
Dedifferentiation of Human Cardiac Myofibroblasts Is Independent of Activation of COX-2/PGE2 Pathway


Abstract
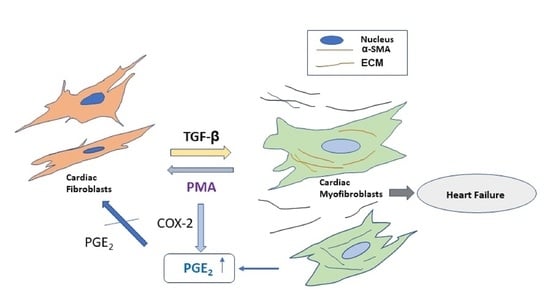
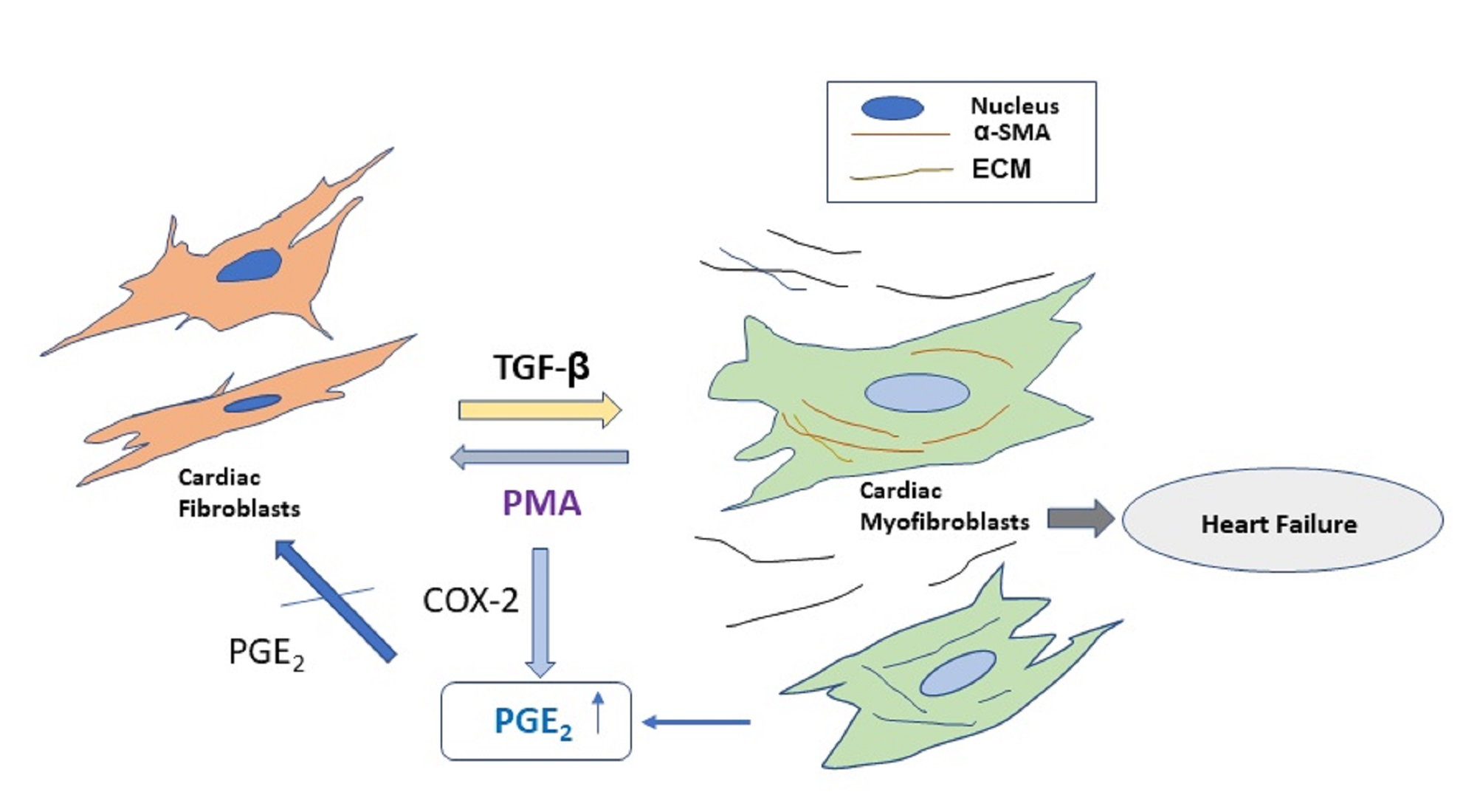{kind=link}
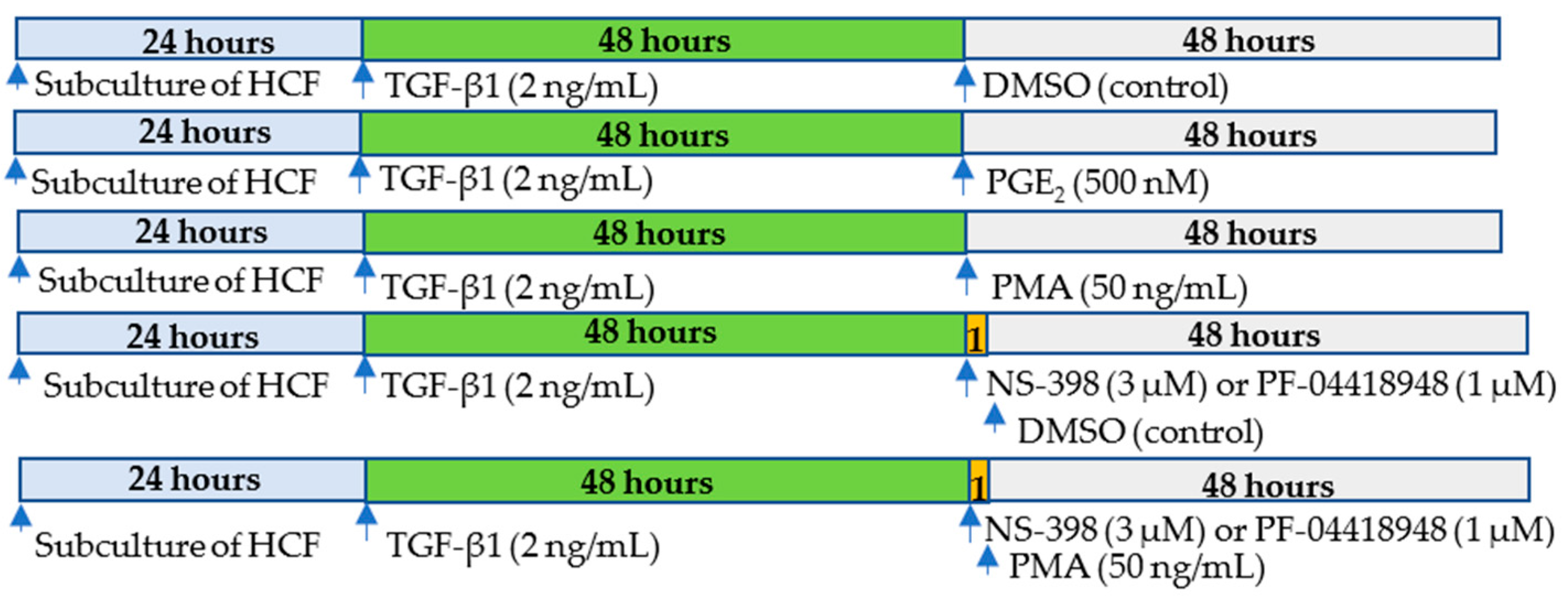{kind=link}
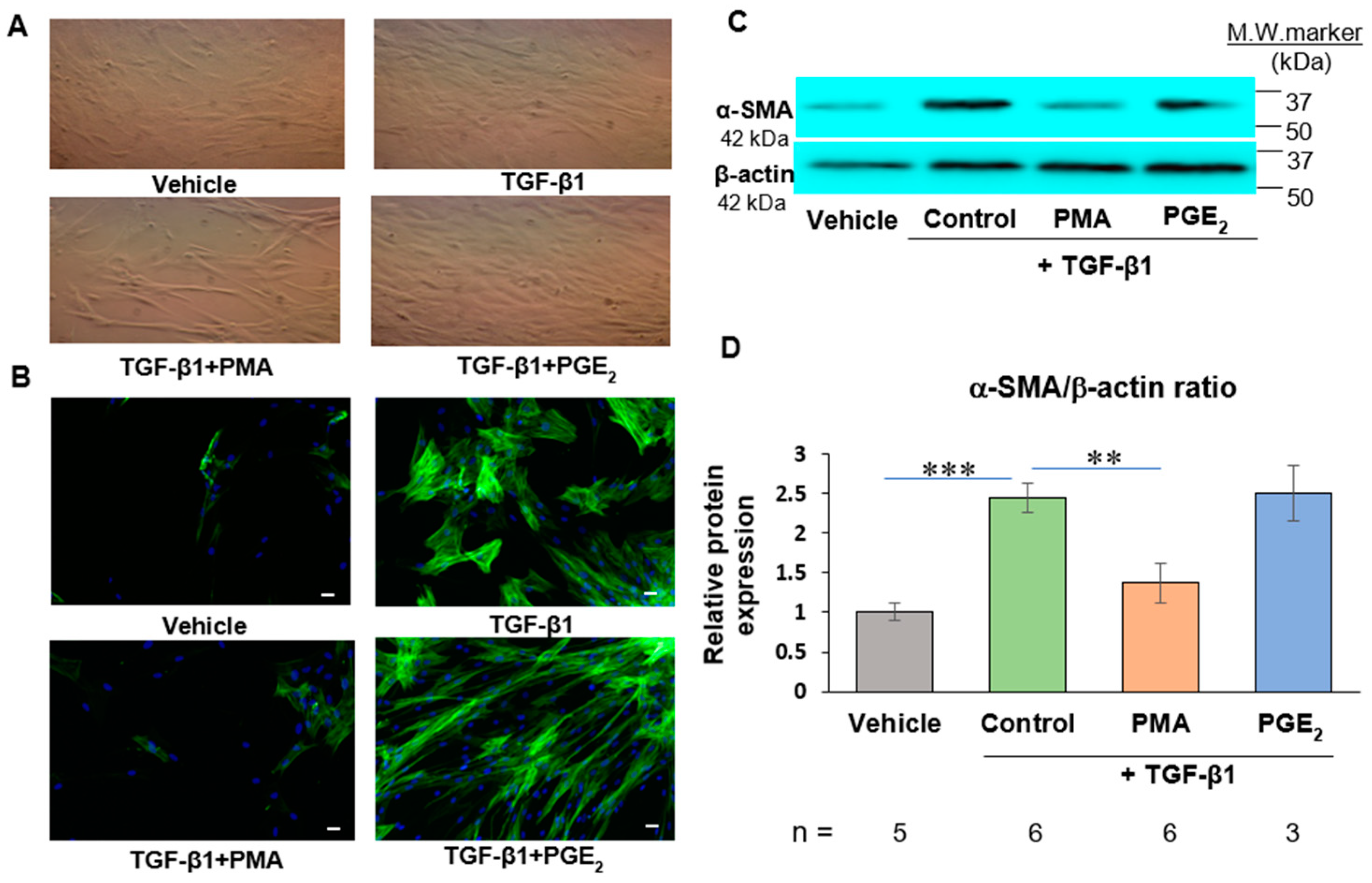{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. PMA but Not PGE2 Dedifferentiates De Novo Human Cardiac Myofibroblasts
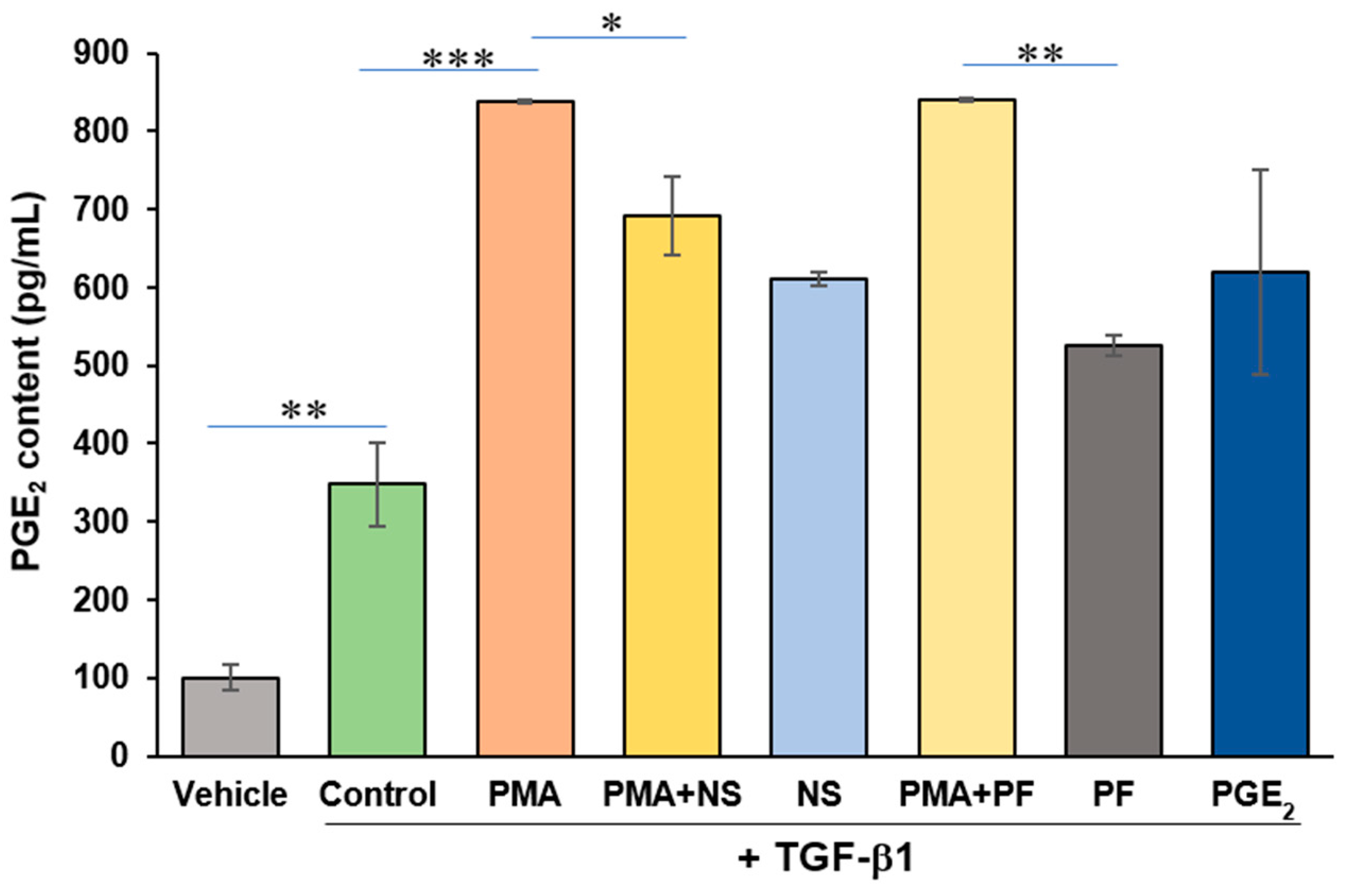
2.2. PMA Enhances PGE2 Content from Human Cardiac Myofibroblasts
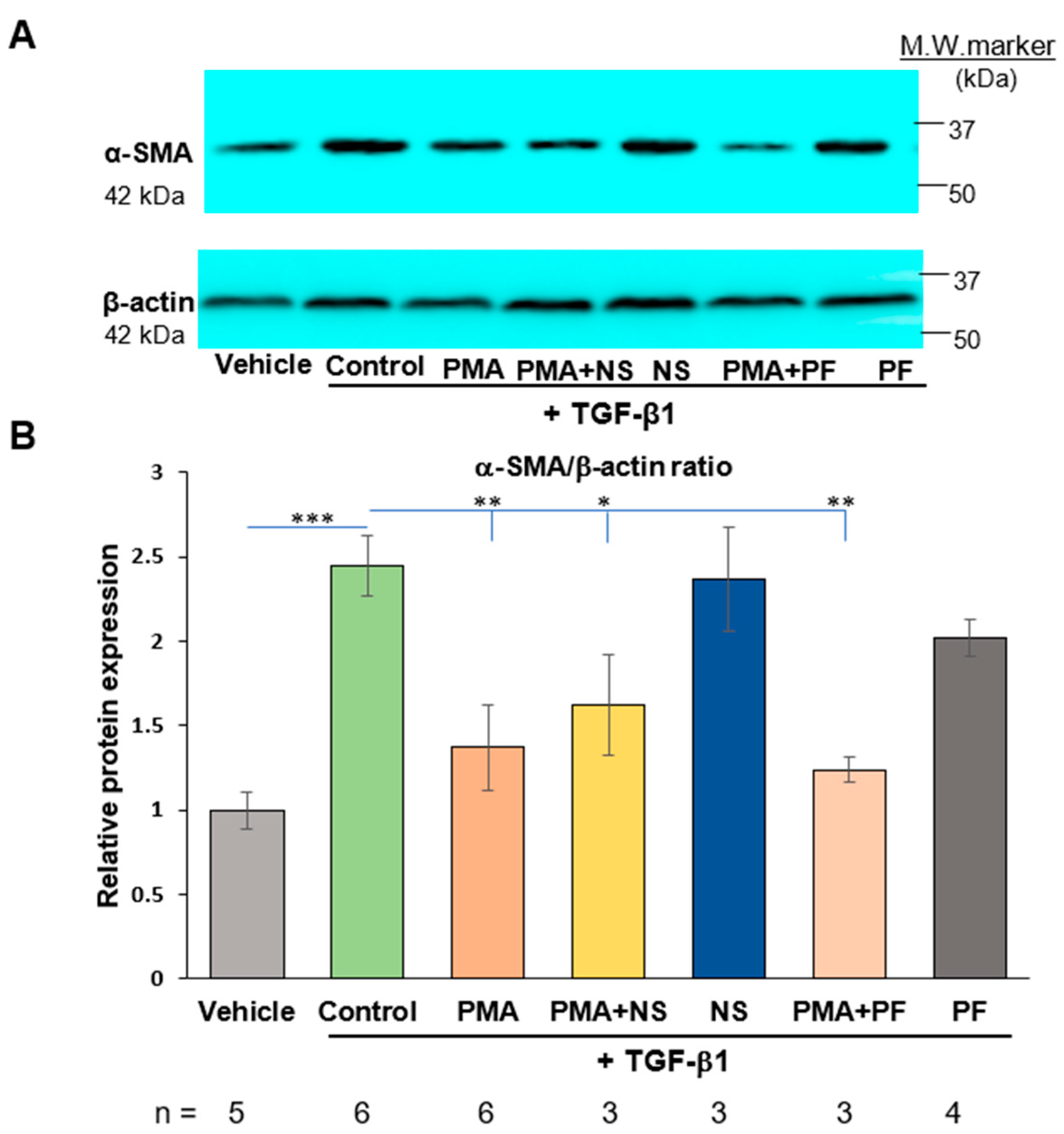
2.3. PMA-Induced Dedifferentiation of Human Cardiac Myofibroblasts Does Not Depend on Activation of COX-2/PGE2 Pathway
2.4. Activation of COX-2/PGE2 Does Not Induce Dedifferentiation of Human Cardiac Myofibroblasts
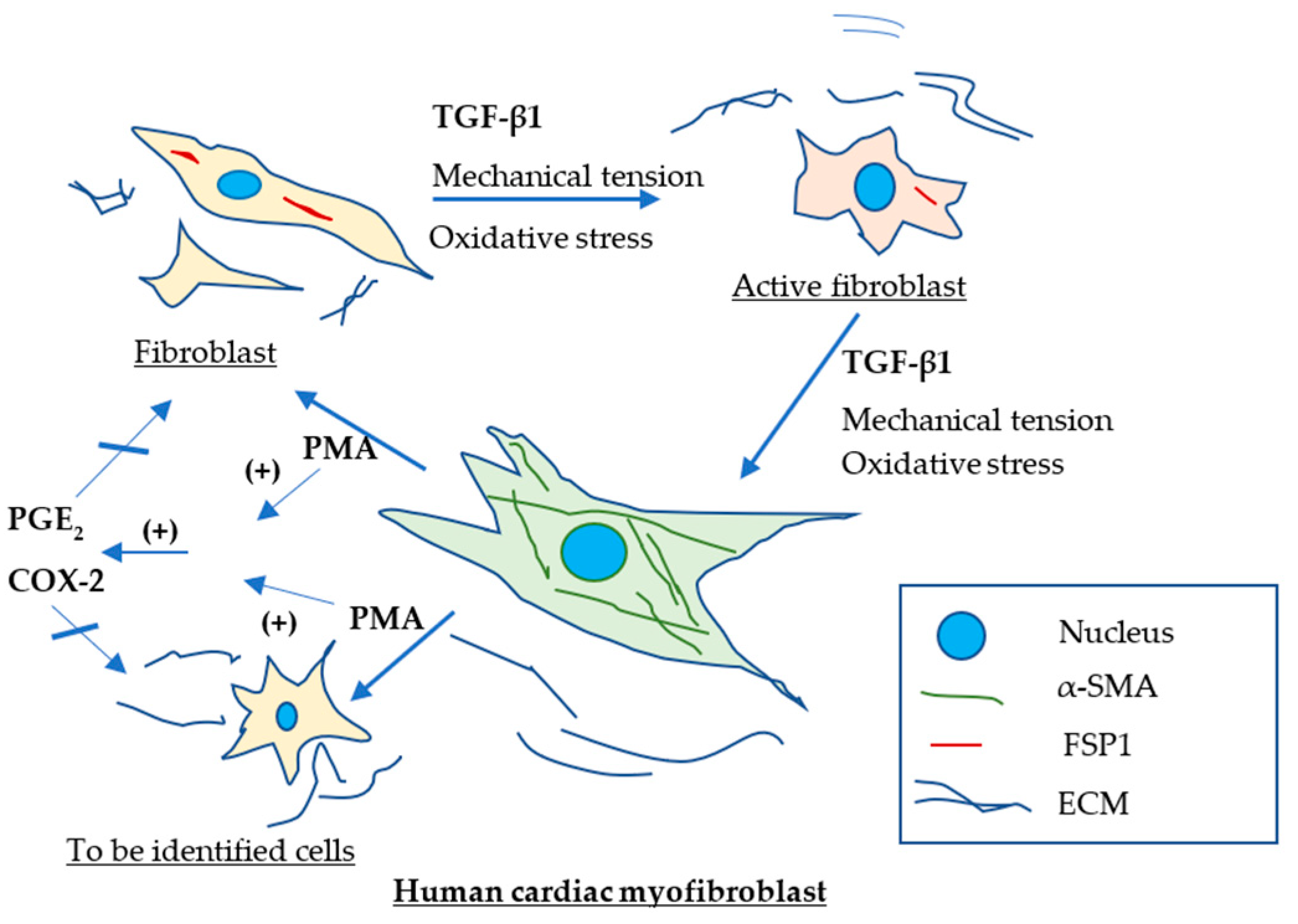
3. Discussion
4. Materials and Methods
4.1. Antibodies and Regeagents
4.2. Cell Culture and Treatment
4.3. Western Blotting
4.4. Immunofluorescence Co-Staining
4.5. Measurement of PGE2 Content
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wynn, T.A. Fibrotic disease and the TH1/TH2 paradigm. Nat. Rev. Immunol. 2004, 4, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef] [PubMed]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Gibb, A.A.; Lazaropoulos, M.P.; Elrod, J.W. Myofibroblasts and Fibrosis: Mitochondrial and Metabolic Control of Cellular Differentiation. Circ. Res. 2020, 127, 427–447. [Google Scholar] [CrossRef]
- Li, R.K.; Li, G.; Mickle, D.A.; Weisel, R.D.; Merante, F.; Luss, H.; Rao, V.; Christakis, G.T.; Williams, W.G. Overexpression of transforming growth factor-beta1 and insulin-like growth factor-I in patients with idiopathic hypertrophic cardiomyopathy. Circulation 1997, 96, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Pauschinger, M.; Knopf, D.; Petschauer, S.; Doerner, A.; Poller, W.; Schwimmbeck, P.L.; Kühl, U.; Schultheiss, H.P. Dilated cardiomyopathy is associated with significant changes in collagen type I/III ratio. Circulation 1999, 99, 2750–2756. [Google Scholar] [CrossRef]
- Rosenkranz, S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc. Res. 2004, 63, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. Resolution of organ fibrosis. J. Clin. Investig. 2018, 128, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, C.K.; Robinson, E.L.; Abdesselem, M.; Trenson, S.; Dries, E.; Gilbert, G.; Janssens, S.; Van Cleemput, J.; Rega, F.; Meyns, B.; et al. Myofibroblast Phenotype and Reversibility of Fibrosis in Patients with End-Stage Heart Failure. J. Am. Coll. Cardiol. 2019, 73, 2267–2282. [Google Scholar] [CrossRef]
- Emelyanova, L.; Sra, A.; Schmuck, E.G.; Raval, A.N.; Downey, F.X.; Jahangir, A.; Rizvi, F.; Ross, G.R. Impact of statins on cellular respiration and de-differentiation of myofibroblasts in human failing hearts. ESC Heart Fail. 2019, 6, 1027–1040. [Google Scholar] [CrossRef] [PubMed]
- Fortier, S.M.; Penke, L.R.; King, D.; Pham, T.X.; Ligresti, G.; Peters-Golden, M. Myofibroblast dedifferentiation proceeds via distinct transcriptomic and phenotypic transitions. JCI Insight 2021, 6, e144799. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Logsdon, N.J.; Shin, Y.J.; Palumbo, S.; Knox, A.; Irish, J.D.; Rounseville, S.P.; Rummel, S.R.; Mohamed, M.; Ahmad, K.; et al. Impaired Myofibroblast Dedifferentiation Contributes to Nonresolving Fibrosis in Aging. Am. J. Respir. Cell. Mol. Biol. 2020, 62, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Wettlaufer, S.H.; Scott, J.P.; McEachin, R.C.; Peters-Golden, M.; Huang, S.K. Reversal of the Transcriptome by Prostaglandin E2 during Myofibroblast Dedifferentiation. Am. J. Respir. Cell. Mol. Biol. 2016, 54, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Garrison, G.; Huang, S.K.; Okunishi, K.; Scott, J.P.; Kumar Penke, L.R.; Scruggs, A.M.; Peters-Golden, M. Reversal of myofibroblast differentiation by prostaglandin E(2). Am. J. Respir. Cell. Mol. Biol. 2013, 48, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Maltseva, O.; Folger, P.; Zekaria, D.; Petridou, S.; Masur, S.K. Fibroblast growth factor reversal of the corneal myofibroblast phenotype. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2490–2495. [Google Scholar]
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef] [PubMed]
- Schuette, R.; LaPointe, M.C. Phorbol ester stimulates cyclooxygenase-2 expression and prostanoid production in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H719–H725. [Google Scholar] [CrossRef] [PubMed]
- Hellweg, C.E.; Arenz, A.; Bogner, S.; Schmitz, C.; Baumstark-Khan, C. Activation of nuclear factor kappa B by different agents: Influence of culture conditions in a cell-based assay. Ann. N. Y. Acad. Sci. 2006, 1091, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, T.; Cao, X.; Zou, J.; Ding, X.; Shen, B.; Lv, W. Prostaglandin E2 induced cardiac hypertrophy through EP2 receptor-dependent activation of β-catenin in 5/6 nephrectomy rats. ESC Heart Fail. 2021, 8, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.S.; Chen, B.C.; Yu, M.T.; Sheu, J.R.; Chen, T.F.; Lin, C.H. Phorbol 12-myristate 13-acetate upregulates cyclooxygenase-2 expression in human pulmonary epithelial cells via Ras, Raf-1, ERK, and NF-kappaB, but not p38 MAPK, pathways. Cell. Signal. 2005, 17, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Mih, J.D.; Shea, B.S.; Kho, A.T.; Sharif, A.S.; Tager, A.M.; Tschumperlin, D.J. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J. Cell. Biol. 2010, 190, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Imanishi, Y.; Ozawa, H.; Sakamoto, K.; Fujii, R.; Shigetomi, S.; Habu, N.; Otsuka, K.; Sato, Y.; Sekimizu, M.; et al. Selective EP2 and Cox-2 inhibition suppresses cell migration by reversing epithelial-to-mesenchymal transition and Cox-2 overexpression and E-cadherin downregulation are implicated in neck metastasis of hypopharyngeal cancer. Am. J. Transl. Res. 2020, 12, 1096–1113. [Google Scholar] [PubMed]
- Mayorga, M.; Bahi, N.; Ballester, M.; Comella, J.X.; Sanchis, D. Bcl-2 is a key factor for cardiac fibroblast resistance to programmed cell death. J. Biol. Chem. 2004, 279, 34882–34889. [Google Scholar] [CrossRef]
- Muhl, L.; Genové, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V.; et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat. Commun. 2020, 11, 3953. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.D.; Watt, F.M. Fibroblast heterogeneity: Implications for human disease. J. Clin. Investig. 2018, 128, 26–35. [Google Scholar] [CrossRef]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Neilson, E.G. Origin and functional heterogeneity of fibroblasts. FASEB J. 2020, 34, 3519–3536. [Google Scholar] [CrossRef] [PubMed]
- Bozyk, P.D.; Moore, B.B. Prostaglandin E2 and the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell. Mol. Biol. 2011, 45, 445–452. [Google Scholar] [CrossRef] [PubMed]
- LaPointe, M.C.; Mendez, M.; Leung, A.; Tao, Z.; Yang, X.P. Inhibition of cyclooxygenase-2 improves cardiac function after myocardial infarction in the mouse. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1416–H1424. [Google Scholar] [CrossRef] [PubMed]
- Harding, P.; LaPointe, M.C. Prostaglandin E2 increases cardiac fibroblast proliferation and increases cyclin D expression via EP1 receptor. Prostaglandins Leukot. Essent. Fat. Acids. 2011, 84, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yue, Z.; Zhang, B.; Yang, M.; Lao, H.; Lai, W.; Zeng, Y.; Chen, S.; Liu, P. Calcium Signal Pathway is Involved in Prostaglandin E2 Induced Cardiac Fibrosis in Cardiac Fibroblasts. J. Pharm. Pharm. Sci. 2018, 21, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.Y.; El-Baz, L.M.; House, S.; Cilvik, S.N.; Dorry, S.J.; Shoukry, N.M.; Salem, M.L.; Hafez, H.S.; Dulin, N.O.; Ornitz, D.M.; et al. Fibroblast growth factor 2 decreases bleomycin-induced pulmonary fibrosis and inhibits fibroblast collagen production and myofibroblast differentiation. J. Pathol. 2018, 246, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Skaletz-Rorowski, A.; Waltenberger, J.; Müller, J.G.; Pawlus, E.; Pinkernell, K.; Breithardt, G. Protein kinase C mediates basic fibroblast growth factor-induced proliferation through mitogen-activated protein kinase in coronary smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1608–1614. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Forselles, K.J.; Root, J.; Clarke, T.; Davey, D.; Aughton, K.; Dack, K.; Pullen, N. In vitro and in vivo characterization of PF-04418948, a novel, potent and selective prostaglandin EP2; receptor antagonist. Br. J. Pharmacol. 2011, 164, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- Alexanian, M.; Przytycki, P.F.; Micheletti, R.; Padmanabhan, A.; Ye, L.; Travers, J.G.; Gonzalez-Teran, B.; Silva, A.C.; Duan, Q.; Ranade, S.S.; et al. A transcriptional switch governs fibroblast activation in heart disease. Nature 2021, 595, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Futaki, N.; Takahashi, S.; Yokoyama, M.; Arai, I.; Higuchi, S.; Otomo, S. NS-398, a new anti-inflammatory agent, selectively inhibits prostaglandin G/H synthase/cyclooxygenase (COX-2) activity in vitro. Prostaglandins 1994, 47, 55–59. [Google Scholar] [CrossRef]
- Abdullah, C.S.; Jin, Z.Q. Targeted deletion of T-cell S1P receptor 1 ameliorates cardiac fibrosis in streptozotocin-induced diabetic mice. FASEB J. 2018, 32, 5426–5435. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luu, V.T.; Phan, S.; Jin, Z.-Q. Dedifferentiation of Human Cardiac Myofibroblasts Is Independent of Activation of COX-2/PGE2 Pathway. Int. J. Mol. Sci. 2022, 23, 3023. https://doi.org/10.3390/ijms23063023
Luu VT, Phan S, Jin Z-Q. Dedifferentiation of Human Cardiac Myofibroblasts Is Independent of Activation of COX-2/PGE2 Pathway. International Journal of Molecular Sciences. 2022; 23(6):3023. https://doi.org/10.3390/ijms23063023
Chicago/Turabian StyleLuu, Vy Tran, Sang Phan, and Zhu-Qiu Jin. 2022. "Dedifferentiation of Human Cardiac Myofibroblasts Is Independent of Activation of COX-2/PGE2 Pathway" International Journal of Molecular Sciences 23, no. 6: 3023. https://doi.org/10.3390/ijms23063023
APA StyleLuu, V. T., Phan, S., & Jin, Z.-Q. (2022). Dedifferentiation of Human Cardiac Myofibroblasts Is Independent of Activation of COX-2/PGE2 Pathway. International Journal of Molecular Sciences, 23(6), 3023. https://doi.org/10.3390/ijms23063023