar] [CrossRef]
- Laskowski, T.; Borzyszkowska, J.; Grynda, J.; Mazerski, J. C-1311 (Symadex), a potential anti-cancer drug, intercalates into DNA between A and G moieties. NMR-derived and MD-refined stereostructure of the d (GAGGCCTC) 2: C-1311 complex. J. Mol. Struct. 2017, 1141, 357–367. [Google Scholar] [CrossRef]
- Pawłowska, M.; Kwaśniewska, A.; Mazerska, Z.; Augustin, E. Enhanced Activity of P4503A4 and UGT1A10 Induced by Acridinone Derivatives C-1305 and C-1311 in MCF-7 and HCT116 Cancer Cells: Consequences for the Drugs’ Cytotoxicity, Metabolism and Cellular Response. Int. J. Mol. Sci. 2020, 21, 3954. [Google Scholar] [CrossRef]
- Polewska, J.; Skwarska, A.; Augustin, E.; Konopa, J. DNA-damaging imidazoacridinone C-1311 induces autophagy followed by irreversible growth arrest and senescence in human lung cancer cells. J. Pharmacol. Exp. Ther. 2013, 346, 393–405. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Drug Dose [µM] | Cell Line | |||
|---|---|---|---|---|---|
| HCT116 | H460 | CCD 841 CoN | MRC-5 | ||
| C-2028 | IC50 | 0.010 ± 0.001 | 0.016 ± 0.002 | 0.025 ± 0.007 | 0.020 ± 0.004 |
| IC90 | 0.044 ± 0.005 | 0.046 ± 0.004 | 3.69 ± 1.08 | 2.69 ± 0.64 | |
| C-2041 | IC50 | 0.008 ± 0.002 | 0.015 ± 0.002 | 0.036 ± 0.005 | 0.023 ± 0.004 |
| IC90 | 0.049 ± 0.005 | 0.046 ± 0.005 | 5.12 ± 1.09 | 6.16 ± 2.06 | |
| C-2045 | IC50 | 0.059 ± 0.008 | 0.095 ± 0.038 | 0.338 ± 0.027 | 0.218 ± 0.037 |
| IC90 | 0.455 ± 0.026 | 0.399 ± 0.052 | 5.34 ± 1.85 | 6.13 ± 2.35 | |
| C-2053 | IC50 | 0.048 ± 0.014 | 0.025 ± 0.006 | 0.240 ± 0.040 | 0.120 ± 0.026 |
| IC90 | 0.195 ± 0.075 | 0.184 ± 0.022 | 4.10 ± 0.47 | 3.80 ± 1.34 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawłowska, M.; Kulesza, J.; Augustin, E. c-Myc Protein Level Affected by Unsymmetrical Bisacridines Influences Apoptosis and Senescence Induced in HCT116 Colorectal and H460 Lung Cancer Cells. Int. J. Mol. Sci. 2022, 23, 3061. https://doi.org/10.3390/ijms23063061
Pawłowska M, Kulesza J, Augustin E. c-Myc Protein Level Affected by Unsymmetrical Bisacridines Influences Apoptosis and Senescence Induced in HCT116 Colorectal and H460 Lung Cancer Cells. International Journal of Molecular Sciences. 2022; 23(6):3061. https://doi.org/10.3390/ijms23063061
Chicago/Turabian StylePawłowska, Monika, Jolanta Kulesza, and Ewa Augustin. 2022. "c-Myc Protein Level Affected by Unsymmetrical Bisacridines Influences Apoptosis and Senescence Induced in HCT116 Colorectal and H460 Lung Cancer Cells" International Journal of Molecular Sciences 23, no. 6: 3061. https://doi.org/10.3390/ijms23063061
APA StylePawłowska, M., Kulesza, J., & Augustin, E. (2022). c-Myc Protein Level Affected by Unsymmetrical Bisacridines Influences Apoptosis and Senescence Induced in HCT116 Colorectal and H460 Lung Cancer Cells. International Journal of Molecular Sciences, 23(6), 3061. https://doi.org/10.3390/ijms23063061