
SARS-CoV-2 Membrane Protein: From Genomic Data to Structural New Insights

 ,
,  , and
, and 
Abstract
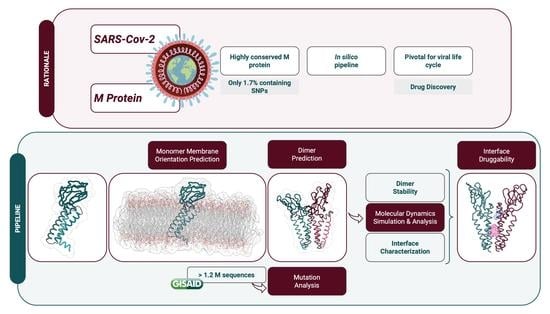
:
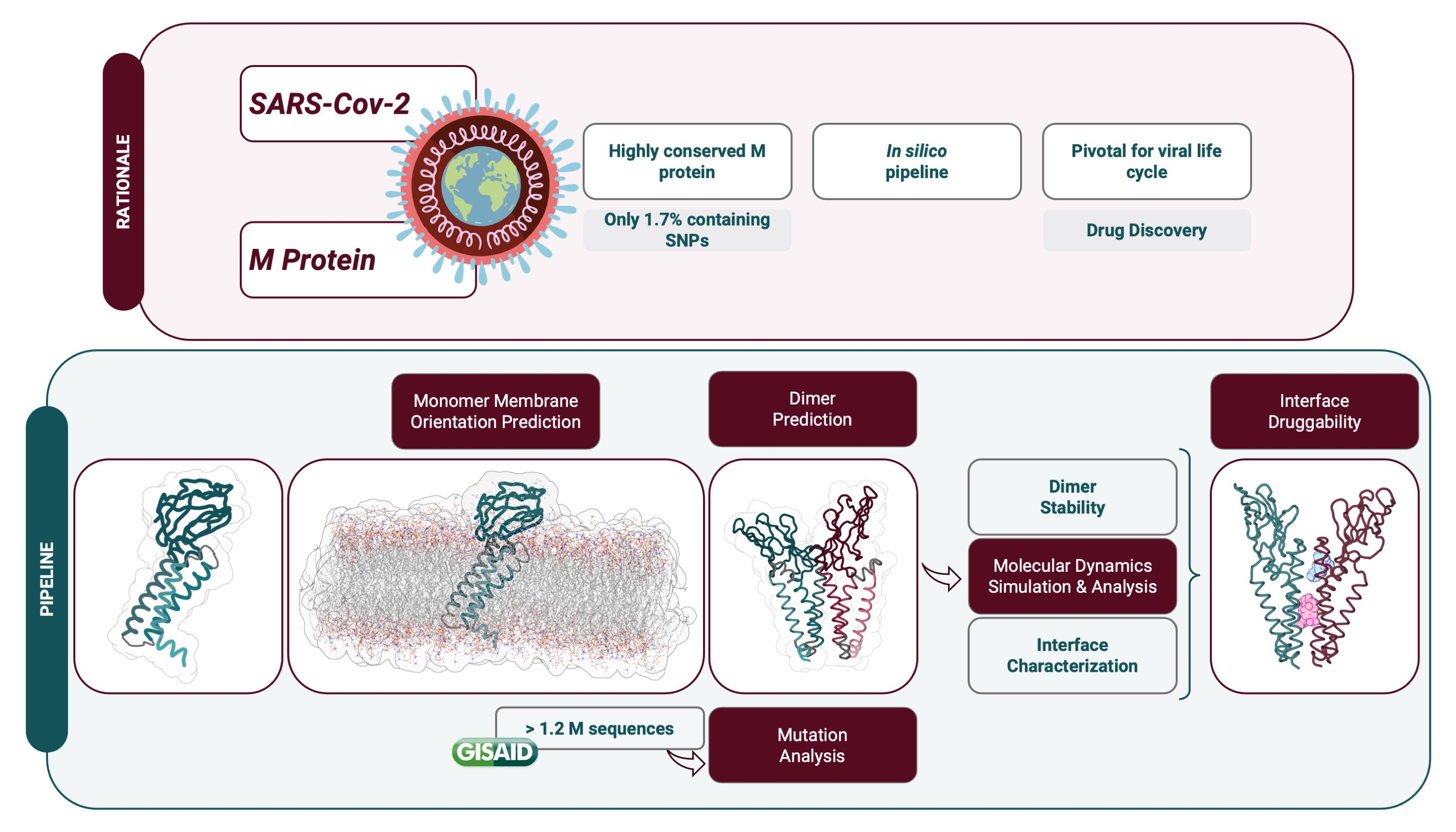
1. Introduction
2. Results
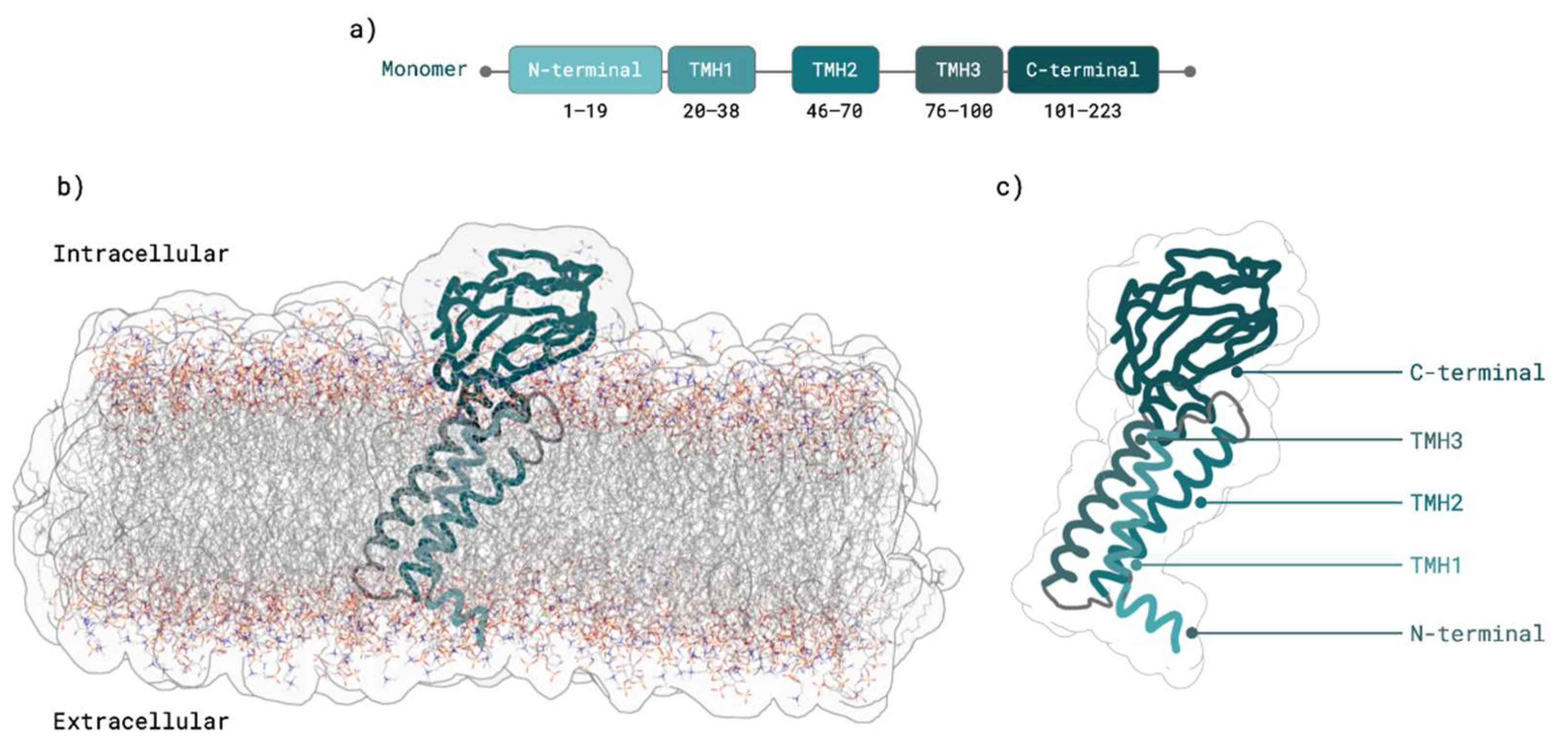
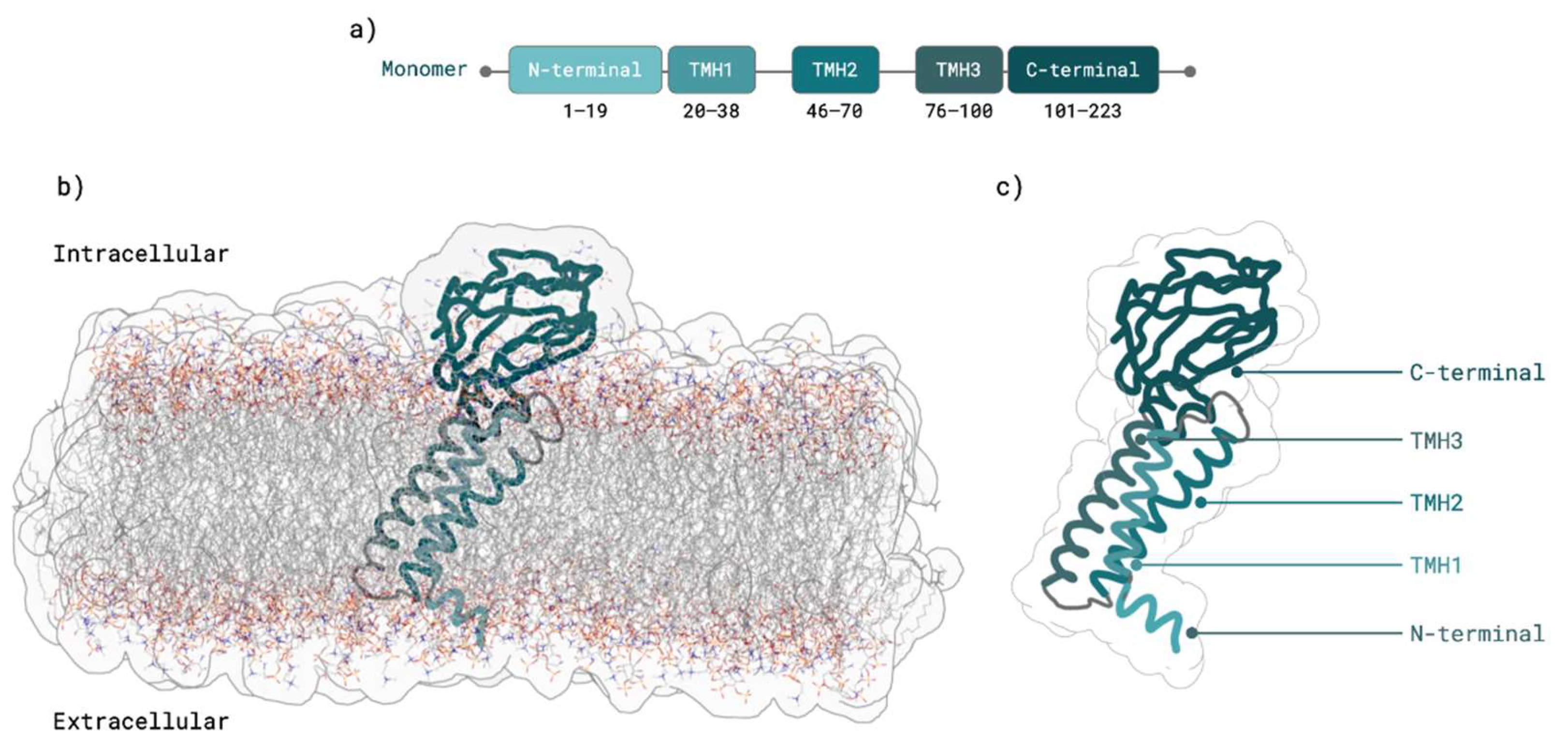
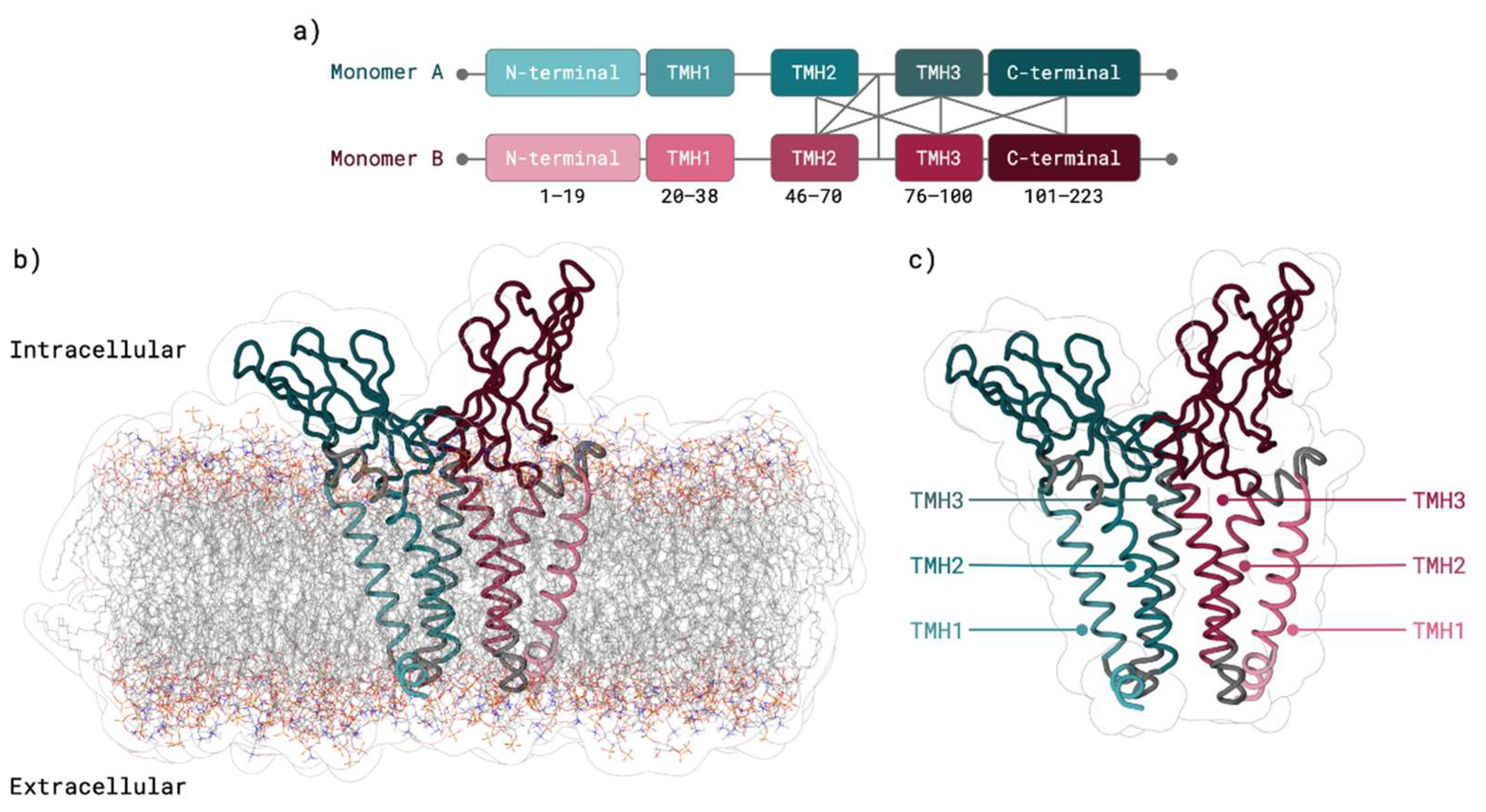
2.1. M Protein Monomer Structure and Membrane Orientation
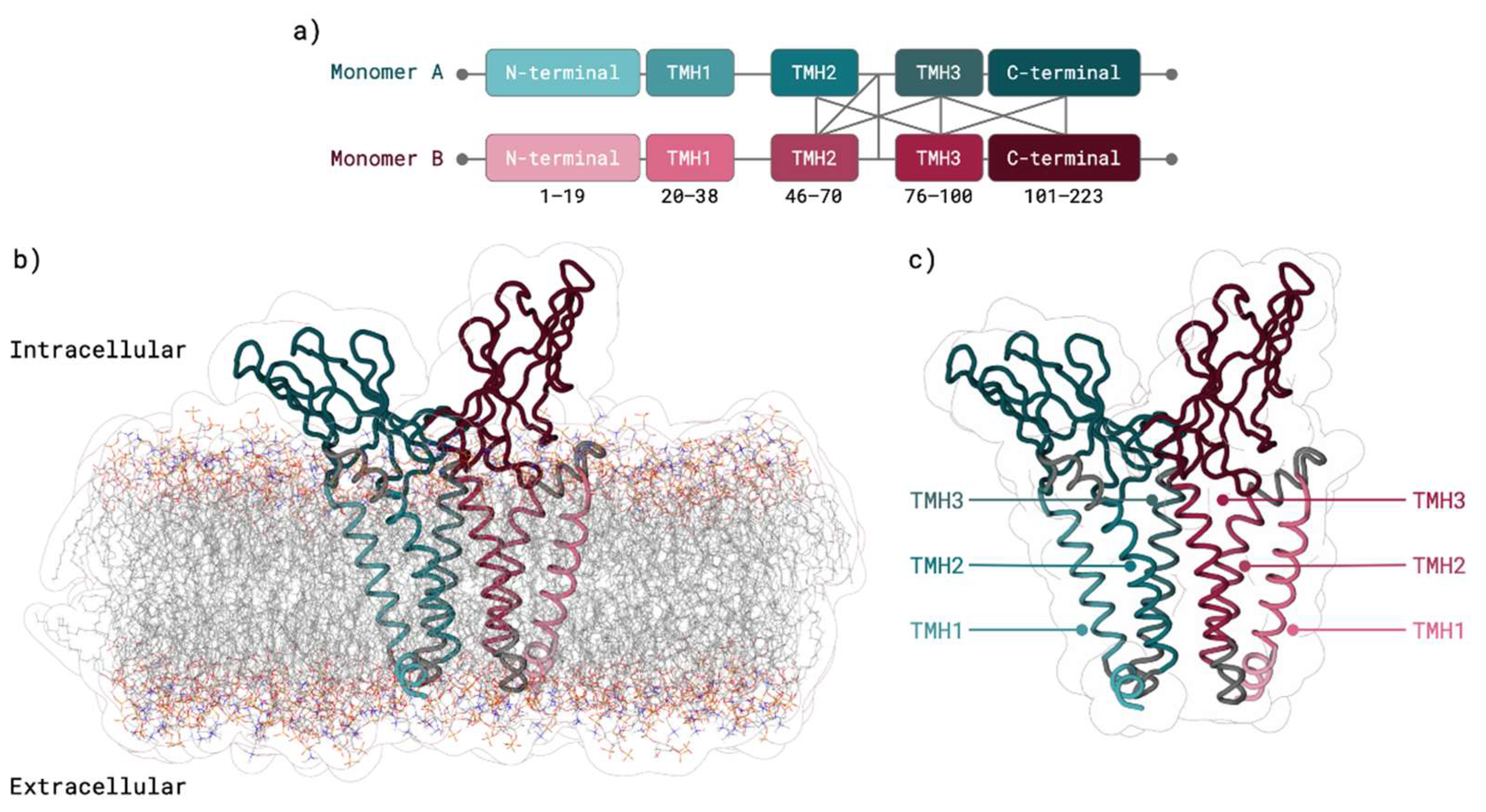
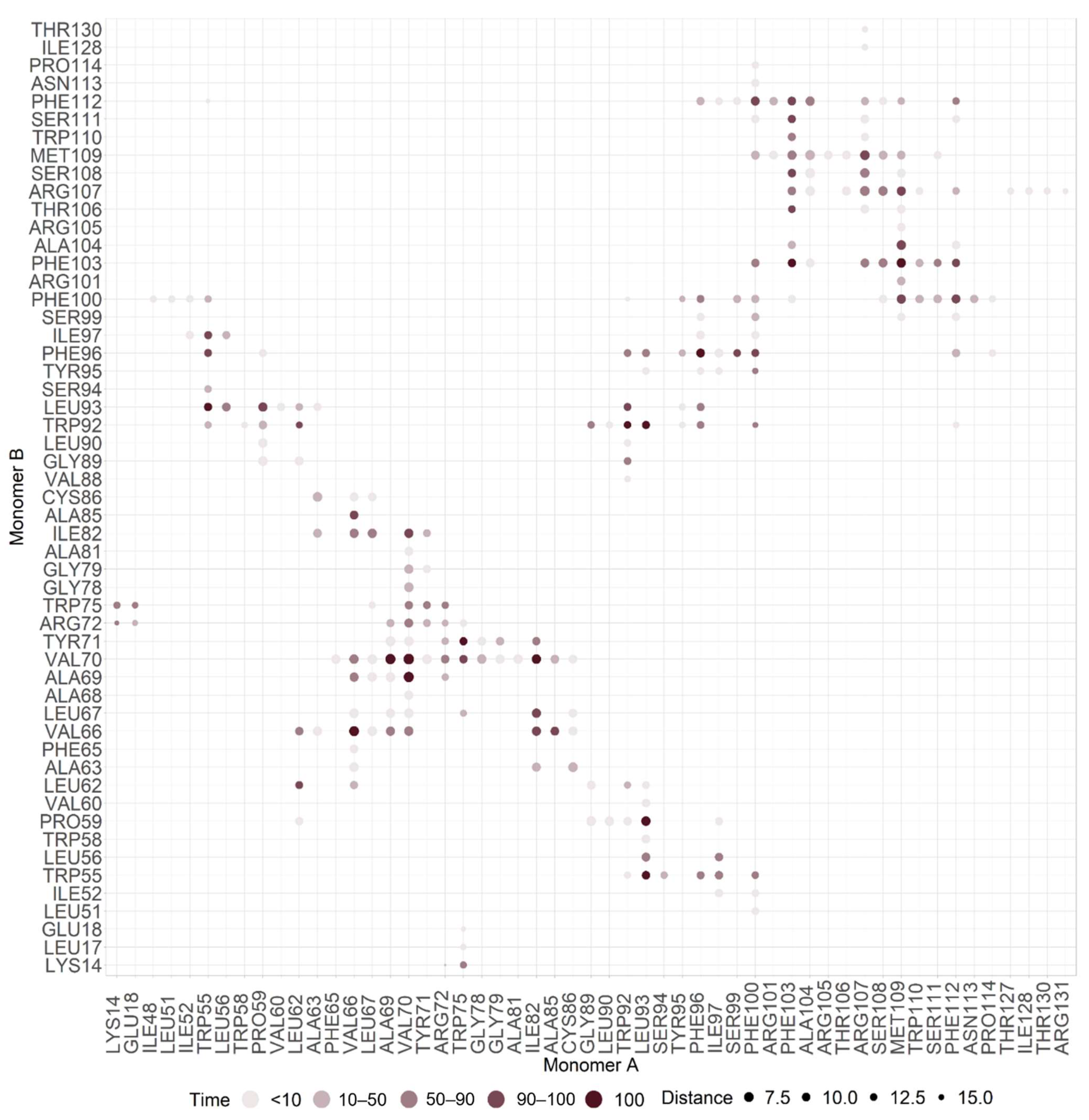
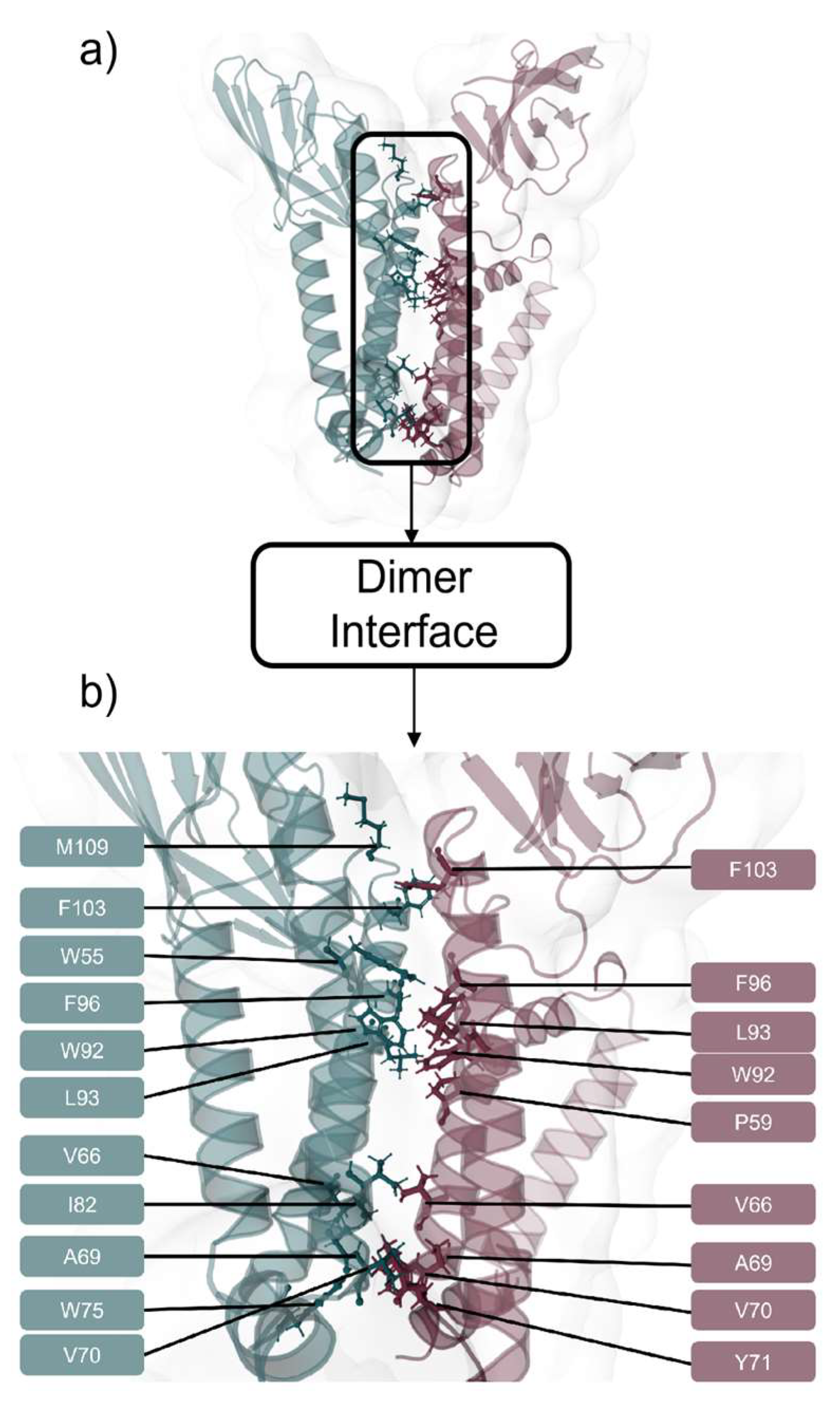
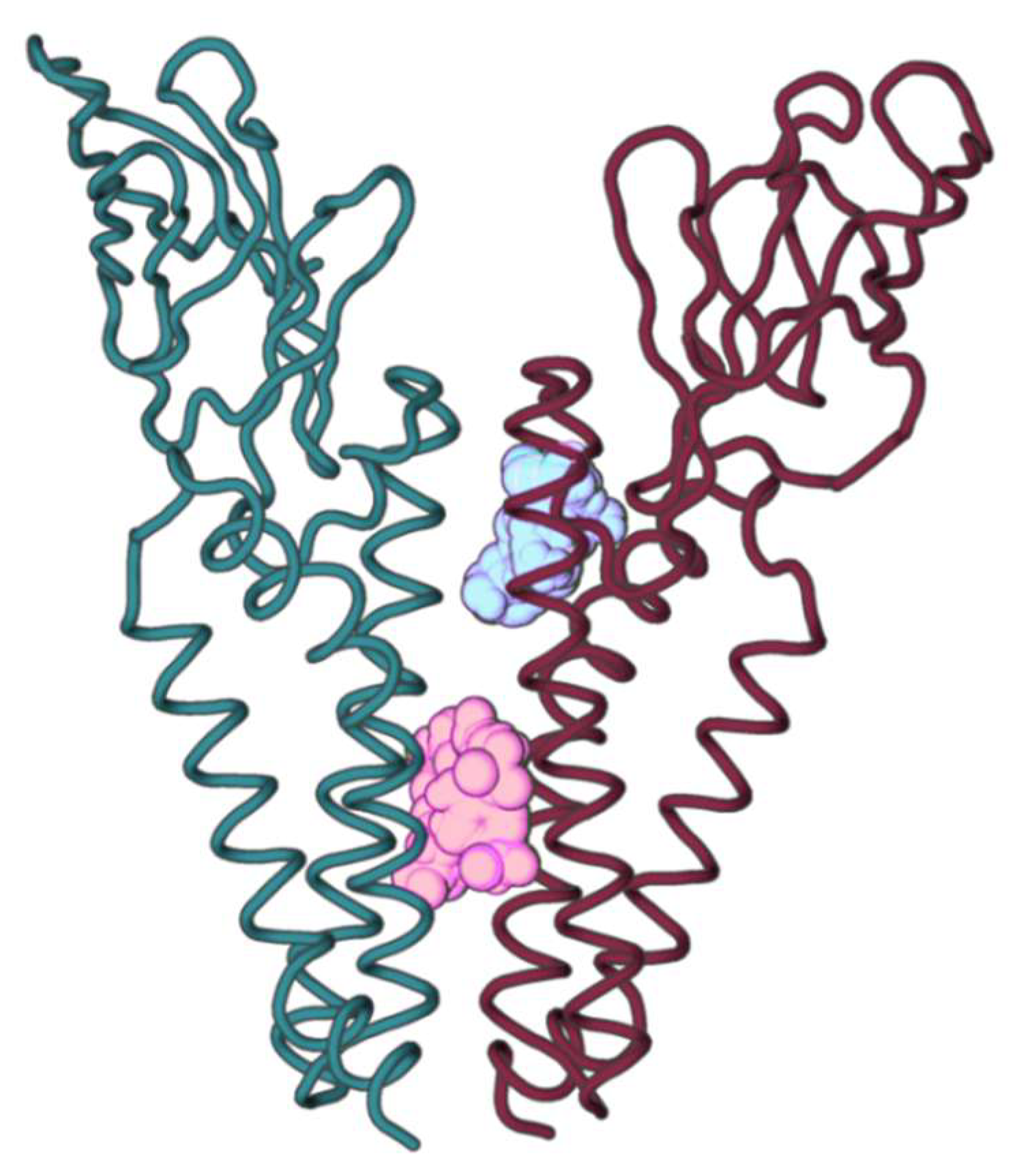
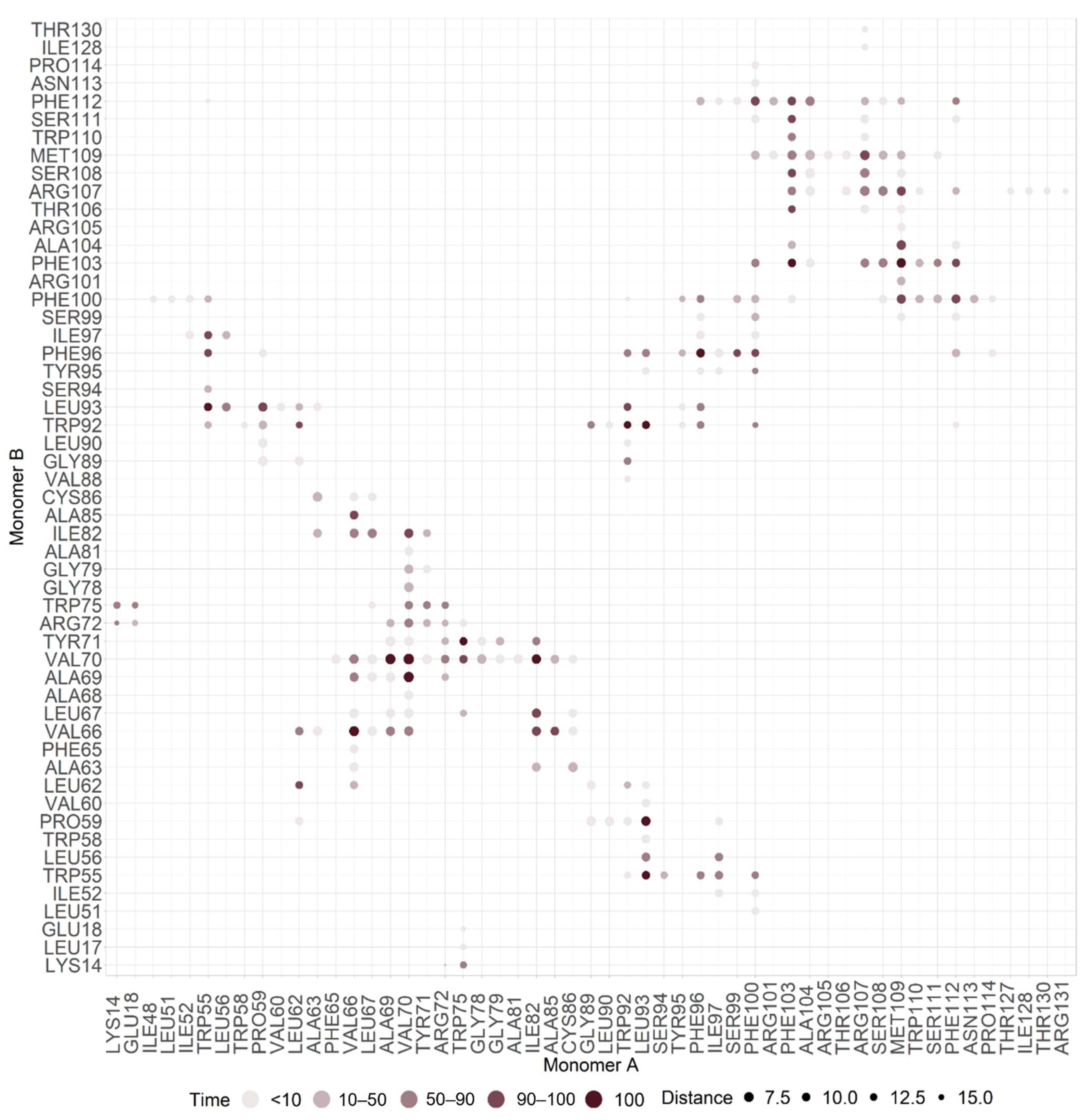
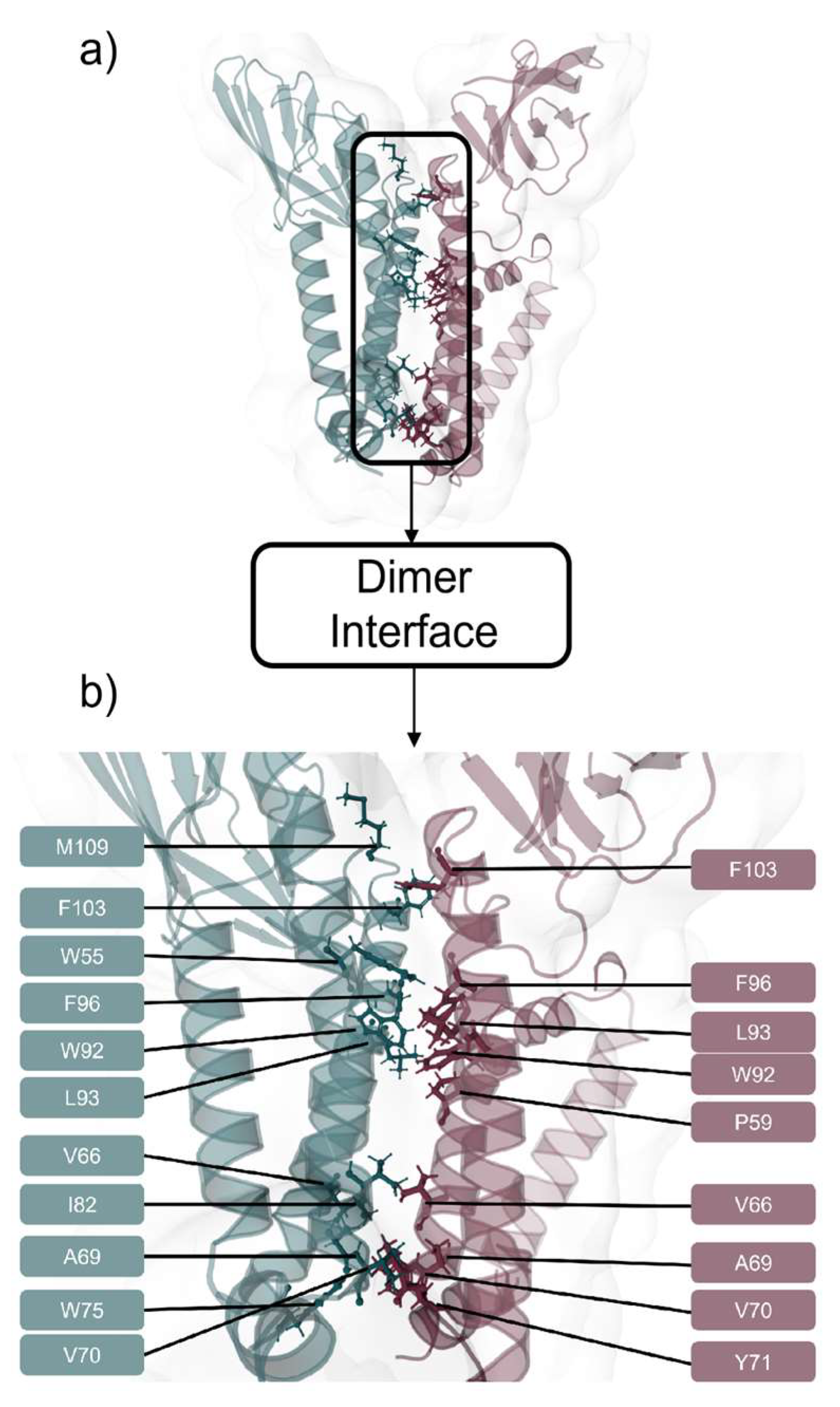
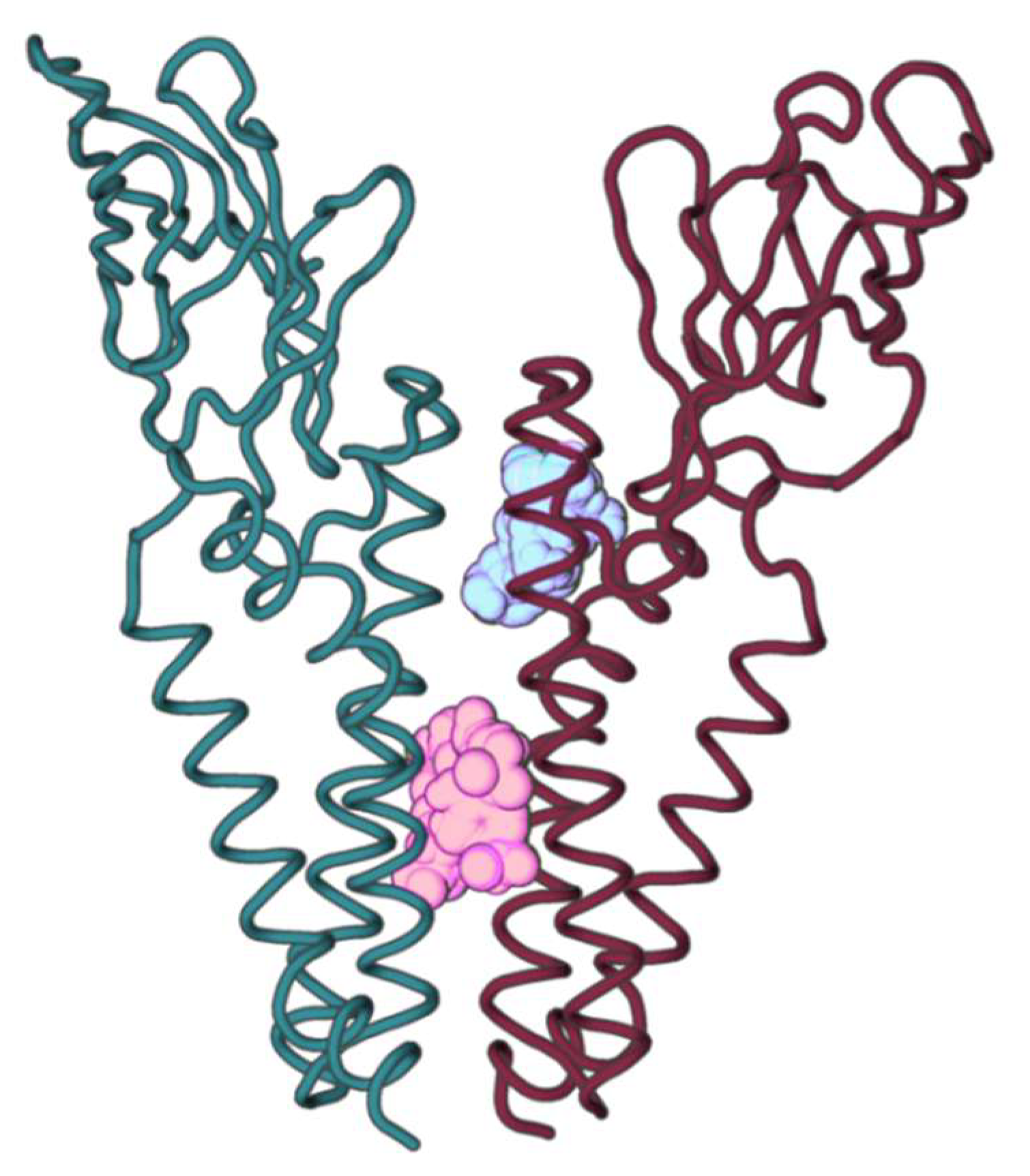
2.2. M Protein Dimer and Interface Prediction
2.2.1. Dimer Prediction
2.2.2. Interface Analysis
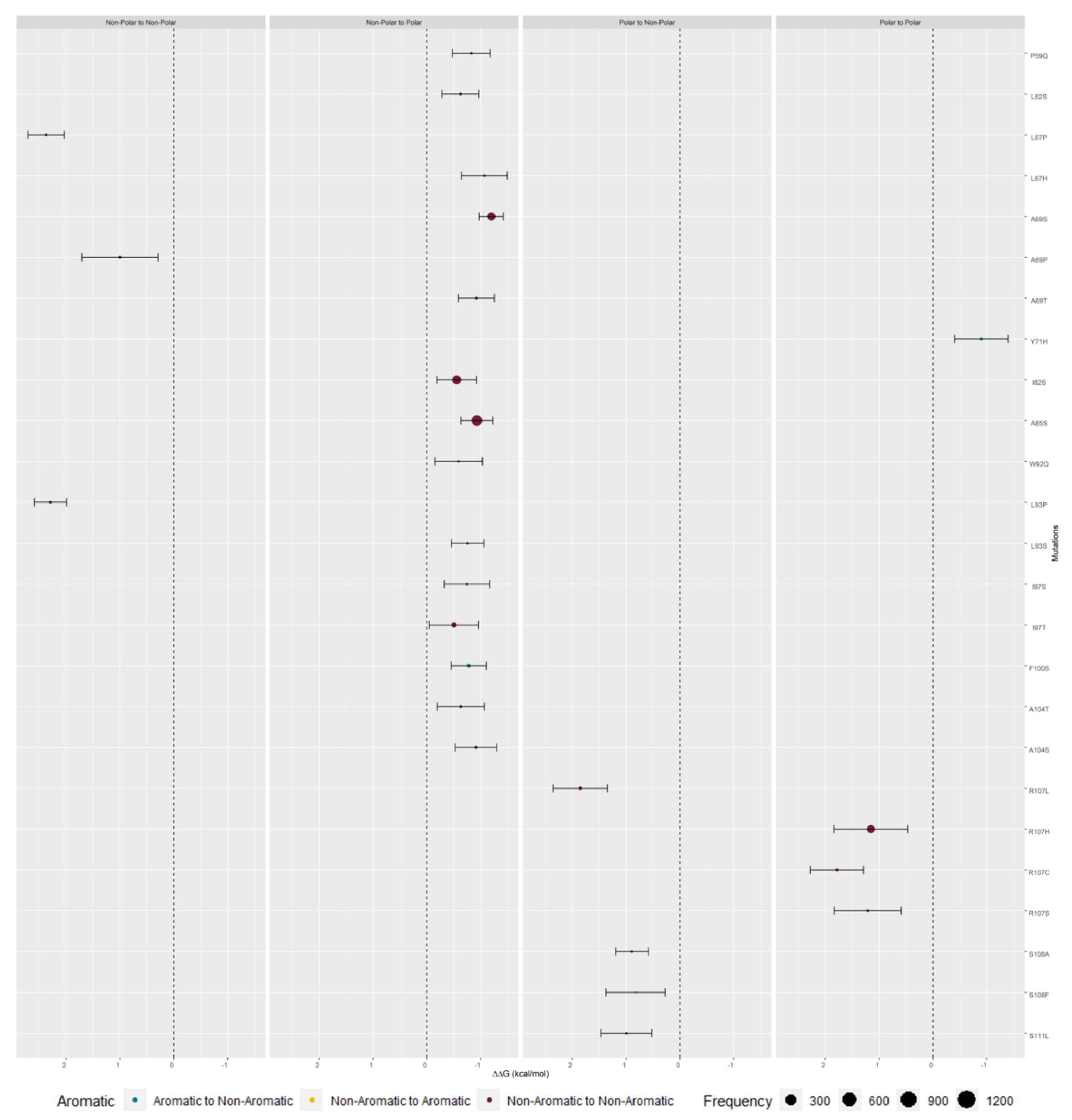
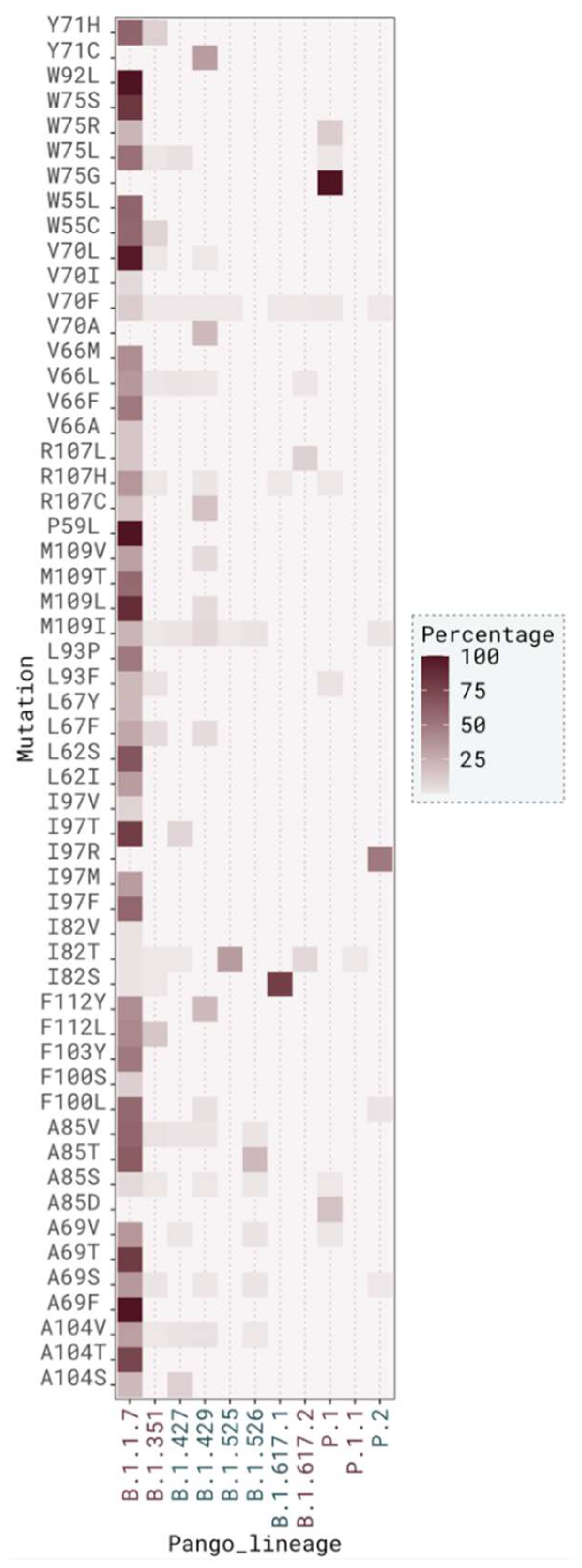
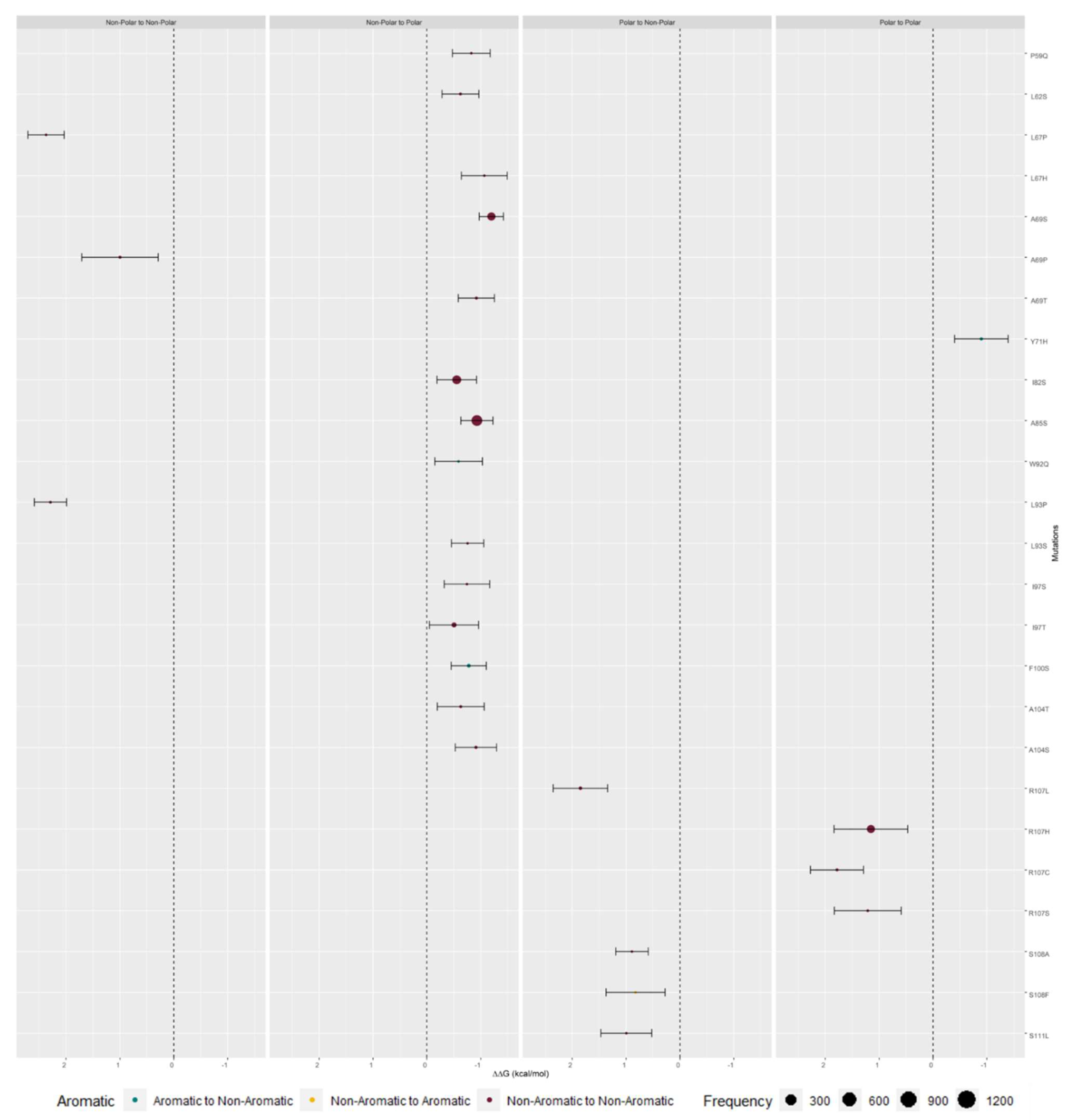
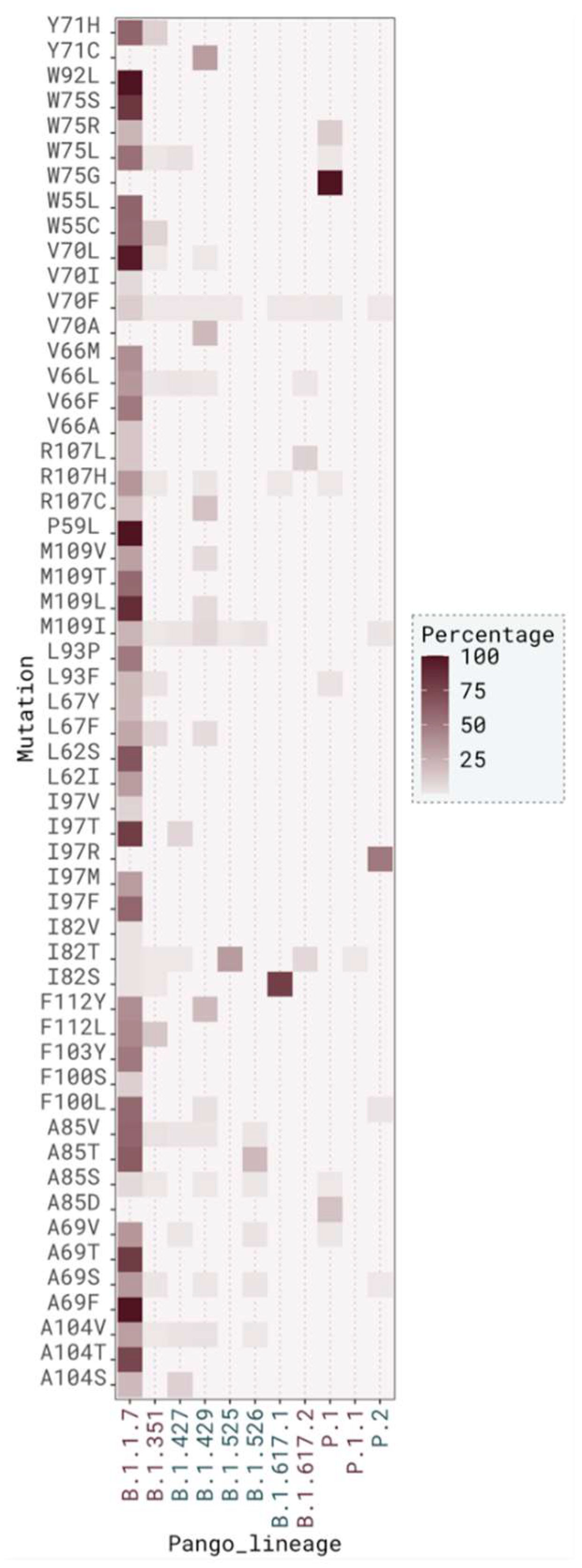
2.3. M Protein Mutation Analysis
2.3.1. Single Mutation Analysis
2.3.2. Mutation Distribution in Variants
2.3.3. Druggable Hot Spots
3. Discussion
3.1. M Protein Monomer Structure and Membrane Orientation
3.2. M Protein Dimer and Interface Prediction
3.3. M Protein Mutation Analysis
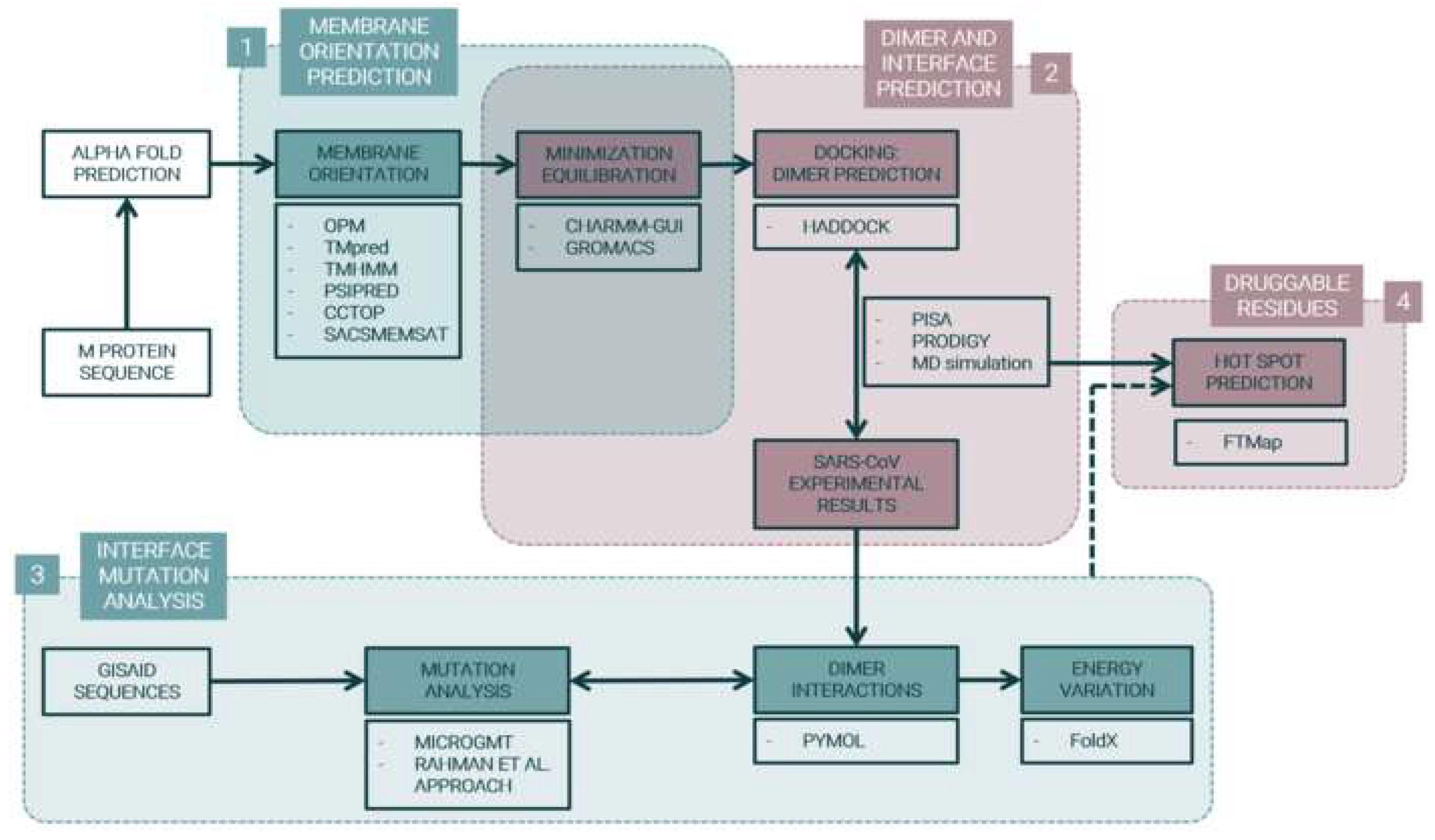
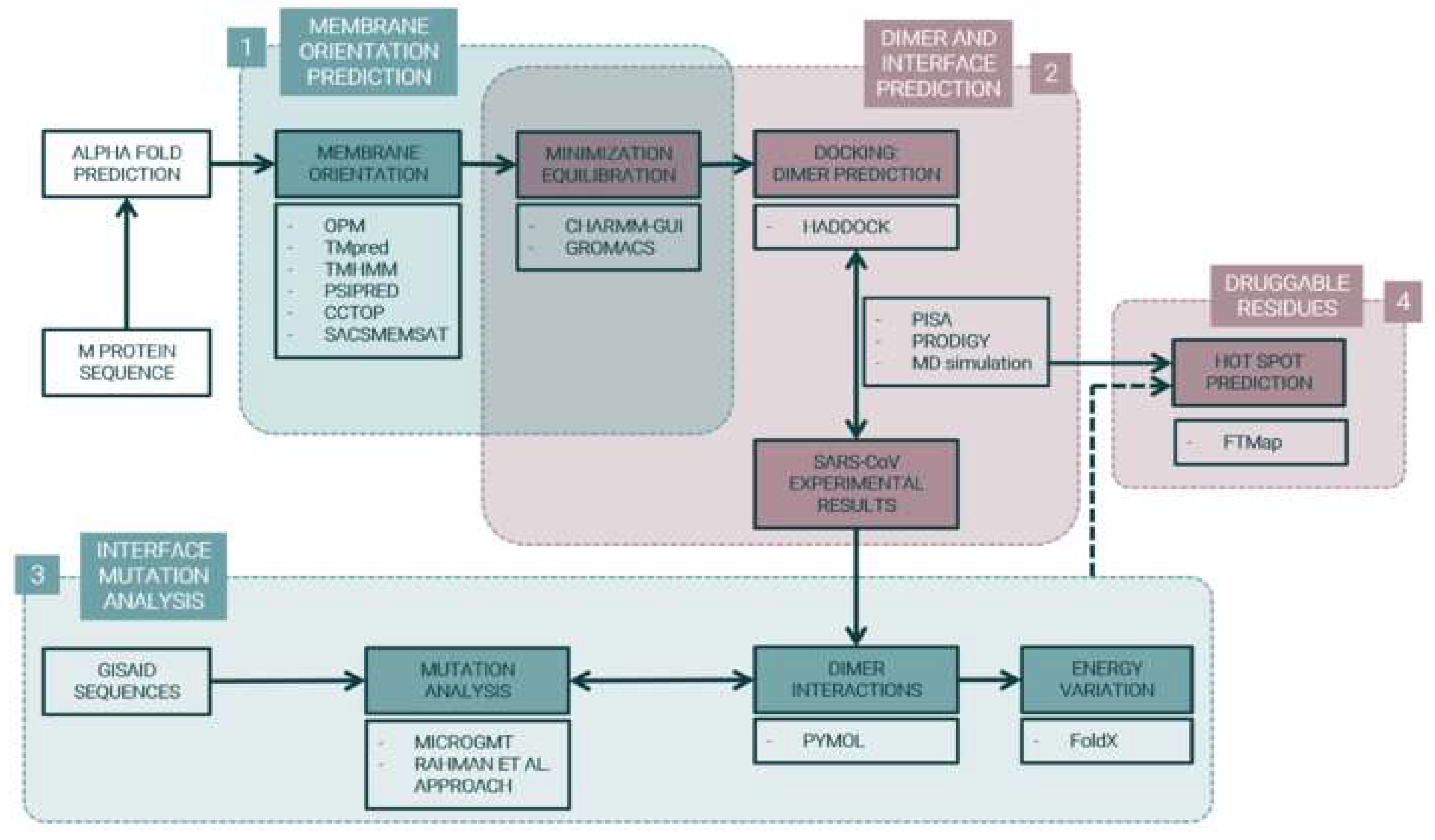
4. Materials and Methods
4.1. M Protein Monomer Structure and Membrane Orientation
4.2. M Protein Dimer and Interface Prediction
4.3. M protein Mutation Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Director-General’s Opening Remarks at the Media Briefing on COVID-19—11 March 2020; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.-Y.; Zhao, R.; Gao, L.-J.; Gao, X.-F.; Wang, D.-P.; Cao, J.-M. SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. Front. Cell. Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef] [PubMed]
- GISAID—Clade and Lineage Nomenclature Aids in Genomic Epidemiology of Active hCoV-19 Viruses. Available online: https://www.gisaid.org/resources/statements-clarifications/clade-and-lineage-nomenclature-aids-in-genomic-epidemiology-of-active-hcov-19-viruses/ (accessed on 4 May 2021).
- SeyedAlinaghi, S.; Mirzapour, P.; Dadras, O.; Pashaei, Z.; Karimi, A.; MohsseniPour, M.; Soleymanzadeh, M.; Barzegary, A.; Afsahi, A.M.; Vahedi, F.; et al. Characterization of SARS-CoV-2 different variants and related morbidity and mortality: A systematic review. Eur. J. Med. Res. 2021, 26, 51. [Google Scholar] [CrossRef] [PubMed]
- Hamed, S.M.; Elkhatib, W.F.; Khairalla, A.S.; Noreddin, A.M. Global dynamics of SARS-CoV-2 clades and their relation to COVID-19 epidemiology. Sci. Rep. 2021, 11, 8435. [Google Scholar] [CrossRef] [PubMed]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Benvenuto, D.; Giovanetti, M.; Angeletti, S.; Ciccozzi, M.; Pascarella, S. SARS-CoV-2 Envelope and membrane proteins: Structural differences linked to virus characteristics? Biomed. Res. Int. 2020, 2020, 4389089. [Google Scholar] [CrossRef] [PubMed]
- Neuman, B.W.; Kiss, G.; Kunding, A.; Bhella, D.; Baksh, M.F.; Connelly, S.; Droese, B.; Klaus, J.P.; Makino, S.; Sawicki, S.G.; et al. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 2011, 174, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Cagliani, R.; Forni, D.; Clerici, M.; Sironi, M. Computational inference of selection underlying the evolution of the novel coronavirus, severe acute respiratory syndrome coronavirus 2. J. Virol. 2020, 94, e00411-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ujike, M.; Taguchi, F. Incorporation of spike and membrane glycoproteins into coronavirus virions. Viruses 2015, 7, 1700–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Haan, C.A.; Vennema, H.; Rottier, P.J. Assembly of the coronavirus envelope: Homotypic interactions between the M proteins. J. Virol. 2000, 74, 4967–4978. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.-T.; Chang, C.-H.; Wang, S.-M.; Huang, K.-J.; Wang, C.-T. Identifying SARS-CoV membrane protein amino acid residues linked to virus-like particle assembly. PLoS ONE 2013, 8, e64013. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Pak, A.J.; He, P.; Monje-Galvan, V.; Casalino, L.; Gaieb, Z.; Dommer, A.C.; Amaro, R.E.; Voth, G.A. A multiscale coarse-grained model of the SARS-CoV-2 virion. Biophys. J. 2021, 120, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Ouzounis, C.A. A recent origin of Orf3a from M protein across the coronavirus lineage arising by sharp divergence. Comput. Struct. Biotechnol. J. 2020, 18, 4093–4102. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kumar, A.; Garg, N.; Giri, R. An insight into SARS-CoV-2 membrane protein interaction with spike, envelope, and nucleocapsid proteins. J. Biomol. Struct. Dyn. 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-K.; Lin, S.-M.; Satange, R.; Lin, S.-C.; Sun, S.-C.; Wu, H.-Y.; Kehn-Hall, K.; Hou, M.-H. Targeting protein–protein interaction interfaces in COVID-19 drug discovery. Comput. Struct. Biotechnol. J. 2021, 19, 2246–2255. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.A.; Saier, M.H., Jr. The SARS-coronavirus infection cycle: A survey of viral membrane proteins, their functional interactions and pathogenesis. Int. J. Mol. Sci. 2021, 22, 1308. [Google Scholar] [CrossRef] [PubMed]
- Arndt, A.L.; Larson, B.J.; Hogue, B.G. A conserved domain in the coronavirus membrane protein tail is important for virus assembly. J. Virol. 2010, 84, 11418–11428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klumperman, J.; Locker, J.K.; Meijer, A.; Horzinek, M.C.; Geuze, H.J.; Rottier, P.J. Coronavirus M proteins accumulate in the Golgi complex beyond the site of virion budding. J. Virol. 1994, 68, 6523–6534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Shi, M.; Li, J.; Song, P.; Li, N. Construction of SARS-CoV-2 virus-like particles by mammalian expression system. Front. Bioeng. Biotechnol. 2020, 8, 862. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, E.P.; Beis, K.; Cameron, A.D.; Iwata, S. Overcoming the challenges of membrane protein crystallography. Curr. Opin. Struct. Biol. 2008, 18, 581–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariano, G.; Farthing, R.J.; Lale-Farjat, S.L.M.; Bergeron, J.R.C. Structural characterization of SARS-CoV-2: Where we are, and where we need to be. Front. Mol. Biosci. 2020, 7, 605236. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Zhong, Q.; Gao, G.F. Overview of SARS-CoV-2 genome-encoded proteins. Sci. China Life Sci. 2022, 65, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Almeida, J.G.; Preto, A.J.; Koukos, P.I.; Bonvin, A.M.J.J.; Moreira, I.S. Membrane proteins structures: A review on computational modeling tools. Biochim. Biophys. Acta Biomembr. 2017, 1859, 2021–2039. [Google Scholar] [CrossRef] [PubMed]
- Satarker, S.; Nampoothiri, M. Structural proteins in severe acute respiratory syndrome coronavirus-2. Arch. Med. Res. 2020, 51, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yang, R.; Wang, W.; Jiang, S.; Yang, C.; Liu, N.; Dai, H.; Lee, I.; Meng, X.; Yuan, Z. Probing the formation, structure and free energy relationships of M protein dimers of SARS-CoV-2. Comput. Struct. Biotechnol. J. 2022, 20, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S. The structure of the membrane protein of SARS-CoV-2 resembles the sugar transporter SemiSWEET. Pathog. Immun. 2020, 5, 342–363. [Google Scholar] [CrossRef] [PubMed]
- Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell. Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Marques-Pereira, C.; Pires, M.; Moreira, I.S. Discovery of Virus-Host interactions using bioinformatic tools. Methods Cell Biol. 2022. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Zidek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef] [PubMed]
- Hofman, K. TMbase-a database of membrane spanning proteins segments. Biol. Chem. Hoppe Seyler. 1993, 374, 166. [Google Scholar]
- Sonnhammer, E.L.; von Heijne, G.; Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998, 6, 175–182. [Google Scholar] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, L.; Reményi, I.; Tusnády, G.E. The human transmembrane proteome. Biol. Direct. 2015, 10, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, L.; Reményi, I.; Tusnády, G.E. CCTOP: A Consensus Constrained TOPology prediction web server. Nucleic Acids Res. 2015, 43, W408–W412. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry 1994, 33, 3038–3049. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Bekker, H.; Berendsen, H.J.C.; Dijkstra, E.J.; Achterop, S.; Vondrumen, R.; Vanderspoel, D.; Sijbers, A.; Keegstra, H.; Renardus, M.K.R. Gromacs-a Parallel Computer for Molecular-Dynamics Simulations. 4th International Conference on Computational Physics (PC 92); World Scientific Publishing: Singapore, 1993; pp. 252–256. [Google Scholar]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; De Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 web server: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.; Li, X.; Gao, X.; Dong, Q. MicroGMT: A mutation tracker for SARS-CoV-2 and other microbial genome sequences. Front. Microbiol. 2020, 11, 1502. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Islam, M.R.; Hoque, M.N.; Alam, A.S.M.R.U.; Akther, M.; Puspo, J.A.; Akter, S.; Anwar, A.; Sultana, M.; Hossain, M.A. Comprehensive annotations of the mutational spectra of SARS-CoV-2 spike protein: A fast and accurate pipeline. Transbound. Emerg. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, L.; Feig, M. Modeling of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) proteins by machine learning and physics-based refinement. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—from vision to reality. Eur. Surveill. 2017, 22. [Google Scholar] [CrossRef] [Green Version]
- Preto, A.J.; Moreira, I.S. SPOTONE: Hot Spots on protein complexes with extremely randomized trees via sequence-only features. Int. J. Mol. Sci. 2020, 21, 7281. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S. The role of water occlusion for the definition of a protein binding hot-spot. Curr. Top Med. Chem. 2015, 15, 2068–2079. [Google Scholar] [CrossRef]
- Munteanu, C.R.; Pimenta, A.C.; Fernandez-Lozano, C.; Melo, A.; Cordeiro, M.N.D.S.; Moreira, I.S. Solvent accessible surface area-based hot-spot detection methods for protein–protein and protein–nucleic acid interfaces. J. Chem. Inf. Model. 2015, 55, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.M.; Ramos, R.M.; Pimenta, A.C.; Moreira, I.S. Solvent-accessible surface area: How well can be applied to hot-spot detection? Proteins 2014, 82, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Ramos, R.M.; Martins, J.M.; Fernandes, P.A.; Ramos, M.J. Are hot-spots occluded from water? J. Biomol. Struct. Dyn. 2014, 32, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Bogan, A.A.; Thorn, K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenke, R.; Kozakov, D.; Chuang, G.-Y.; Beglov, D.; Hall, D.; Landon, M.R.; Mattos, C.; Vajda, S. Fragment-based identification of druggable “hot spots” of proteins using Fourier domain correlation techniques. Bioinformatics 2009, 25, 621–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Hall, D.R.; Chuang, G.-Y.; Cencic, R.; Brenke, R.; Grove, L.E.; Beglov, D.; Pelletier, J.; Whitty, A.; Vajda, S. Structural conservation of druggable hot spots in protein–protein interfaces. Proc. Natl. Acad. Sci. USA 2011, 108, 13528–13533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohnuud, T.; Beglov, D.; Ngan, C.H.; Zerbe, B.; Hall, D.R.; Brenke, R.; Vajda, S.; Frank-Kamenetskii, M.D.; Kozakov, D. Computational mapping reveals dramatic effect of Hoogsteen breathing on duplex DNA reactivity with formaldehyde. Nucleic Acids Res. 2012, 40, 7644–7652. [Google Scholar] [CrossRef]
- Zhang, C.; Zheng, W.; Huang, X.; Bell, E.W.; Zhou, X.; Zhang, Y. Protein structure and sequence reanalysis of 2019-nCoV genome refutes snakes as its intermediate host and the unique similarity between its spike protein insertions and HIV-1. J. Proteome Res. 2020, 19, 1351–1360. [Google Scholar] [CrossRef] [Green Version]
- Computational Predictions of Protein Structures Associated with COVID-19. Available online: https://deepmind.com/research/open-source/computational-predictions-of-protein-structures-associated-with-COVID-19 (accessed on 2 September 2021).
- Collins, L.T.; Elkholy, T.; Mubin, S.; Hill, D.; Williams, R.; Ezike, K.; Singhal, A. Elucidation of SARS-CoV-2 budding mechanisms through molecular dynamics simulations of M and E protein complexes. J. Phys. Chem. Lett. 2021, 12, 12249–12255. [Google Scholar] [CrossRef] [PubMed]
- Kouyama, T.; Kanada, S.; Takeguchi, Y.; Narusawa, A.; Murakami, M.; Ihara, K. Crystal structure of the light-driven chloride pump halorhodopsin from Natronomonas pharaonis. J. Mol. Biol. 2010, 396, 564–579. [Google Scholar] [CrossRef]
- Osipiuk, J.; Ma, X.; Ton-That, H.; Anderson, W.F.; Joachimiak, A.; Center for structural genomics of infectious diseases (CSGID). SrtA sortase from actinomyces oris. Worldwide Protein Data Bank 2017. [Google Scholar] [CrossRef]
- Halfon, Y.; Jimenez-Fernande, A.; La Ros, R.; Espinos, R.; Krogh Johansen, H.; Matzov, D.; Eyal, Z.; Bashan, A.; Zimmerman, E.; Belousoff, M.; et al. Pseudomonas aeruginosa 50s ribosome from a clinical isolate with a mutation in uL6. Worldwide Protein Data Bank 2019. [Google Scholar] [CrossRef]
- Kern, D.M.; Sorum, B.; Mali, S.S.; Hoel, C.M.; Sridharan, S.; Remis, J.P.; Toso, D.B.; Kotecha, A.; Bautista, D.M.; Brohawn, S.G. Cryo-EM structure of SARS-CoV-2 ORF3a in lipid nanodiscs. Nat. Struct. Mol. Biol. 2021, 28, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Alsulami, A.F.; Thomas, S.E.; Jamasb, A.R.; Beaudoin, C.A.; Moghul, I.; Bannerman, B.; Copoiu, L.; Vedithi, S.C.; Torres, P.; Blundell, T.L. SARS-CoV-2 3D database: Understanding the coronavirus proteome and evaluating possible drug targets. Brief Bioinform. 2021, 22, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Monje-Galvan, V.; Voth, G.A. Molecular interactions of the M and E integral membrane proteins of SARS-CoV-2. Faraday Discuss. 2021, 232, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Feig, M. SARS-Cov-2 Protein Structure Models. Available online: https://github.com/feiglab/sars-cov-2-proteins (accessed on 19 February 2022).
- Majumdar, P.; Niyogi, S. SARS-CoV-2 mutations: The biological trackway towards viral fitness. Epidemiol. Infect. 2021, 149, e110. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Bard, J.D.; Triche, T.J.; Judkins, A.R.; Biegel, J.A.; Gai, X. Emerging variants of concern in SARS-CoV-2 membrane protein: A highly conserved target with potential pathological and therapeutic implications. Emerg. Microbes. Infect. 2021, 10, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, C.; Appert-Collin, A.; Bagnard, D.; Blaise, S.; Romier-Crouzet, B.; Efremov, R.G.; Sartelet, H.; Duca, L.; Maurice, P.; Bennasroune, A. Transmembrane peptides as inhibitors of protein–protein interactions: An efficient strategy to target cancer cells? Front. Oncol. 2020, 10, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AlQuraishi, M. Machine learning in protein structure prediction. Curr. Opin. Chem. Biol. 2021, 65, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Liu, D.X. Post-translational modifications of coronavirus proteins: Roles and function. Future Virol. 2018, 13, 405–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Yamada, Y.; Fung, T.S.; Huang, M.; Chia, R.; Liu, D.X. Identification of N-linked glycosylation sites in the spike protein and their functional impact on the replication and infectivity of coronavirus infectious bronchitis virus in cell culture. Virology 2018, 513, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Shajahan, A.; Pepi, L.E.; Rouhani, D.S.; Heiss, C.; Azadi, P. Glycosylation of SARS-CoV-2: Structural and functional insights. Anal. Bioanal Chem. 2021, 413, 7179–7193. [Google Scholar] [CrossRef] [PubMed]
- Hogue, B.G.; Machamer, C.E. Coronavirus structural proteins and virus assembly. In Nidoviruses; Perlman, S., Gallagher, T., Snijder, E.J., Eds.; ASM Press: Washington, DC, USA, 2014; pp. 179–200. [Google Scholar]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meer, G. Lipids of the golgi membrane. Trends Cell Biol. 1998, 8, 29–33. [Google Scholar] [CrossRef]
- Rath, S.L.; Tripathy, M.; Mandal, N. How does temperature affect the dynamics of SARS-CoV-2 M proteins? Insights from molecular dynamics simulations. bioRxiv 2021. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Di Nola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Blundell, T.L.; Srinivasan, N. Symmetry, stability, and dynamics of multidomain and multicomponent protein systems. Proc. Natl. Acad. Sci. USA 1996, 93, 14243–14248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, S.J.; Bonvin, A.M.J.J. CPORT: A consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PLoS ONE 2011, 6, e17695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Vangone, A.; Bonvin, A.M. Contacts-based prediction of binding affinity in protein–protein complexes. Elife 2015, 4, e07454. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasello, G.; Armenia, I.; Molla, G. The protein imager: A full-featured online molecular viewer interface with server-side HQ-rendering capabilities. Bioinformatics 2020, 36, 2909–2911. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, L. ggplot2: Elegant Graphics for Data Analysis by WICKHAM, H. Biometrics 2011, 678–679. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monomer A | ΔSASA A (Å2) | relSASA A | Monomer B | ΔSASA B | relSASA B | Percentage (%) | Cα Distance |
|---|---|---|---|---|---|---|---|
| (Å2) | (Å) | ||||||
| W55 | 69.00 ± 23.91 | 0.62 ± 0.16 | L93 | 70.51 ± 16.96 | 0.67 ± 0.14 | 100.00 | 10.93 ± 0.65 |
| V66 | 57.26 ± 11.49 | 0.80 ± 0.13 | V66 | 58.18 ± 11.81 | 0.80 ± 0.10 | 100.00 | 7.11 ± 0.32 |
| A69 | 15.96 ± 6.92 | 0.93 ± 0.15 | V70 | 87.47 ± 11.74 | 0.88 ± 0.08 | 100.00 | 6.31 ± 0.41 |
| V70 | 83.79 ± 16.05 | 0.81 ± 0.16 | A69 | 14.78 ± 9.16 | 0.78 ± 0.43 | 100.00 | 6.70 ± 0.50 |
| V70 | 83.79 ± 16.05 | 0.81 ± 0.16 | V70 | 87.47 ± 11.74 | 0.88 ± 0.08 | 100.00 | 5.25 ± 0.51 |
| W75 | 66.06 ± 38.00 | 0.33 ± 0.18 | Y71 | 8.53 ± 40.92 | 0.09 ± 0.57 | 100.00 | 11.42 ± 0.74 |
| I82 | 63.02 ± 18.62 | 0.65 ± 0.14 | V70 | 87.47 ± 11.74 | 0.88 ± 0.08 | 100.00 | 8.62 ± 0.65 |
| W92 | 64.08 ± 12.99 | 0.87 ± 0.10 | W92 | 48.02 ± 16.01 | 0.76 ± 0.17 | 100.00 | 12.58 ± 0.49 |
| L93 | 67.87 ± 23.73 | 0.62 ± 0.20 | P59 | 11.83 ± 23.49 | 0.20 ± 0.53 | 100.00 | 8.62 ± 0.61 |
| F96 | 67.33 ± 16.53 | 0.90 ± 0.09 | F96 | 52.22 ± 15.81 | 0.89 ± 0.12 | 100.00 | 9.67 ± 0.65 |
| F103 | 66.38 ± 15.37 | 0.88 ± 0.10 | F103 | 78.66 ± 15.91 | 0.95 ± 0.07 | 100.00 | 10.79 ± 0.58 |
| M109 | 89.25 ± 27.84 | 0.54 ± 0.14 | F103 | 78.66 ± 15.91 | 0.95 ± 0.07 | 100.00 | 8.31 ± 0.44 |
| P59 | 32.47 ± 25.51 | 0.50 ± 0.27 | L93 | 70.51 ± 16.96 | 0.67 ± 0.14 | 99.67 | 09.01 ± 0.62 |
| F112 | 76.09 ± 25.84 | 0.84 ± 0.08 | F100 | 64.39 ± 26.02 | 0.50 ± 0.19 | 99.67 | 9.13 ± 0.49 |
| V70 | 83.79 ± 16.05 | 0.81 ± 0.16 | I82 | 45.82 ± 20.01 | 0.50 ± 0.20 | 99.34 | 9.08 ± 0.66 |
| F100 | 83.51 ± 28.35 | 0.62 ± 0.14 | F112 | 38.18 ± 31.03 | 0.52 ± 0.41 | 99.34 | 9.16 ± 0.55 |
| W55 | 69.00 ± 23.91 | 0.62 ± 0.16 | I97 | 22.90 ± 22.99 | 0.23 ± 0.24 | 99.01 | 11.45 ± 0.68 |
| W92 | 64.08 ± 12.99 | 0.87 ± 0.10 | L93 | 70.51 ± 16.96 | 0.67 ± 0.14 | 99.01 | 11.78 ± 0.60 |
| R107 | 71.92 ± 29.68 | 0.36 ± 0.13 | M109 | 86.63 ± 28.09 | 0.49 ± 0.14 | 99.01 | 7.72 ± 0.77 |
| L62 | 24.35 ± 18.78 | 0.44 ± 0.30 | L62 | 17.37 ± 14.70 | 0.34 ± 0.30 | 98.35 | 11.78 ± 0.44 |
| M109 | 89.25 ± 27.84 | 0.54 ± 0.14 | F100 | 64.39 ± 26.02 | 0.50 ± 0.19 | 97.36 | 8.9 ± 0.57 |
| M109 | 89.25 ± 27.84 | 0.54 ± 0.14 | A104 | 17.84 ± 14.34 | 0.26 ± 0.21 | 97.36 | 7.78 ± 0.50 |
| I82 | 63.02 ± 18.62 | 0.65 ± 0.14 | L67 | 14.01 ± 19.51 | 0.17 ± 0.24 | 96.37 | 8.69 ± 0.55 |
| F103 | 66.38 ± 15.37 | 0.88 ± 0.10 | S108 | 9.79 ± 12.38 | 0.28 ± 0.76 | 95.05 | 10.8 ± 0.67 |
| F112 | 76.09 ± 25.84 | 0.84 ± 0.08 | F103 | 78.66 ± 15.91 | 0.95 ± 0.07 | 94.72 | 11.13 ± 0.55 |
| W75 | 66.06 ± 38.00 | 0.33 ± 0.18 | V70 | 87.47 ± 11.74 | 0.88 ± 0.08 | 94.39 | 10.58 ± 0.65 |
| F103 | 66.38 ± 15.37 | 0.88 ± 0.10 | F112 | 38.18 ± 31.03 | 0.52 ± 0.41 | 94.39 | 10.28 ± 0.69 |
| I82 | 63.02 ± 18.62 | 0.65 ± 0.14 | V66 | 58.18 ± 11.81 | 0.80 ± 0.10 | 93.07 | 9.02 ± 0.49 |
| W55 | 69.00 ± 23.91 | 0.62 ± 0.16 | F96 | 52.22 ± 15.81 | 0.89 ± 0.12 | 93.07 | 11.66 ± 0.63 |
| V66 | 57.26 ± 11.49 | 0.80 ± 0.13 | A85 | 0.92 ± 6.38 | 0.00 ± 0.00 | 92.08 | 9.78 ± 0.44 |
| F103 | 66.38 ± 15.37 | 0.88 ± 0.10 | S111 | −3.07 ± 3.85 | 0.00 ± 0.00 | 92.08 | 11.02 ± 0.76 |
| A85 | 1.49 ± 6.45 | 0.00 ± 0.00 | V66 | 58.18 ± 11.81 | 0.80 ± 0.10 | 91.42 | 9.62 ± 0.45 |
| F100 | 83.51 ± 28.35 | 0.62 ± 0.14 | F96 | 52.22 ± 15.81 | 0.89 ± 0.12 | 91.42 | 11.66 ± 0.72 |
| M109 | 89.25 ± 27.84 | 0.54 ± 0.14 | R107 | 53.28 ± 38.80 | 0.26 ± 0.18 | 91.42 | 8.96 ± 0.68 |
| ΔΔGbinding > 0.5 kcal/mol | ΔΔGbinding < −0.5 kcal/mol | −0.5 kcal/mol < ΔΔGbinding |
|---|---|---|
| <0.5 kcal/mol | ||
| 606 (2.77%) | 2683 (12.27%) | 18,579 (84.96%) |
| Polar Residue to Non-Polar Residue | Remained a Polar Residue | Non-Polar to a Polar Residue | Remained a Non-Polar Residue | |
|---|---|---|---|---|
| Frequency | 0.11% | 2.68% | 41.68% | 55.53% |
| ΔΔGbinding (kcal/mol) | 1.14 ± 0.48 | 0.65 ± 1.07 | −0.42 ± 0.36 | 0.14 ± 0.49 |
| Remained an Aromatic Residue | Aromatic to a Non-Aromatic Residue | Non-Aromatic to an Aromatic Residue | Remained a Non-Aromatic Residue | |
|---|---|---|---|---|
| Frequency | 0.03% | 2.69% | 7.27% | 90.01% |
| ΔΔGbinding (kcal/mol) | −0.20 ± 0.15 | −0.11 ± 0.30 | 0.09 ± 0.29 | 0.04 ± 0.77 |
| GRY | GH | G | GR | GV | S | O | L | V | |
|---|---|---|---|---|---|---|---|---|---|
| Frequency | 36.69% | 21.25% | 19.06% | 17.27% | 4.36% | 0.90% | 0.38% | 0.05% | 0.05% |
| Most frequent mutation in clade (percentage) | V70L (73.30%) | I82T (47.23%) | I82T (71.26%) | V70F (26.32%) | - | - | - | - | - |
| ΔΔGbinding for most frequent mutation (kcal/mol) | −0.02 ± 0.22 | −0.49 ± 0.38 | −0.49 ± 0.38 | 0.17 ± 0.47 | - | - | - | - | - |
| Residue | ΔSASA (Å2) | relSASA |
|---|---|---|
| I82 | 54.42 ± 13.27 | 0.58 ± 0.12 |
| V70 | 85.63 ± 11.01 | 0.84 ± 0.10 |
| A69 | 15.37 ± 4.26 | 0.90 ± 0.12 |
| M109 | 87.94 ± 14.46 | 0.52 ± 0.07 |
| A104 | 13.69 ± 8.89 | 0.21 ± 0.13 |
| R107 | 62.60 ± 21.03 | 0.32 ± 0.10 |
| W75 | 49.28 ± 20.33 | 0.27 ± 0.11 |
| Residue | Number of Mutations | ΔΔGbinding < −0.5 kcal/mol | −0.5 kcal/mol < ΔΔGbinding < 0.5 kcal/mol | ΔΔGbinding > 0.5 kcal/mol |
|---|---|---|---|---|
| F96 | 1 | 0% | 100% | 0% |
| S111 | 1 | 0% | 0% | 100% |
| S108 | 2 | 0% | 0% | 100% |
| F103 | 7 | 0% | 100% | 0% |
| F112 | 14 | 0% | 100% | 0% |
| W55 | 32 | 0% | 100% | 0% |
| F100 | 98 | 30% | 70% | 0% |
| I97 | 228 | 50% | 50% | 0% |
| M109 | 1088 | 0% | 100% | 0% |
| Residue | Number of Mutations | ΔΔGbinding < −0.5 kcal/mol | −0.5 kcal/mol < ΔΔGbinding < 0.5 kcal/mol | ΔΔGbinding > 0.5 kcal/mol |
|---|---|---|---|---|
| P59 | 4 | 25% | 75% | 0% |
| W92 | 4 | 25% | 75% | 0% |
| L62 | 6 | 50% | 50% | 0% |
| L67 | 40 | 17% | 66% | 17% |
| L93 | 61 | 1% | 98% | 1% |
| V66 | 502 | 0% | 100% | 0% |
| A85 | 1604 | 75% | 25% | 0% |
| I82 | 7094 | 10% | 90% | 0% |
| Mutation | ΔΔGbinding (kcal/mol) |
|---|---|
| I82T | −0.49 |
| I97T | −0.5 |
| I82S | −0.55 |
| W92Q | −0.59 |
| L62S | −0.62 |
| A104T | −0.63 |
| I97S | −0.74 |
| L93S | −0.76 |
| F100S | −0.78 |
| P59Q | −0.83 |
| Y71H | −0.9 |
| A104S | −0.91 |
| A69T | −0.92 |
| A85S | −0.93 |
| L67H | −1.07 |
| A69S | −1.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marques-Pereira, C.; Pires, M.N.; Gouveia, R.P.; Pereira, N.N.; Caniceiro, A.B.; Rosário-Ferreira, N.; Moreira, I.S. SARS-CoV-2 Membrane Protein: From Genomic Data to Structural New Insights. Int. J. Mol. Sci. 2022, 23, 2986. https://doi.org/10.3390/ijms23062986
Marques-Pereira C, Pires MN, Gouveia RP, Pereira NN, Caniceiro AB, Rosário-Ferreira N, Moreira IS. SARS-CoV-2 Membrane Protein: From Genomic Data to Structural New Insights. International Journal of Molecular Sciences. 2022; 23(6):2986. https://doi.org/10.3390/ijms23062986
Chicago/Turabian StyleMarques-Pereira, Catarina, Manuel N. Pires, Raquel P. Gouveia, Nádia N. Pereira, Ana B. Caniceiro, Nícia Rosário-Ferreira, and Irina S. Moreira. 2022. "SARS-CoV-2 Membrane Protein: From Genomic Data to Structural New Insights" International Journal of Molecular Sciences 23, no. 6: 2986. https://doi.org/10.3390/ijms23062986
APA StyleMarques-Pereira, C., Pires, M. N., Gouveia, R. P., Pereira, N. N., Caniceiro, A. B., Rosário-Ferreira, N., & Moreira, I. S. (2022). SARS-CoV-2 Membrane Protein: From Genomic Data to Structural New Insights. International Journal of Molecular Sciences, 23(6), 2986. https://doi.org/10.3390/ijms23062986