
Mitochondrial Ca2+ Homeostasis: Emerging Roles and Clinical Significance in Cardiac Remodeling


Abstract
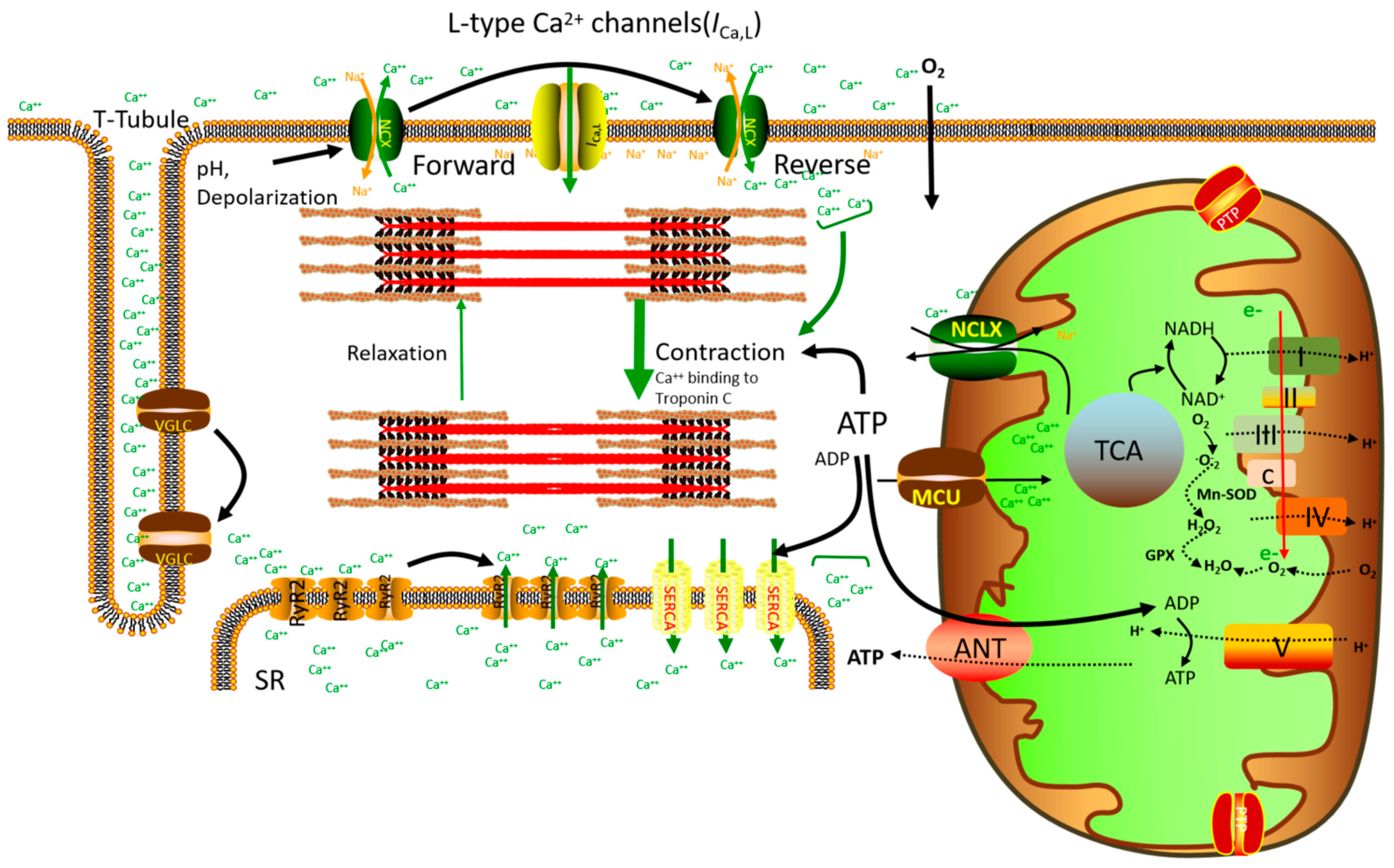
1. Introduction
2. Mitochondrial Ca2+ Homeostasis and Cardiac Energy Regulation
3. Mitochondrial Ca2+ Homeostasis and the Regulation of Cardiac Cell Death
4. Mitochondrial Ca2+ Homeostasis and Mitochondrial ROS Emission
5. Mitochondrial Ca2+ Signaling and Autophagy
6. Mitochondrial Ca2+ Uniporter (mtCU) Protein Complex and Mitochondrial Ca2+ Homeostasis
7. Other Mitochondrial Ca2+ Influx Mechanisms on Mitochondrial Ca2+ Homeostasis
8. MAMs and Mitochondrial Ca2+ Homeostasis
9. Molecular Mechanism Underlying Mitochondrial Calcium Efflux Regulation
10. Mitochondrial Ca2+ Dyshomeostasis and Cardiac Pathological Remodeling
11. Targeting Mitochondria: Mitochondrial Ca2+ as Drug Targets in the Treatment of Cardiovascular Diseases
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Milane, L.; Trivedi, M.; Singh, A.; Talekar, M.; Amiji, M. Mitochondrial biology, targets, and drug delivery. J. Control. Release Off. J. Control. Release Soc. 2015, 207, 40–58. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, H.; Hoppel, C.L. Mitochondria in the human heart. J. Bioenerg. Biomembr. 2009, 41, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Kalkhoran, S.B.; Hernandez-Resendiz, S.; Samangouei, P.; Ong, S.G.; Hausenloy, D.J. Mitochondrial Shaping Proteins in Cardiac Health and Disease—The Long and the Short of It! Cardiovasc. Drugs Ther. 2017, 31, 87–107. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.W.; Tandler, B.; Hoppel, C.L. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J. Biol. Chem. 1977, 252, 8731–8739. [Google Scholar] [CrossRef]
- Kurian, G.A.; Berenshtein, E.; Saada, A.; Chevion, M. Rat cardiac mitochondrial sub-populations show distinct features of oxidative phosphorylation during ischemia, reperfusion and ischemic preconditioning. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2012, 30, 83–94. [Google Scholar] [CrossRef]
- Hollander, J.M.; Thapa, D.; Shepherd, D.L. Physiological and structural differences in spatially distinct subpopulations of cardiac mitochondria: Influence of cardiac pathologies. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1–H14. [Google Scholar] [CrossRef]
- Rosca, M.G.; Hoppel, C.L. Mitochondria in heart failure. Cardiovasc. Res. 2010, 88, 40–50. [Google Scholar] [CrossRef]
- Hoppel, C.L.; Tandler, B.; Fujioka, H.; Riva, A. Dynamic organization of mitochondria in human heart and in myocardial disease. Int. J. Biochem. Cell Biol. 2009, 41, 1949–1956. [Google Scholar] [CrossRef]
- Vasileiou, P.V.S.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686. [Google Scholar] [CrossRef]
- Dietl, A.; Maack, C. Targeting Mitochondrial Calcium Handling and Reactive Oxygen Species in Heart Failure. Curr. Heart Fail. Rep. 2017, 14, 338–349. [Google Scholar] [CrossRef]
- Vasquez-Trincado, C.; Garcia-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, M.; He, S.; Sun, G.; Sun, X. Targeting Calcium Homeostasis in Myocardial Ischemia/Reperfusion Injury: An Overview of Regulatory Mechanisms and Therapeutic Reagents. Front. Pharmacol. 2020, 11, 872. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Macdonald, W.A.; Tang, H.; Hool, L.C. The L-type Ca2+ channel as a therapeutic target in heart disease. Curr. Med. Chem. 2009, 16, 3341–3358. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.N.; Zhang, Y.; Ren, J. Mitophagy, Mitochondrial Dynamics, and Homeostasis in Cardiovascular Aging. Oxidative Med. Cell. Longev. 2019, 2019, 9825061. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M.; McCormack, J.G. Ca2+ as a second messenger within mitochondria of the heart and other tissues. Annu. Rev. Physiol. 1990, 52, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef]
- Dominguez-Rodriguez, A.; Ruiz-Hurtado, G.; Benitah, J.P.; Gomez, A.M. The other side of cardiac Ca2+ signaling: Transcriptional control. Front. Physiol. 2012, 3, 452. [Google Scholar] [CrossRef]
- Fabiato, A.; Fabiato, F. Calcium-induced release of calcium from the sarcoplasmic reticulum of skinned cells from adult human, dog, cat, rabbit, rat, and frog hearts and from fetal and new-born rat ventricles. Ann. N. Y. Acad. Sci. 1978, 307, 491–522. [Google Scholar] [CrossRef]
- Hulme, J.T.; Orchard, C.H. Effect of acidosis on Ca2+ uptake and release by sarcoplasmic reticulum of intact rat ventricular myocytes. Am. J. Physiol. 1998, 275, H977–H987. [Google Scholar] [CrossRef]
- Karlstad, J.; Sun, Y.; Singh, B.B. Ca2+ signaling: An outlook on the characterization of Ca2+ channels and their im-portance in cellular functions. Adv. Exp. Med. Biol. 2012, 740, 143–157. [Google Scholar]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Bittremieux, M.; Missiroli, S.; Morganti, C.; Patergnani, S.; Sbano, L.; Rimessi, A.; Kerkhofs, M.; Parys, J.B.; Bultynck, G.; et al. Endoplasmic Reticulum-Mitochondria Communication Through Ca2+ Signaling: The Importance of Mitochondria-Associated Membranes (MAMs). Adv. Exp. Med. Biol. 2017, 997, 49–67. [Google Scholar] [PubMed]
- Haworth, R.A.; Hunter, D.R. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch. Biochem. Biophys. 1979, 195, 460–467. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Molkentin, J.D. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Kohlhaas, M.; Liu, T.; Knopp, A.; Zeller, T.; Ong, M.F.; Bohm, M.; O’Rourke, B.; Maack, C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 2010, 121, 1606–1613. [Google Scholar] [CrossRef]
- Lambert, J.P.; Murray, E.K.; Elrod, J.W. MCUB and mitochondrial calcium uptake—Modeling, function, and therapeutic potential. Expert Opin. Ther. Targets 2020, 24, 163–169. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Fabiato, A. Simulated calcium current can both cause calcium loading in and trigger calcium release from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J. Gen. Physiol. 1985, 85, 291–320. [Google Scholar] [CrossRef]
- Bers, D.M.; Bassani, J.W.; Bassani, R.A. Na-Ca exchange and Ca fluxes during contraction and relaxation in mammalian ventricular muscle. Ann. N. Y. Acad. Sci. 1996, 779, 430–442. [Google Scholar] [CrossRef]
- Suk, J.Y.; Kim, Y.S.; Park, W.J. HRC (histidine-rich Ca2+ binding protein) resides in the lumen of sarcoplasmic reticulum as a multimer. Biochem. Biophys. Res. Commun. 1999, 263, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Kelley, J.; Schmeisser, G.; Kobayashi, Y.M.; Jones, L.R. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J. Biol. Chem. 1997, 272, 23389–23397. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, G.T.; Ding, S.; Chen, F. Molecular cloning of junctin from human and developing rabbit heart. Mol. Genet. Metab. 2000, 69, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.F.; Bolck, B.; Erdmann, E.; Schwinger, R.H. Sarcoplasmic reticulum Ca2+-ATPase modulates cardiac contraction and relaxation. Cardiovasc. Res. 2003, 57, 20–27. [Google Scholar] [CrossRef]
- Harris, D.A.; Das, A.M. Control of mitochondrial ATP synthesis in the heart. Biochem. J. 1991, 280 Pt 3, 561–573. [Google Scholar] [CrossRef]
- Piquereau, J.; Caffin, F.; Novotova, M.; Lemaire, C.; Veksler, V.; Garnier, A.; Ventura-Clapier, R.; Joubert, F. Mitochondrial dynamics in the adult cardiomyocytes: Which roles for a highly specialized cell? Front. Physiol. 2013, 4, 102. [Google Scholar] [CrossRef]
- Kohlhaas, M.; Maack, C. Interplay of defective excitation-contraction coupling, energy starvation, and oxidative stress in heart failure. Trends Cardiovasc. Med. 2011, 21, 69–73. [Google Scholar] [CrossRef]
- Cortassa, S.; Aon, M.A.; Marban, E.; Winslow, R.L.; O’Rourke, B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys. J. 2003, 84, 2734–2755. [Google Scholar] [CrossRef]
- Viola, H.M.; Hool, L.C. How does calcium regulate mitochondrial energetics in the heart?—New insights. Heart Lung Circ. 2014, 23, 602–609. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Hansford, R.G.; Zorov, D. Role of mitochondrial calcium transport in the control of substrate oxidation. Mol. Cell Biochem. 1998, 184, 359–369. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 1990, 70, 391–425. [Google Scholar] [CrossRef] [PubMed]
- Nichols, B.J.; Rigoulet, M.; Denton, R.M. Comparison of the effects of Ca2+, adenine nucleotides and pH on the kinetic properties of mitochondrial NAD+-isocitrate dehydrogenase and oxoglutarate dehydrogenase from the yeast Saccharomyces cerevisiae and rat heart. Biochem. J. 1994, 303 Pt 2, 461–465. [Google Scholar] [CrossRef]
- McCormack, J.G.; Denton, R.M. Role of Ca2+ ions in the regulation of intramitochondrial metabolism in rat heart. Evidence from studies with isolated mitochondria that adrenaline activates the pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase complexes by increasing the intramitochondrial concentration of Ca2+. Biochem. J. 1984, 218, 235–247. [Google Scholar]
- Rutter, G.A.; Denton, R.M. Regulation of NAD+-linked isocitrate dehydrogenase and 2-oxoglutarate dehydrogenase by Ca2+ ions within toluene-permeabilized rat heart mitochondria. Interactions with regulation by adenine nucleotides and NADH/NAD+ ratios. Biochem. J. 1988, 252, 181–189. [Google Scholar] [CrossRef]
- Balaban, R.S. Cardiac energy metabolism homeostasis: Role of cytosolic calcium. J. Mol. Cell. Cardiol. 2002, 34, 1259–1271. [Google Scholar] [CrossRef]
- Maack, C.; Cortassa, S.; Aon, M.A.; Ganesan, A.N.; Liu, T.; O’Rourke, B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 2006, 99, 172–182. [Google Scholar] [CrossRef]
- Aon, M.A.; Cortassa, S.; Marban, E.; O’Rourke, B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 2003, 278, 44735–44744. [Google Scholar] [CrossRef]
- Rostovtseva, T.; Colombini, M. ATP flux is controlled by a voltage-gated channel from the mitochondrial outer membrane. J. Biol. Chem. 1996, 271, 28006–28008. [Google Scholar] [CrossRef]
- Sampson, M.J.; Lovell, R.S.; Craigen, W.J. The murine voltage-dependent anion channel gene family. Conserved structure and function. J. Biol. Chem. 1997, 272, 18966–18973. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M.; Virji, S.; Ward, J.M. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 1998, 258, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Sheu, S.S.; Nauduri, D.; Anders, M.W. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim. Biophys. Acta 2006, 1762, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Gunter, T.E.; Sheu, S.S. Characteristics and possible functions of mitochondrial Ca2+ transport mechanisms. Biochim. Biophys. Acta 2009, 1787, 1291–1308. [Google Scholar] [CrossRef] [PubMed]
- Liochev, S.I.; Fridovich, I. Superoxide and iron: Partners in crime. IUBMB Life 1999, 48, 157–161. [Google Scholar] [CrossRef]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef]
- Viola, H.M.; Arthur, P.G.; Hool, L.C. Transient exposure to hydrogen peroxide causes an increase in mitochondria-derived superoxide as a result of sustained alteration in L-type Ca2+ channel function in the absence of apoptosis in ventricular myocytes. Circ. Res. 2007, 100, 1036–1044. [Google Scholar] [CrossRef]
- Viola, H.M.; Davies, S.M.; Filipovska, A.; Hool, L.C. L-type Ca2+ channel contributes to alterations in mitochondrial calcium handling in the mdx ventricular myocyte. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H767–H775. [Google Scholar] [CrossRef]
- Viola, H.M.; Arthur, P.G.; Hool, L.C. Evidence for regulation of mitochondrial function by the L-type Ca2+ channel in ventricular myocytes. J. Mol. Cell. Cardiol. 2009, 46, 1016–1026. [Google Scholar] [CrossRef]
- Bers, D.M. Altered cardiac myocyte Ca regulation in heart failure. Physiology 2006, 21, 380–387. [Google Scholar] [CrossRef]
- Kohlhaas, M.; Nickel, A.G.; Maack, C. Mitochondrial energetics and calcium coupling in the heart. J. Physiol. 2017, 595, 3753–3763. [Google Scholar] [CrossRef] [PubMed]
- Cortassa, S.; Aon, M.A.; O’Rourke, B.; Jacques, R.; Tseng, H.J.; Marban, E.; Winslow, R.L. A computational model integrating electrophysiology, contraction, and mitochondrial bioenergetics in the ventricular myocyte. Biophys. J. 2006, 91, 1564–1589. [Google Scholar] [CrossRef] [PubMed]
- Stout, A.K.; Raphael, H.M.; Kanterewicz, B.I.; Klann, E.; Reynolds, I.J. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat. Neurosci. 1998, 1, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, A.; Janke, J.; Doring, H.J.; Leder, O. Myocardial fiber necrosis due to intracellular Ca overload a new principle in cardiac pathophysiology. Recent Adv. Stud. Card. Struct. Metab. 1974, 4, 563–580. [Google Scholar]
- Bernardi, P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef]
- Nieminen, A.L.; Byrne, A.M.; Herman, B.; Lemasters, J.J. Mitochondrial permeability transition in hepatocytes induced by t-BuOOH: NAD(P)H and reactive oxygen species. Am. J. Physiol. 1997, 272 Pt 1, C1286–C1294. [Google Scholar] [CrossRef]
- Vercesi, A.E.; Kowaltowski, A.J.; Grijalba, M.T.; Meinicke, A.R.; Castilho, R.F. The role of reactive oxygen species in mitochondrial permeability transition. Biosci. Rep. 1997, 17, 43–52. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, C.M.; Fogarty, K.E.; Drummond, R.M.; Tuft, R.A.; Walsh, J.V., Jr. Quantitative analysis of spontaneous mitochondrial depolarizations. Biophys. J. 2003, 85, 3350–3357. [Google Scholar] [CrossRef]
- Vergun, O.; Votyakova, T.V.; Reynolds, I.J. Spontaneous changes in mitochondrial membrane potential in single isolated brain mitochondria. Biophys. J. 2003, 85, 3358–3366. [Google Scholar] [CrossRef]
- Huser, J.; Blatter, L.A. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem. J. 1999, 343 Pt 2, 311–317. [Google Scholar] [CrossRef]
- Al-Nasser, I.; Crompton, M. The reversible Ca2+-induced permeabilization of rat liver mitochondria. Biochem. J. 1986, 239, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Al-Nasser, I.; Crompton, M. The entrapment of the Ca2+ indicator arsenazo III in the matrix space of rat liver mitochondria by permeabilization and resealing. Na+-dependent and -independent effluxes of Ca2+ in arsenazo III-loaded mitochondria. Biochem. J. 1986, 239, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Vergun, O.; Reynolds, I.J. Fluctuations in mitochondrial membrane potential in single isolated brain mitochondria: Modulation by adenine nucleotides and Ca2+. Biophys. J. 2004, 87, 3585–3593. [Google Scholar] [CrossRef]
- Bernardi, P.; Broekemeier, K.M.; Pfeiffer, D.R. Recent progress on regulation of the mitochondrial permeability transition pore; a cyclosporin-sensitive pore in the inner mitochondrial membrane. J. Bioenerg. Biomembr. 1994, 26, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Gunter, T.E.; Pfeiffer, D.R. Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 1990, 258 Pt 1, C755–C786. [Google Scholar] [CrossRef] [PubMed]
- Altschuld, R.A.; Hohl, C.M.; Castillo, L.C.; Garleb, A.A.; Starling, R.C.; Brierley, G.P. Cyclosporin inhibits mitochondrial calcium efflux in isolated adult rat ventricular cardiomyocytes. Am. J. Physiol. 1992, 262 Pt 2, H1699–H1704. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Reed, J.C. Mitochondrial control of cell death. Nat. Med. 2000, 6, 513–519. [Google Scholar] [CrossRef]
- Giorgi, C.; Romagnoli, A.; Pinton, P.; Rizzuto, R. Ca2+ signaling, mitochondria and cell death. Curr. Mol. Med. 2008, 8, 119–130. [Google Scholar]
- Pinton, P.; Ferrari, D.; Rapizzi, E.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: Significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001, 20, 2690–2701. [Google Scholar] [CrossRef]
- Szalai, G.; Krishnamurthy, R.; Hajnoczky, G. Apoptosis driven by IP3-linked mitochondrial calcium signals. EMBO J. 1999, 18, 6349–6361. [Google Scholar] [CrossRef]
- Blackshaw, S.; Sawa, A.; Sharp, A.H.; Ross, C.A.; Snyder, S.H.; Khan, A.A. Type 3 inositol 1,4,5-trisphosphate receptor modulates cell death. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2000, 14, 1375–1379. [Google Scholar]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Beutner, G.; Ruck, A.; Riede, B.; Welte, W.; Brdiczka, D. Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Lett. 1996, 396, 189–195. [Google Scholar] [CrossRef]
- McEnery, M.W.; Snowman, A.M.; Trifiletti, R.R.; Snyder, S.H. Isolation of the mitochondrial benzodiazepine receptor: Association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc. Natl. Acad. Sci. USA 1992, 89, 3170–3174. [Google Scholar] [CrossRef] [PubMed]
- Capano, M.; Crompton, M. Biphasic translocation of Bax to mitochondria. Biochem. J. 2002, 367 Pt 1, 169–178. [Google Scholar] [CrossRef]
- Beutner, G.; Ruck, A.; Riede, B.; Brdiczka, D. Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases. Biochim. Et Biophys. Acta 1998, 1368, 7–18. [Google Scholar] [CrossRef]
- Brustovetsky, N.; Klingenberg, M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+. Biochemistry 1996, 35, 8483–8488. [Google Scholar] [CrossRef]
- Kokoszka, J.E.; Waymire, K.G.; Levy, S.E.; Sligh, J.E.; Cai, J.; Jones, D.P.; MacGregor, G.R.; Wallace, D.C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004, 427, 461–465. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007, 9, 550–555. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Ramachandran, A.; Lebofsky, M.; Baines, C.P.; Lemasters, J.J.; Jaeschke, H. Cyclophilin D deficiency protects against acetaminophen-induced oxidant stress and liver injury. Free Radic. Res. 2011, 45, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Ichas, F.; Jouaville, L.S.; Mazat, J.P. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 1997, 89, 1145–1153. [Google Scholar] [CrossRef]
- Elrod, J.W.; Wong, R.; Mishra, S.; Vagnozzi, R.J.; Sakthievel, B.; Goonasekera, S.A.; Karch, J.; Gabel, S.; Farber, J.; Force, T.; et al. Cyclophilin D controls mitochondrial pore-dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Investig. 2010, 120, 3680–3687. [Google Scholar] [CrossRef]
- Brookes, P.S.; Levonen, A.L.; Shiva, S.; Sarti, P.; Darley-Usmar, V.M. Mitochondria: Regulators of signal transduction by reactive oxygen and nitrogen species. Free Radic. Biol. Med. 2002, 33, 755–764. [Google Scholar] [CrossRef]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement from the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Chen, Y.R.; Zweier, J.L. Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 2014, 114, 524–537. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Nickel, A.G.; von Hardenberg, A.; Hohl, M.; Loffler, J.R.; Kohlhaas, M.; Becker, J.; Reil, J.C.; Kazakov, A.; Bonnekoh, J.; Stadelmaier, M.; et al. Reversal of Mitochondrial Transhydrogenase Causes Oxidative Stress in Heart Failure. Cell Metab. 2015, 22, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wei, C.L.; Zhang, W.R.; Cheng, H.P.; Liu, J. Cross-talk between calcium and reactive oxygen species signaling. Acta Pharmacol. Sin. 2006, 27, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Wan, B.; LaNoue, K.F.; Cheung, J.Y.; Scaduto, R.C., Jr. Regulation of citric acid cycle by calcium. J. Biol. Chem. 1989, 264, 13430–13439. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357 Pt 3, 593–615. [Google Scholar] [CrossRef]
- Grijalba, M.T.; Vercesi, A.E.; Schreier, S. Ca2+-induced increased lipid packing and domain formation in submitochondrial particles. A possible early step in the mechanism of Ca2+-stimulated generation of reactive oxygen species by the respiratory chain. Biochemistry 1999, 38, 13279–13287. [Google Scholar] [CrossRef]
- Bathori, G.; Csordas, G.; Garcia-Perez, C.; Davies, E.; Hajnoczky, G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J. Biol. Chem. 2006, 281, 17347–17358. [Google Scholar] [CrossRef]
- Feissner, R.F.; Skalska, J.; Gaum, W.E.; Sheu, S.S. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. 2009, 14, 1197–1218. [Google Scholar] [CrossRef]
- Marcil, M.; Bourduas, K.; Ascah, A.; Burelle, Y. Exercise training induces respiratory substrate-specific decrease in Ca2+-induced permeability transition pore opening in heart mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1549–H1557. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Starkov, A.A.; Fiskum, G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J. Neurochem. 2003, 86, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Kembro, J.M.; Aon, M.A.; Winslow, R.L.; O’Rourke, B.; Cortassa, S. Integrating mitochondrial energetics, redox and ROS metabolic networks: A two-compartment model. Biophys. J. 2013, 104, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.D.; Greenstein, J.L.; Cortassa, S.; O’Rourke, B.; Winslow, R.L. A computational model of reactive oxygen species and redox balance in cardiac mitochondria. Biophys. J. 2013, 105, 1045–1056. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aon, M.A.; Cortassa, S.; O’Rourke, B. Redox-optimized ROS balance: A unifying hypothesis. Biochim. Biophys. Acta 2010, 1797, 865–877. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, L.; Yu, C.A. Generation of superoxide anion by succinate-cytochrome c reductase from bovine heart mitochondria. J. Biol. Chem. 1998, 273, 33972–33976. [Google Scholar] [CrossRef]
- Tretter, L.; Adam-Vizi, V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 7771–7778. [Google Scholar] [CrossRef]
- Cardenas, C.; Foskett, J.K. Mitochondrial Ca2+ signals in autophagy. Cell Calcium 2012, 52, 44–51. [Google Scholar] [CrossRef]
- La Rovere, R.M.; Roest, G.; Bultynck, G.; Parys, J.B. Intracellular Ca2+ signaling and Ca2+ microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 2016, 60, 74–87. [Google Scholar] [CrossRef]
- East, D.A.; Campanella, M. Ca2+ in quality control: An unresolved riddle critical to autophagy and mitophagy. Autophagy 2013, 9, 1710–1719. [Google Scholar] [CrossRef]
- Cardenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgo, J.; Muller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef]
- MacVicar, T.D.; Mannack, L.V.; Lees, R.M.; Lane, J.D. Targeted siRNA Screens Identify ER-to-Mitochondrial Calcium Exchange in Autophagy and Mitophagy Responses in RPE1 Cells. Int. J. Mol. Sci. 2015, 16, 13356–13380. [Google Scholar] [CrossRef] [PubMed]
- Granatiero, V.; Giorgio, V.; Cali, T.; Patron, M.; Brini, M.; Bernardi, P.; Tiranti, V.; Zeviani, M.; Pallafacchina, G.; De Stefani, D.; et al. Reduced mitochondrial Ca2+ transients stimulate autophagy in human fibroblasts carrying the 13514A>G mutation of the ND5 subunit of NADH dehydrogenase. Cell Death Differ. 2016, 23, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Ahumada-Castro, U.; Silva-Pavez, E.; Lovy, A.; Pardo, E.; Molgomicron, J.; Cardenas, C. MTOR-independent autophagy induced by interrupted endoplasmic reticulum-mitochondrial Ca2+ communication: A dead end in cancer cells. Autophagy 2019, 15, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.; Cheng, X.T.; Lin, M.Y.; Huang, N.; Sheng, Z.H. Mul1 restrains Parkin-mediated mitophagy in mature neurons by maintaining ER-mitochondrial contacts. Nat. Commun. 2019, 10, 3645. [Google Scholar] [CrossRef]
- Kania, E.; Roest, G.; Vervliet, T.; Parys, J.B.; Bultynck, G. IP3 Receptor-Mediated Calcium Signaling and Its Role in Autophagy in Cancer. Front. Oncol. 2017, 7, 140. [Google Scholar] [CrossRef]
- Gherardi, G.; Di Marco, G.; Rizzuto, R.; Mammucari, C. Crosstalk between Mitochondrial Ca2+ Uptake and Autophagy in Skeletal Muscle. Oxidative Med. Cell. Longev. 2019, 2019, 1845321. [Google Scholar] [CrossRef]
- Mallilankaraman, K.; Cardenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenar, T.; Csordas, G.; Madireddi, P.; Yang, J.; Muller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Parys, J.B.; Bultynck, G. ITPRs/inositol 1,4,5-trisphosphate receptors in autophagy: From enemy to ally. Autophagy 2015, 11, 1944–1948. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C.J. The ER-Mitochondria Tethering Complex VAPB-PTPIP51 Regulates Autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef]
- Pacher, P.; Thomas, A.P.; Hajnoczky, G. Ca2+ marks: Miniature calcium signals in single mitochondria driven by ryanodine receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 2380–2385. [Google Scholar] [CrossRef]
- Collins, T.J.; Lipp, P.; Berridge, M.J.; Bootman, M.D. Mitochondrial Ca2+ uptake depends on the spatial and temporal profile of cytosolic Ca2+ signals. J. Biol. Chem. 2001, 276, 26411–26420. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.L.; Adaniya, S.M.; Cypress, M.W.; Suzuki, Y.; Kusakari, Y.; Jhun, B.S.; Jin, O. Role of mitochondrial Ca2+ homeostasis in cardiac muscles. Arch. Biochem. Biophys. 2019, 663, 276–287. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef] [PubMed]
- Mammucari, C.; Raffaello, A.; Vecellio Reane, D.; Rizzuto, R. Molecular structure and pathophysiological roles of the Mitochondrial Calcium Uniporter. Biochim. Biophys. Acta 2016, 1863, 2457–2464. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A. VDAC1 functions in Ca2+ homeostasis and cell life and death in health and disease. Cell Calcium 2018, 69, 81–100. [Google Scholar] [CrossRef]
- Deluca, H.F.; Engstrom, G.W. Calcium uptake by rat kidney mitochondria. Proc. Natl. Acad. Sci. USA 1961, 47, 1744–1750. [Google Scholar] [CrossRef]
- Vasington, F.D.; Murphy, J.V. Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J. Biol. Chem. 1962, 237, 2670–2677. [Google Scholar] [CrossRef]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 2010, 467, 291–296. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Sho-shan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabo, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [PubMed]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 2013, 8, e55785. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovacs-Bogdan, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.E.; Chandramoorthy, H.C.; Shanmughapriya, S.; Zhang, X.Q.; Vallem, S.; Doonan, P.J.; Mallianka-raman, K.; Guo, S.; Rajan, S.; Elrod, J.W.; et al. SLC25A23 augments mitochondrial Ca2+ uptake, interacts with MCU, and induces oxidative stress-mediated cell death. Mol. Biol. Cell 2014, 25, 936–947. [Google Scholar] [CrossRef]
- Fan, M.; Zhang, J.; Tsai, C.W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.F.; Feng, L. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 2020, 582, 129–133. [Google Scholar] [CrossRef]
- Yoo, J.; Wu, M.; Yin, Y.; Herzik, M.A., Jr.; Lander, G.C.; Lee, S.Y. Cryo-EM structure of a mitochondrial calcium uniporter. Science 2018, 361, 506–511. [Google Scholar] [CrossRef]
- Fan, C.; Fan, M.; Orlando, B.J.; Fastman, N.M.; Zhang, J.; Xu, Y.; Chambers, M.G.; Xu, X.; Perry, K.; Liao, M.; et al. X-ray and cryo-EM structures of the mitochondrial calcium uniporter. Nature 2018, 559, 575–579. [Google Scholar] [CrossRef]
- Carafoli, E.; Lehninger, A.L. A survey of the interaction of calcium ions with mitochondria from different tissues and species. Biochem. J. 1971, 122, 681–690. [Google Scholar] [CrossRef]
- Rottenberg, H.; Scarpa, A. Calcium uptake and membrane potential in mitochondria. Biochemistry 1974, 13, 4811–4817. [Google Scholar] [CrossRef]
- Xie, A.; Zhou, A.; Liu, H.; Shi, G.; Liu, M.; Boheler, K.R.; Dudley, S.C., Jr. Mitochondrial Ca2+ flux modulates spontaneous electrical activity in ventricular cardiomyocytes. PLoS ONE 2018, 13, e0200448. [Google Scholar] [CrossRef]
- Zhao, Z.; Gordan, R.; Wen, H.; Fefelova, N.; Zang, W.J.; Xie, L.H. Modulation of intracellular calcium waves and triggered activities by mitochondrial ca flux in mouse cardiomyocytes. PLoS ONE 2013, 8, e80574. [Google Scholar] [CrossRef] [PubMed]
- Haumann, J.; Camara, A.K.S.; Gadicherla, A.K.; Navarro, C.D.; Boelens, A.D.; Blomeyer, C.A.; Dash, R.K.; Boswell, M.R.; Kwok, W.M.; Stowe, D.F. Slow Ca2+ Efflux by Ca2+/H+ Exchange in Cardiac Mitochondria Is Modulated by Ca2+ Re-uptake via MCU, Extra-Mitochondrial pH, and H+ Pumping by F0F1-ATPase. Front. Physiol. 2018, 9, 1914. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, E.J.; Stathopulos, P.B.; Madesh, M. Regulation of Ca2+ exchanges and signaling in mitochondria. Curr. Opin. Physiol. 2020, 17, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Bick, A.G.; Calvo, S.E.; Mootha, V.K. Evolutionary diversity of the mitochondrial calcium uniporter. Science 2012, 336, 886. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Mootha, V.K. The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Biol. 2015, 16, 545–553. [Google Scholar] [CrossRef]
- Drago, I.; De Stefani, D.; Rizzuto, R.; Pozzan, T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 12986–12991. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Lu, X.; Correll, R.N.; Schwanekamp, J.A.; Vagnozzi, R.J.; Sargent, M.A.; York, A.J.; Zhang, J.; Bers, D.M.; Molkentin, J.D. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015, 12, 15–22. [Google Scholar] [CrossRef]
- Luongo, T.S.; Lambert, J.P.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef]
- Uchi, J.; Jhun, B.S.; Xu, S.; Hurst, S.; Raffaello, A.; Liu, X.; Yi, B.; Zhang, H.; Gross, P.; Mishra, J.; et al. Adrenergic signaling regulates mitochondrial Ca2+ uptake through Pyk2-dependent tyrosine phosphorylation of the mitochondrial Ca2+ uniporter. Antioxid. Redox Signal. 2014, 21, 863–879. [Google Scholar] [CrossRef]
- Lambert, J.P.; Luongo, T.S.; Tomar, D.; Jadiya, P.; Gao, E.; Zhang, X.; Lucchese, A.M.; Kolmetzky, D.W.; Shah, N.S.; Elrod, J.W. MCUB Regulates the Molecular Composition of the Mitochondrial Calcium Uniporter Channel to Limit Mitochondrial Calcium Overload During Stress. Circulation 2019, 140, 1720–1733. [Google Scholar] [CrossRef]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Vecellio Reane, D.; Mantoan, M.; Granatiero, V.; Szabo, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.E.; Chandramoorthy, H.C.; Shamugapriya, S.; Zhang, X.; Rajan, S.; Mallilankaraman, K.; Gandhirajan, R.K.; Vagnozzi, R.J.; Ferrer, L.M.; Sreekrishnanilayam, K.; et al. MICU1 motifs define mitochondrial calcium uniporter binding and activity. Cell Rep. 2013, 5, 1576–1588. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.R.; Groschner, L.N.; Parichatikanond, W.; Kuo, L.; Bondarenko, A.I.; Rost, R.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial Ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic beta-cells. J. Biol. Chem. 2012, 287, 34445–34454. [Google Scholar] [CrossRef]
- Bick, A.G.; Wakimoto, H.; Kamer, K.J.; Sancak, Y.; Goldberger, O.; Axelsson, A.; DeLaughter, D.M.; Gorham, J.M.; Mootha, V.K.; Seidman, J.G.; et al. Cardiovascular homeostasis dependence on MICU2, a regulatory subunit of the mitochondrial calcium uniporter. Proc. Natl. Acad. Sci. USA 2017, 114, E9096–E9104. [Google Scholar] [CrossRef] [PubMed]
- Waldeck-Weiermair, M.; Malli, R.; Parichatikanond, W.; Gottschalk, B.; Madreiter-Sokolowski, C.T.; Klec, C.; Rost, R.; Graier, W.F. Rearrangement of MICU1 multimers for activation of MCU is solely controlled by cytosolic Ca2+. Sci. Rep. 2015, 5, 15602. [Google Scholar] [CrossRef]
- Vais, H.; Mallilankaraman, K.; Mak, D.D.; Hoff, H.; Payne, R.; Tanis, J.E.; Foskett, J.K. EMRE Is a Matrix Ca2+ Sensor that Governs Gatekeeping of the Mitochondrial Ca2+ Uniporter. Cell Rep. 2016, 14, 403–410. [Google Scholar] [CrossRef]
- Tomar, D.; Dong, Z.; Shanmughapriya, S.; Koch, D.A.; Thomas, T.; Hoffman, N.E.; Timbalia, S.A.; Goldman, S.J.; Breves, S.L.; Corbally, D.P.; et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 2016, 15, 1673–1685. [Google Scholar] [CrossRef]
- Nemani, N.; Shanmughapriya, S.; Madesh, M. Molecular regulation of MCU: Implications in physiology and disease. Cell Calcium 2018, 74, 86–93. [Google Scholar] [CrossRef]
- Chaudhuri, D.; Artiga, D.J.; Abiria, S.A.; Clapham, D.E. Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc. Natl. Acad. Sci. USA 2016, 113, E1872–E1880. [Google Scholar] [CrossRef]
- Dong, Z.; Shanmughapriya, S.; Tomar, D.; Siddiqui, N.; Lynch, S.; Nemani, N.; Breves, S.L.; Zhang, X.; Tripathi, A.; Palaniappan, P.; et al. Mitochondrial Ca2+ Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol. Cell 2017, 65, 1014–1028.e7. [Google Scholar] [CrossRef]
- Jhun, B.S.; Mishra, J.; Monaco, S.; Fu, D.; Jiang, W.; Sheu, S.S.; O-Uchi, J. The mitochondrial Ca2+ uniporter: Regulation by auxiliary subunits and signal transduction pathways. Am. J. Physiol. Cell Physiol. 2016, 311, C67–C80. [Google Scholar] [CrossRef] [PubMed]
- Beutner, G.; Sharma, V.K.; Lin, L.; Ryu, S.Y.; Dirksen, R.T.; Sheu, S.S. Type 1 ryanodine receptor in cardiac mitochondria: Transducer of excitation-metabolism coupling. Biochim. Biophys. Acta 2005, 1717, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jin, O.; Jhun, B.S.; Hurst, S.; Bisetto, S.; Gross, P.; Chen, M.; Kettlewell, S.; Park, J.; Oyamada, H.; Smith, G.L.; et al. Overexpression of ryanodine receptor type 1 enhances mitochondrial fragmentation and Ca2+-induced ATP production in cardiac H9c2 myoblasts. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1736–H1751. [Google Scholar]
- Buntinas, L.; Gunter, K.K.; Sparagna, G.C.; Gunter, T.E. The rapid mode of calcium uptake into heart mitochondria (RaM): Comparison to RaM in liver mitochondria. Biochim. Biophys. Acta 2001, 1504, 248–261. [Google Scholar] [CrossRef]
- Gunter, T.E.; Gunter, K.K.; Sheu, S.S.; Gavin, C.E. Mitochondrial calcium transport: Physiological and pathological relevance. Am. J. Physiol. 1994, 267 Pt 1, C313–C339. [Google Scholar] [CrossRef] [PubMed]
- Motloch, L.J.; Larbig, R.; Gebing, T.; Reda, S.; Schwaiger, A.; Leitner, J.; Wolny, M.; Eckardt, L.; Hoppe, U.C. By Regulating Mitochondrial Ca2+-Uptake UCP2 Modulates Intracellular Ca2+. PLoS ONE 2016, 11, e0148359. [Google Scholar] [CrossRef] [PubMed]
- Michels, G.; Khan, I.F.; Endres-Becker, J.; Rottlaender, D.; Herzig, S.; Ruhparwar, A.; Wahlers, T.; Hoppe, U.C. Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage-gated Ca2+ channels. Circulation 2009, 119, 2435–2443. [Google Scholar] [CrossRef] [PubMed]
- Motloch, L.J.; Gebing, T.; Reda, S.; Schwaiger, A.; Wolny, M.; Hoppe, U.C. UCP3 Regulates Single-Channel Activity of the Cardiac mCa1. J. Membr. Biol. 2016, 249, 577–584. [Google Scholar] [CrossRef]
- Bogeski, I.; Gulaboski, R.; Kappl, R.; Mirceski, V.; Stefova, M.; Petreska, J.; Hoth, M. Calcium binding and transport by coenzyme Q. J. Am. Chem. Soc. 2011, 133, 9293–9303. [Google Scholar] [CrossRef]
- Waldeck-Weiermair, M.; Jean-Quartier, C.; Rost, R.; Khan, M.J.; Vishnu, N.; Bondarenko, A.I.; Imamura, H.; Malli, R.; Graier, W.F. Leucine zipper EF hand-containing transmembrane protein 1 (Letm1) and uncoupling proteins 2 and 3 (UCP2/3) contribute to two distinct mitochondrial Ca2+ uptake pathways. J. Biol. Chem. 2011, 286, 28444–28455. [Google Scholar] [CrossRef]
- Ricquier, D.; Bouillaud, F. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem. J. 2000, 345 Pt 2, 161–179. [Google Scholar] [CrossRef] [PubMed]
- Trenker, M.; Malli, R.; Fertschai, I.; Levak-Frank, S.; Graier, W.F. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat. Cell Biol. 2007, 9, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Waldeck-Weiermair, M.; Malli, R.; Naghdi, S.; Trenker, M.; Kahn, M.J.; Graier, W.F. The contribution of UCP2 and UCP3 to mitochondrial Ca2+ uptake is differentially determined by the source of supplied Ca2+. Cell Calcium 2010, 47, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Waldeck-Weiermair, M.; Duan, X.; Naghdi, S.; Khan, M.J.; Trenker, M.; Malli, R.; Graier, W.F. Uncoupling protein 3 adjusts mitochondrial Ca2+ uptake to high and low Ca2+ signals. Cell Calcium 2010, 48, 288–301. [Google Scholar] [CrossRef]
- Bondarenko, A.I.; Parichatikanond, W.; Madreiter, C.T.; Rost, R.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. UCP2 modulates single-channel properties of a MCU-dependent Ca2+ inward current in mitochondria. Pflug. Arch. Eur. J. Physiol. 2015, 467, 2509–2518. [Google Scholar] [CrossRef]
- De Marchi, U.; Castelbou, C.; Demaurex, N. Uncoupling protein 3 (UCP3) modulates the activity of Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) by decreasing mitochondrial ATP production. J. Biol. Chem. 2011, 286, 32533–32541. [Google Scholar] [CrossRef]
- Mehta, S.L.; Li, P.A. Neuroprotective role of mitochondrial uncoupling protein 2 in cerebral stroke. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2009, 29, 1069–1078. [Google Scholar] [CrossRef]
- Larbig, R.; Reda, S.; Paar, V.; Trost, A.; Leitner, J.; Weichselbaumer, S.; Motloch, K.A.; Wernly, B.; Arrer, A.; Strauss, B.; et al. Through modulation of cardiac Ca2+ handling, UCP2 affects cardiac electrophysiology and influences the susceptibility for Ca2+ -mediated arrhythmias. Exp. Physiol. 2017, 102, 650–662. [Google Scholar] [CrossRef]
- Koch, G.L. The endoplasmic reticulum and calcium storage. Bioessays 1990, 12, 527–531. [Google Scholar] [CrossRef]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef]
- Copeland, D.E.; Dalton, A.J. An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J. Biophys. Biochem. Cytol. 1959, 5, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Hayashi, T. New insights into the role of mitochondria-associated endoplasmic reticulum membrane. Inter-Natl. Rev. Cell Mol. Biol. 2011, 292, 73–117. [Google Scholar]
- Area-Gomez, E.; Schon, E.A. Mitochondria-associated ER membranes and Alzheimer disease. Curr. Opin. Genet. Dev. 2016, 38, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Marchi, S.; Bonora, M.; Aguiari, P.; Bononi, A.; De Stefani, D.; Giorgi, C.; Leo, S.; Rimessi, A.; Siviero, R.; et al. Ca2+ transfer from the ER to mitochondria: When, how and why. Biochim. Biophys. Acta 2009, 1787, 1342–1351. [Google Scholar] [CrossRef]
- Csordas, G.; Weaver, D.; Hajnoczky, G. Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Lam, A.K.; Galione, A. The endoplasmic reticulum and junctional membrane communication during calcium signaling. Biochim. Biophys. Acta 2013, 1833, 2542–2559. [Google Scholar] [CrossRef]
- Raturi, A.; Simmen, T. Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (MAM). Biochim. Biophys. Acta 2013, 1833, 213–224. [Google Scholar] [CrossRef]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.P. MAM: More than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef]
- D’Eletto, M.; Rossin, F.; Occhigrossi, L.; Farrace, M.G.; Faccenda, D.; Desai, R.; Marchi, S.; Refolo, G.; Falasca, L.; Antonioli, M.; et al. Transglutaminase Type 2 Regulates ER-Mitochondria Contact Sites by Interacting with GRP75. Cell Rep. 2018, 25, 3573–3581.e4. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Varnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Xu, H.; Guan, N.; Ren, Y.L.; Wei, Q.J.; Tao, Y.H.; Yang, G.S.; Liu, X.Y.; Bu, D.F.; Zhang, Y.; Zhu, S.N. IP3R-Grp75-VDAC1-MCU calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an Adriamycin nephropathy rat model. BMC Nephrol. 2018, 19, 140. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. USA 2019, 116, 25322–25328. [Google Scholar] [CrossRef] [PubMed]
- Paillard, M.; Tubbs, E.; Thiebaut, P.A.; Gomez, L.; Fauconnier, J.; Da Silva, C.C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A.; et al. Depressing mitochondria-reticulum inter-actions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation 2013, 128, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Simmen, T.; Aslan, J.E.; Blagoveshchenskaya, A.D.; Thomas, L.; Wan, L.; Xiang, Y.; Feliciangeli, S.F.; Hung, C.H.; Crump, C.M.; Thomas, G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005, 24, 717–729. [Google Scholar] [CrossRef]
- Myhill, N.; Lynes, E.M.; Nanji, J.A.; Blagoveshchenskaya, A.D.; Fei, H.; Carmine Simmen, K.; Cooper, T.J.; Thomas, G.; Simmen, T. The subcellular distribution of calnexin is mediated by PACS-2. Mol. Biol. Cell 2008, 19, 2777–2788. [Google Scholar] [CrossRef]
- Wu, Z.; Bowen, W.D. Role of sigma-1 receptor C-terminal segment in inositol 1,4,5-trisphosphate receptor activation: Constitutive enhancement of calcium signaling in MCF-7 tumor cells. J. Biol. Chem. 2008, 283, 28198–28215. [Google Scholar] [CrossRef]
- Su, T.P.; Su, T.C.; Nakamura, Y.; Tsai, S.Y. The Sigma-1 Receptor as a Pluripotent Modulator in Living Systems. Trends Pharm. Sci 2016, 37, 262–278. [Google Scholar] [CrossRef]
- Matsuzaki, H.; Fujimoto, T.; Tanaka, M.; Shirasawa, S. Tespa1 is a novel component of mitochondria-associated endoplasmic reticulum membranes and affects mitochondrial calcium flux. Biochem. Biophys. Res. Commun. 2013, 433, 322–326. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, D.; Zhou, L.; Pei, Y.; Zhuang, Y.; Cui, W.; Chen, J. FUN14 domain-containing 1 promotes breast cancer proliferation and migration by activating calcium-NFATC1-BMI1 axis. EBioMedicine 2019, 41, 384–394. [Google Scholar] [CrossRef]
- Ren, J.; Sun, M.; Zhou, H.; Ajoolabady, A.; Zhou, Y.; Tao, J.; Sowers, J.R.; Zhang, Y. FUNDC1 interacts with FBXL2 to govern mitochondrial integrity and cardiac function through an IP3R3-dependent manner in obesity. Sci. Adv. 2020, 6, eabc8561. [Google Scholar] [CrossRef]
- Wu, W.; Lin, C.; Wu, K.; Jiang, L.; Wang, X.; Li, W.; Zhuang, H.; Zhang, X.; Chen, H.; Li, S.; et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 2016, 35, 1368–1384. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007, 8, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell Rep. 2016, 15, 2226–2238. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [PubMed]
- Drago, I.; Pizzo, P.; Pozzan, T. After half a century mitochondrial calcium in- and efflux machineries reveal themselves. EMBO J. 2011, 30, 4119–4125. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Tsai, M.F.; Jiang, D.; Zhao, L.; Clapham, D.; Miller, C. Functional reconstitution of the mitochondrial Ca2+/H+ antiporter Letm1. J. Gen. Physiol. 2014, 143, 67–73. [Google Scholar] [CrossRef]
- Palty, R.; Ohana, E.; Hershfinkel, M.; Volokita, M.; Elgazar, V.; Beharier, O.; Silverman, W.F.; Argaman, M.; Sekler, I. Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J. Biol. Chem. 2004, 279, 25234–25240. [Google Scholar] [CrossRef]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef]
- Morciano, G.; Bonora, M.; Campo, G.; Aquila, G.; Rizzo, P.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Mechanistic Role of mPTP in Ischemia-Reperfusion Injury. Adv. Exp. Med. Biol. 2017, 982, 169–189. [Google Scholar]
- De Marchi, E.; Bonora, M.; Giorgi, C.; Pinton, P. The mitochondrial permeability transition pore is a dispensable element for mitochondrial calcium efflux. Cell Calcium 2014, 56, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Saotome, M.; Katoh, H.; Satoh, H.; Nagasaka, S.; Yoshihara, S.; Terada, H.; Hayashi, H. Mitochondrial membrane potential modulates regulation of mitochondrial Ca2+ in rat ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1820–H1828. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Liu, X.; Wang, W. Regulation of metabolism in individual mitochondria during excitation-contraction coupling. J. Mol. Cell. Cardiol. 2014, 76, 235–246. [Google Scholar] [CrossRef]
- Akopova, O.V. The role of mitochondrial permeability transition pore in transmembrane Ca2+-exchange in mitochondria. Ukrains’ kyi Biokhimichnyi Zhurnal (1999) 2008, 80, 40–47. [Google Scholar]
- Bhosale, G.; Sharpe, J.A.; Sundier, S.Y.; Duchen, M.R. Calcium signaling as a mediator of cell energy demand and a trigger to cell death. Ann. N. Y. Acad. Sci. 2015, 1350, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, A.B.; Gottlieb, R.A. Heart mitochondria: Gates of life and death. Cardiovasc. Res. 2008, 77, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.C.; Son, M.J.; Woo, S.H. Regulation of cardiac calcium by mechanotransduction: Role of mitochondria. Arch. Biochem. Biophys. 2018, 659, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Qiu, H. The Physiological and Pathological Roles of Mitochondrial Calcium Uptake in Heart. Int. J. Mol. Sci. 2020, 21, 7689. [Google Scholar] [CrossRef]
- Gong, Y.; Lin, J.; Ma, Z.; Yu, M.; Wang, M.; Lai, D.; Fu, G. Mitochondria-associated membrane-modulated Ca2+ transfer: A potential treatment target in cardiac ischemia reperfusion injury and heart failure. Life Sci. 2021, 278, 119511. [Google Scholar] [CrossRef]
- Luo, M.; Anderson, M.E. Mechanisms of altered Ca2+ handling in heart failure. Circ. Res. 2013, 113, 690–708. [Google Scholar] [CrossRef]
- Marks, A.R. Calcium cycling proteins and heart failure: Mechanisms and therapeutics. J. Clin. Investig. 2013, 123, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Belevych, A.; Kubalova, Z.; Terentyev, D.; Hamlin, R.L.; Carnes, C.A.; Gyorke, S. Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys. J. 2007, 93, 4083–4092. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Karamanlidis, G.; Nascimben, L.; Couper, G.S.; Shekar, P.S.; del Monte, F.; Tian, R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ. Res. 2010, 106, 1541–1548. [Google Scholar] [CrossRef]
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintron, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W., II.; Kang, Y.J.; Prolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef]
- Huang, Q.; Zhou, H.J.; Zhang, H.; Huang, Y.; Hinojosa-Kirschenbaum, F.; Fan, P.; Yao, L.; Belardinelli, L.; Tellides, G.; Giordano, F.J.; et al. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation 2015, 131, 1082–1097. [Google Scholar] [CrossRef]
- Graham, D.; Huynh, N.N.; Hamilton, C.A.; Beattie, E.; Smith, R.A.; Cocheme, H.M.; Murphy, M.P.; Dominiczak, A.F. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension 2009, 54, 322–328. [Google Scholar] [CrossRef]
- Solaini, G.; Harris, D.A. Biochemical dysfunction in heart mitochondria exposed to ischaemia and reperfusion. Biochem. J. 2005, 390 Pt 2, 377–394. [Google Scholar] [CrossRef]
- Jassem, W.; Fuggle, S.V.; Rela, M.; Koo, D.D.; Heaton, N.D. The role of mitochondria in ischemia/reperfusion injury. Transplantation 2002, 73, 493–499. [Google Scholar] [CrossRef]
- Miura, T.; Tanno, M.; Sato, T. Mitochondrial kinase signalling pathways in myocardial protection from ischae-mia/reperfusion-induced necrosis. Cardiovasc. Res. 2010, 88, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Venardos, K.M.; Perkins, A.; Headrick, J.; Kaye, D.M. Myocardial ischemia-reperfusion injury, antioxidant enzyme systems, and selenium: A review. Curr. Med. Chem. 2007, 14, 1539–1549. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, K.; Vandecasteele, G.; Carli, C.; Romagnoli, A.; Szabadkai, G.; Rizzuto, R. Regulation of Ca2+ signalling and Ca2+-mediated cell death by the transcriptional coactivator PGC-1alpha. Cell Death Differ. 2006, 13, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Di, W.; Lv, J.; Jiang, S.; Lu, C.; Yang, Z.; Ma, Z.; Hu, W.; Yang, Y.; Xu, B. PGC-1: The Energetic Regulator in Cardiac Metabolism. Curr. Issues Mol. Biol. 2018, 28, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.G. Mitochondrial dysfunction in diabetic cardiomyopathy. Biochim. Biophys. Acta 2011, 1813, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Flarsheim, C.E.; Grupp, I.L.; Matlib, M.A. Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am. J. Physiol. 1996, 271 Pt 2, H192–H202. [Google Scholar] [CrossRef]
- Tanaka, Y.; Konno, N.; Kako, K.J. Mitochondrial dysfunction observed in situ in cardiomyocytes of rats in experimental diabetes. Cardiovasc. Res. 1992, 26, 409–414. [Google Scholar] [CrossRef]
- Riojas-Hernandez, A.; Bernal-Ramirez, J.; Rodriguez-Mier, D.; Morales-Marroquin, F.E.; Dominguez-Barragan, E.M.; Borja-Villa, C.; Rivera-Alvarez, I.; Garcia-Rivas, G.; Altamirano, J.; Garcia, N. Enhanced oxidative stress sensitizes the mitochondrial permeability transition pore to opening in heart from Zucker Fa/fa rats with type 2 diabetes. Life Sci. 2015, 141, 32–43. [Google Scholar] [CrossRef]
- Fauconnier, J.; Lanner, J.T.; Zhang, S.J.; Tavi, P.; Bruton, J.D.; Katz, A.; Westerblad, H. Insulin and inositol 1,4,5-trisphosphate trigger abnormal cytosolic Ca2+ transients and reveal mitochondrial Ca2+ handling defects in cardiomyocytes of ob/ob mice. Diabetes 2005, 54, 2375–2381. [Google Scholar] [CrossRef]
- Belke, D.D.; Swanson, E.A.; Dillmann, W.H. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes 2004, 53, 3201–3208. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Chen, X.; Baines, C.P.; Klevitsky, R.; Zhang, X.; Zhang, H.; Jaleel, N.; Chua, B.H.; Hewett, T.E.; Robbins, J.; et al. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J. Clin. Investig. 2007, 117, 2431–2444. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.H.; Delannoy, M.; Hullihen, J.; Chiu, W.; Pedersen, P.L. Mitochondrial ATP synthasome. Cristae-enriched membranes and a multiwell detergent screening assay yield dispersed single complexes containing the ATP synthase and carriers for Pi and ADP/ATP. J. Biol. Chem. 2003, 278, 12305–12309. [Google Scholar] [CrossRef]
- Graham, B.H.; Waymire, K.G.; Cottrell, B.; Trounce, I.A.; MacGregor, G.R.; Wallace, D.C. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat. Genet. 1997, 16, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Bakker, H.D.; Scholte, H.R.; Van den Bogert, C.; Ruitenbeek, W.; Jeneson, J.A.; Wanders, R.J.; Abeling, N.G.; Dorland, B.; Sengers, R.C.; Van Gennip, A.H. Deficiency of the adenine nucleotide translocator in muscle of a patient with myopathy and lactic acidosis: A new mitochondrial defect. Pediatric Res. 1993, 33 Pt 1, 412–417. [Google Scholar] [CrossRef]
- Tatuch, Y.; Robinson, B.H. The mitochondrial DNA mutation at 8993 associated with NARP slows the rate of ATP synthesis in isolated lymphoblast mitochondria. Biochem. Biophys. Res. Commun. 1993, 192, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Echaniz-Laguna, A.; Chassagne, M.; Ceresuela, J.; Rouvet, I.; Padet, S.; Acquaviva, C.; Nataf, S.; Vinzio, S.; Bo-zon, D.; Mousson de Camaret, B. Complete loss of expression of the ANT1 gene causing cardiomyopathy and myopathy. J. Med. Genet. 2012, 49, 146–150. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Davis, J.; Baines, C.P.; Sargent, M.A.; Karch, J.; Wang, X.; Huang, T.; Molkentin, J.D. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 2014, 21, 1209–1217. [Google Scholar] [CrossRef]
- Palmieri, L.; Alberio, S.; Pisano, I.; Lodi, T.; Meznaric-Petrusa, M.; Zidar, J.; Santoro, A.; Scarcia, P.; Fontanesi, F.; Lamantea, E.; et al. Complete loss-of-function of the heart/muscle-specific adenine nucleotide trans-locator is associated with mitochondrial myopathy and cardiomyopathy. Hum. Mol. Genet. 2005, 14, 3079–3088. [Google Scholar] [CrossRef]
- Limongelli, G.; D’Alessandro, R.; Maddaloni, V.; Rea, A.; Sarkozy, A.; McKenna, W.J. Skeletal muscle involvement in cardiomyopathies. J. Cardiovasc. Med. 2013, 14, 837–861. [Google Scholar] [CrossRef]
- Adam-Vizi, V.; Starkov, A.A. Calcium and mitochondrial reactive oxygen species generation: How to read the facts. J. Alzheimer’s Dis. JAD 2010, 20 (Suppl. 2), S413–S426. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Richardson, A.P. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.X.; Cui, S.M.; Zhang, Y.M.; Ren, J. Mitochondrial Ca2+ regulation in the etiology of heart failure: Physiological and pathophysiological implications. Acta Pharmacol. Sin. 2020, 41, 1301–1309. [Google Scholar] [CrossRef]
- Beal, M.F. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann. Neurol. 1995, 38, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Takimoto, E.; Dimaano, V.L.; DeMazumder, D.; Kettlewell, S.; Smith, G.; Sidor, A.; Abraham, T.P.; O’Rourke, B. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ. Res. 2014, 115, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, A.; Matsuoka, S. Physiological and Pathophysiological Roles of Mitochondrial Na+-Ca2+ Exchanger, NCLX, in Hearts. Biomolecules 2021, 11, 1876. [Google Scholar] [CrossRef] [PubMed]
- Marta, K.; Hasan, P.; Rodriguez-Prados, M.; Paillard, M.; Hajnoczky, G. Pharmacological inhibition of the mitochondrial Ca2+ uniporter: Relevance for pathophysiology and human therapy. J. Mol. Cell. Cardiol. 2021, 151, 135–144. [Google Scholar] [CrossRef]
- Diaz-Juarez, J.; Suarez, J.; Cividini, F.; Scott, B.T.; Diemer, T.; Dai, A.; Dillmann, W.H. Expression of the mitochondrial calcium uniporter in cardiac myocytes improves impaired mitochondrial calcium handling and metabolism in simulated hyperglycemia. Am. J. Physiol. Cell Physiol. 2016, 311, C1005–C1013. [Google Scholar] [CrossRef]
- Xie, A.; Song, Z.; Liu, H.; Zhou, A.; Shi, G.; Wang, Q.; Gu, L.; Liu, M.; Xie, L.H.; Qu, Z.; et al. Mitochondrial Ca2+ Influx Contributes to Arrhythmic Risk in Nonischemic Cardiomyopathy. J. Am. Heart Assoc. 2018, 7, e007805. [Google Scholar] [CrossRef]
- Miranda-Silva, D.; Lima, T.; Rodrigues, P.; Leite-Moreira, A.; Falcao-Pires, I. Mechanisms underlying the pathophysiology of heart failure with preserved ejection fraction: The tip of the iceberg. Heart Fail. Rev. 2021, 26, 453–478. [Google Scholar] [CrossRef]
- Chugh, S.S.; Reinier, K.; Teodorescu, C.; Evanado, A.; Kehr, E.; Al Samara, M.; Mariani, R.; Gunson, K.; Jui, J. Epidemiology of sudden cardiac death: Clinical and research implications. Prog. Cardiovasc. Dis. 2008, 51, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.; Veress, R.; Belevych, A.; Terentyev, D. The role of calcium homeostasis remodeling in inherited cardiac arrhythmia syndromes. Pflug. Arch. Eur. J. Physiol. 2021, 473, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rivas Gde, J.; Carvajal, K.; Correa, F.; Zazueta, C. Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br. J. Pharmacol. 2006, 149, 829–837. [Google Scholar] [CrossRef]
- Kwong, J.Q. The mitochondrial calcium uniporter in the heart: Energetics and beyond. J. Physiol. 2017, 595, 3743–3751. [Google Scholar] [CrossRef] [PubMed]
- Bovo, E.; Lipsius, S.L.; Zima, A.V. Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during beta-adrenergic receptor stimulation in rabbit cardiomyocytes. J. Physiol. 2012, 590, 3291–3304. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, J.; Zhang, L.; Xia, H. Experimental study on alleviating atherosclerosis through intervention of mitochondrial calcium transport and calcium-induced membrane permeability transition. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2021, 69, 1156–1160. [Google Scholar] [CrossRef]
- Botker, H.E.; Cabrera-Fuentes, H.A.; Ruiz-Meana, M.; Heusch, G.; Ovize, M. Translational issues for mitoprotective agents as adjunct to reperfusion therapy in patients with ST-segment elevation myocardial infarction. J. Cell Mol. Med. 2020, 24, 2717–2729. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, G.; Matoba, T.; Nakano, Y.; Nagaoka, K.; Ishikita, A.; Nakano, K.; Funamoto, D.; Sunagawa, K.; Egashira, K. Nanoparticle-Mediated Targeting of Cyclosporine A Enhances Cardioprotection Against Ischemia-Reperfusion Injury Through Inhibition of Mitochondrial Permeability Transition Pore Opening. Sci. Rep. 2016, 6, 20467. [Google Scholar] [CrossRef]
- Jahandiez, V.; Cour, M.; Bochaton, T.; Abrial, M.; Loufouat, J.; Gharib, A.; Varennes, A.; Ovize, M.; Argaud, L. Fast therapeutic hypothermia prevents post-cardiac arrest syndrome through cyclophilin D-mediated mitochondrial permeability transition inhibition. Basic Res. Cardiol. 2017, 112, 35. [Google Scholar] [CrossRef]
- Monaco, G.; Decrock, E.; Akl, H.; Ponsaerts, R.; Vervliet, T.; Luyten, T.; De Maeyer, M.; Missiaen, L.; Distelhorst, C.W.; De Smedt, H.; et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2012, 19, 295–309. [Google Scholar] [CrossRef]
- El-Hayek, R.; Antoniu, B.; Wang, J.; Hamilton, S.L.; Ikemoto, N. Identification of calcium release-triggering and blocking regions of the II-III loop of the skeletal muscle dihydropyridine receptor. J. Biol. Chem. 1995, 270, 22116–22118. [Google Scholar] [CrossRef]
- Dulhunty, A.F.; Cengia, L.; Young, J.; Pace, S.M.; Harvey, P.J.; Lamb, G.D.; Chan, Y.N.; Wimmer, N.; Toth, I.; Casarotto, M.G. Functional implications of modifying RyR-activating peptides for membrane permeability. Br. J. Pharmacol. 2005, 144, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Gurrola, G.B.; Zhang, J.; Valdivia, C.R.; SanMartin, M.; Zamudio, F.Z.; Zhang, L.; Possani, L.D.; Valdi-via, H.H. Structure-function relationships of peptides forming the calcin family of ryanodine receptor ligands. J. Gen. Physiol. 2016, 147, 375–394. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Shah, K.; Bradbury, N.A.; Li, C.; White, C. Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell Death Dis. 2014, 5, e1482. [Google Scholar] [CrossRef] [PubMed]
- Keinan, N.; Pahima, H.; Ben-Hail, D.; Shoshan-Barmatz, V. The role of calcium in VDAC1 oligomerization and mitochondria-mediated apoptosis. Biochim. Biophys. Acta 2013, 1833, 1745–1754. [Google Scholar] [CrossRef]
- Monaco, G.; Decrock, E.; Arbel, N.; van Vliet, A.R.; La Rovere, R.M.; De Smedt, H.; Parys, J.B.; Agostinis, P.; Leybaert, L.; Shoshan-Barmatz, V.; et al. The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. J. Biol. Chem. 2015, 290, 9150–9161. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, S.; Hu, S.; Chen, Y.; Ren, J. ER-Mitochondria Microdomains in Cardiac Ischemia-Reperfusion Injury: A Fresh Perspective. Front. Physiol. 2018, 9, 755. [Google Scholar] [CrossRef]
- Naia, L.; Ferreira, I.L.; Ferreiro, E.; Rego, A.C. Mitochondrial Ca2+ handling in Huntington’s and Alzheimer’s diseases—Role of ER-mitochondria crosstalk. Biochem. Biophys. Res. Commun. 2017, 483, 1069–1077. [Google Scholar] [CrossRef]
- Zaichick, S.V.; McGrath, K.M.; Caraveo, G. The role of Ca2+ signaling in Parkinson’s disease. Dis. Models Mech. 2017, 10, 519–535. [Google Scholar] [CrossRef]
- Sun, Y.; Deng, T.; Lu, N.; Yan, M.; Zheng, X. B-type natriuretic peptide protects cardiomyocytes at reperfusion via mitochondrial calcium uniporter. Biomed. Pharmacother. 2010, 64, 170–176. [Google Scholar] [CrossRef]
- Kerkhofs, M.; Bultynck, G.; Vervliet, T.; Monaco, G. Therapeutic implications of novel peptides targeting ER-mitochondria Ca2+-flux systems. Drug Discov. Today 2019, 24, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Hansson, M.J.; Mattiasson, G.; Månsson, R.; Karlsson, J.; Keep, M.F.; Waldmeier, P.; Ruegg, U.T.; Dumont, J.M.; Besseghir, K.; Elmér, E. The nonimmunosuppressive cyclosporin analogs NIM811 and UNIL025 display nanomolar potencies on permeability transition in brain-derived mitochondria. J. Bioenerg. Biomembr. 2004, 36, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Belosludtseva, N.V.; Starinets, V.S.; Mikheeva, I.B.; Serov, D.A.; Astashev, M.E.; Belosludtsev, M.N.; Dubinin, M.V.; Belosludtsev, K.N. Effect of the MPT Pore Inhibitor Alisporivir on the Development of Mitochondrial Dysfunction in the Heart Tissue of Diabetic Mice. Biology. 2021, 10, 839. [Google Scholar] [CrossRef] [PubMed]
- Gadicherla, A.K.; Wang, N.; Bulic, M.; Agullo-Pascual, E.; Lissoni, A.; De Smet, M.; Delmar, M.; Bultynck, G.; Krysko, D.V.; Camara, A.; et al. Mitochondrial Cx43 hemichannels contribute to mitochondrial calcium entry and cell death in the heart. Basic Res. Cardiol. 2017, 112, 27. [Google Scholar] [CrossRef]
- Guo, R.; Si, R.; Scott, B.T.; Makino, A. Mitochondrial connexin40 regulates mitochondrial calcium uptake in coronary endothelial cells. Am. J. Physiol. Cell Physiol. 2017, 312, C398–C406. [Google Scholar] [CrossRef]
- Srisakuldee, W.; Makazan, Z.; Nickel, B.E.; Zhang, F.; Thliveris, J.A.; Pasumarthi, K.B.; Kardami, E. The FGF-2-triggered protection of cardiac subsarcolemmal mitochondria from calcium overload is mitochondrial connexin 43-dependent. Cardiovasc. Res. 2014, 103, 72–80. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Name | Source/Mechanism | Proposed Mode of Action | References |
|---|---|---|---|
| CsA | an inhibitor of mPTP and can bind to CypD | a positive regulator of mPTP | [277,278,279] |
| mtCsA | non-immunosuppressive derivatives of CsA | desensitizes mPTP to Ca2+ and inhibits pore opening | [291] |
| BH4-Bcl-2 | BH4 domain of Bcl-2 | inhibit IP3R-mediated Ca2+ release | [280] |
| Bcl-2/IP3R disruptor 2 (Bird-2) | an IP3R-derived peptide | inhibit IP3R-mediated Ca2+ release | [280] |
| peptide A | a short fragment of the cytosolic II–III loop of the DHPR | induces RyR1-mediated Ca2+ release | [281] |
| Calcins | have a high affinity for RyR1 and specifically bind and stimulate its activity. | have similar effects on RyR1 gating | [283] |
| ANTP-N-TER | N-terminal of VDAC1 | significantly inhibit mitochondrial Ca2+ uptake | [284] |
| ANTP-L14-15 | short peptides from the N-terminal of LP4 | inhibit mitochondrial Ca2+ uptake | [284] |
| R-tf-d-lp4 peptide | the VDAC1-based peptide | significantly increased intracellular Ca2+ levels | [285] |
| BH4-Bcl-XL | an inhibitor of RyR in ER | blocks VDAC1-mediated Ca2+ uptake into mitochondria | [286,287,288,289] |
| Nesiritide, Natrecor | recombinant BNP peptide | inhibiting MCU and affecting mPTP opening | [290] |
| Octapeptide RRNYRRNY (RNY) | inhibits the Cx43 hemichannels in mitochondria | reduces mitochondrial Ca2+ overload | [294] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, D.; Wang, F.; Li, P.; Gao, Y. Mitochondrial Ca2+ Homeostasis: Emerging Roles and Clinical Significance in Cardiac Remodeling. Int. J. Mol. Sci. 2022, 23, 3025. https://doi.org/10.3390/ijms23063025
Zhang D, Wang F, Li P, Gao Y. Mitochondrial Ca2+ Homeostasis: Emerging Roles and Clinical Significance in Cardiac Remodeling. International Journal of Molecular Sciences. 2022; 23(6):3025. https://doi.org/10.3390/ijms23063025
Chicago/Turabian StyleZhang, Dejiu, Fei Wang, Peifeng Li, and Yanyan Gao. 2022. "Mitochondrial Ca2+ Homeostasis: Emerging Roles and Clinical Significance in Cardiac Remodeling" International Journal of Molecular Sciences 23, no. 6: 3025. https://doi.org/10.3390/ijms23063025
APA StyleZhang, D., Wang, F., Li, P., & Gao, Y. (2022). Mitochondrial Ca2+ Homeostasis: Emerging Roles and Clinical Significance in Cardiac Remodeling. International Journal of Molecular Sciences, 23(6), 3025. https://doi.org/10.3390/ijms23063025