
Signaling Induced by Chronic Viral Hepatitis: Dependence and Consequences

 , , and
, , and
Abstract
:1. Introduction
2. Hepatitis B, C, D Viruses
3. Virus-Induced Oxidative Stress Signaling
4. Virus-Induced Pro-Inflammatory Signaling
5. Deregulation of Cellular Metabolism Pathways
6. Virus-Induced Pro-Fibrotic/Pro-Oncogenic Signaling
7. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO Team Global HIV, H. a. S. T. I. P. Global Progress Report on HIV, Viral Hepatitis and Sexually Transmitted Infections, 2021; World Health Organization: Geneva, Switzerland, 2021; p. 108. [Google Scholar]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, E.M.; Sherman, R.L.; Henley, S.J.; Jemal, A.; Siegel, D.A.; Feuer, E.J.; Firth, A.U.; Kohler, B.A.; Scott, S.; Ma, J.; et al. Annual Report to the Nation on the Status of Cancer, Featuring Cancer in Men and Women Age 20–49 Years. J. Natl. Cancer Inst. 2019, 111, 1279–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, M.F.; Chen, D.S.; Dusheiko, G.M.; Janssen, H.L.A.; Lau, D.T.Y.; Locarnini, S.A.; Peters, M.G.; Lai, C.L. Hepatitis B virus infection. Nat. Rev. Dis. Primers 2018, 4, 18035. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Protzer, U. Attacking hepatitis B virus cccDNA—The holy grail to hepatitis B cure. J. Hepatol 2016, 64, S41–S48. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef]
- Mentha, N.; Clement, S.; Negro, F.; Alfaiate, D. A review on hepatitis D: From virology to new therapies. J. Adv. Res. 2019, 17, 3–15. [Google Scholar] [CrossRef]
- Turon-Lagot, V.; Saviano, A.; Schuster, C.; Baumert, T.F.; Verrier, E.R. Targeting the Host for New Therapeutic Perspectives in Hepatitis D. J. Clin. Med. 2020, 9, 222. [Google Scholar] [CrossRef] [Green Version]
- Wedemeyer, H.; Negro, F. Devil hepatitis D: An orphan disease or largely underdiagnosed? Gut 2019, 68, 381–382. [Google Scholar] [CrossRef]
- Dubuisson, J.; Cosset, F.L. Virology and cell biology of the hepatitis C virus life cycle: An update. J. Hepatol. 2014, 61, S3–S13. [Google Scholar] [CrossRef] [Green Version]
- Comparcola, D.; Alisi, A.; Nobili, V. Hepatitis C virus and nonalcoholic Fatty liver disease: Similar risk factors for necroinflammation, fibrosis, and cirrhosis. Clin. Gastroenterol. Hepatol. 2010, 8, 97. [Google Scholar] [CrossRef]
- Lupberger, J.; Croonenborghs, T.; Roca Suarez, A.A.; Van Renne, N.; Juhling, F.; Oudot, M.A.; Virzi, A.; Bandiera, S.; Jamey, C.; Meszaros, G.; et al. Combined Analysis of Metabolomes, Proteomes, and Transcriptomes of Hepatitis C Virus-Infected Cells and Liver to Identify Pathways Associated with Disease Development. Gastroenterology 2019, 157, 537–551.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlotsky, J.M.; Negro, F.; Aghemo, A.; Berenguer, M.; Dalgard, O.; Dusheiko, G.; Marra, F.; Puoti, M.; Wedemeyer, H.; European Association for the Study of the Liver. EASL recommendations on treatment of hepatitis C: Final update of the series. J. Hepatol. 2020, 73, 1170–1218. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.G.; Boyd, A.; Combe, E.; Testoni, B.; Zoulim, F. Covalently closed circular DNA: The ultimate therapeutic target for curing HBV infections. J. Hepatol. 2021, 75, 706–717. [Google Scholar] [CrossRef]
- Roca Suarez, A.A.; Testoni, B.; Zoulim, F. HBV 2021: New therapeutic strategies against an old foe. Liver Int. 2021, 41 (Suppl. S1), 15–23. [Google Scholar] [CrossRef]
- Asselah, T.; Loureiro, D.; Le Gal, F.; Narguet, S.; Brichler, S.; Bouton, V.; Abazid, M.; Boyer, N.; Giuly, N.; Gerber, A.; et al. Early virological response in six patients with hepatitis D virus infection and compensated cirrhosis treated with Bulevirtide in real-life. Liver Int. 2021, 41, 1509–1517. [Google Scholar] [CrossRef]
- Hamdane, N.; Juhling, F.; Crouchet, E.; El Saghire, H.; Thumann, C.; Oudot, M.A.; Bandiera, S.; Saviano, A.; Ponsolles, C.; Roca Suarez, A.A.; et al. HCV-Induced Epigenetic Changes Associated with Liver Cancer Risk Persist after Sustained Virologic Response. Gastroenterology 2019, 156, 2313–2329.e7. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.U.; Seo, Y.S.; Lee, H.A.; Kim, M.N.; Lee, E.J.; Shin, H.J.; Lee, Y.R.; Lee, H.W.; Park, J.Y.; Kim, D.Y.; et al. Hepatocellular Carcinoma Risk Steadily Persists over Time Despite Long-Term Antiviral Therapy for Hepatitis B: A Multicenter Study. Cancer Epidemiol. Biomark. Prev. 2020, 29, 832–837. [Google Scholar] [CrossRef] [Green Version]
- Juhling, F.; Hamdane, N.; Crouchet, E.; Li, S.; El Saghire, H.; Mukherji, A.; Fujiwara, N.; Oudot, M.A.; Thumann, C.; Saviano, A.; et al. Targeting clinical epigenetic reprogramming for chemoprevention of metabolic and viral hepatocellular carcinoma. Gut 2021, 70, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Andrisani, O. Epigenetic mechanisms in hepatitis B virus-associated hepatocellular carcinoma. Hepatoma Res. 2021, 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.X.; Tanaka, L.Y.; Wosniak, J.; Laurindo, F.R. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: Roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal. 2009, 11, 2409–2427. [Google Scholar] [CrossRef] [PubMed]
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, Oxidative Stress and Innate Immunity. Front. Physiol. 2018, 9, 1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Xu, M.; Jeong, S.; Qian, Y.; Wu, H.; Xia, Q.; Kong, X. The Role of Nrf2 in Liver Disease: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 2018, 9, 1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleksunes, L.M.; Manautou, J.E. Emerging role of Nrf2 in protecting against hepatic and gastrointestinal disease. Toxicol. Pathol. 2007, 35, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Spahis, S.; Delvin, E.; Borys, J.M.; Levy, E. Oxidative Stress as a Critical Factor in Nonalcoholic Fatty Liver Disease Pathogenesis. Antioxid. Redox Signal. 2017, 26, 519–541. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget 2017, 8, 3895–3932. [Google Scholar] [CrossRef] [Green Version]
- Block, T.M.; Mehta, A.S.; Fimmel, C.J.; Jordan, R. Molecular viral oncology of hepatocellular carcinoma. Oncogene 2003, 22, 5093–5107. [Google Scholar] [CrossRef] [Green Version]
- Benali-Furet, N.L.; Chami, M.; Houel, L.; De Giorgi, F.; Vernejoul, F.; Lagorce, D.; Buscail, L.; Bartenschlager, R.; Ichas, F.; Rizzuto, R.; et al. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 2005, 24, 4921–4933. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.W.; Egan, P.A. Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J. 2005, 19, 1510–1512. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Ye, L.; Yu, X.; Xu, B.; Li, K.; Zhu, X.; Liu, H.; Wu, X.; Kong, L. Hepatitis C virus NS4B induces unfolded protein response and endoplasmic reticulum overload response-dependent NF-kappaB activation. Virology 2009, 391, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Gao, B.; Ye, L.; Kong, L.; Jing, W.; Yang, X.; Wu, Z.; Ye, L. Hepatitis C virus non-structural protein NS4B can modulate an unfolded protein response. J. Microbiol. 2005, 43, 529–536. [Google Scholar]
- Wang, T.; Campbell, R.V.; Yi, M.K.; Lemon, S.M.; Weinman, S.A. Role of Hepatitis C virus core protein in viral-induced mitochondrial dysfunction. J. Viral Hepat. 2010, 17, 784–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.M.; Kim, S.J.; Kim, J.H.; Lee, W.; Kim, G.W.; Lee, K.H.; Choi, K.Y.; Oh, J.W. Interaction of hepatitis C virus core protein with Hsp60 triggers the production of reactive oxygen species and enhances TNF-alpha-mediated apoptosis. Cancer Lett. 2009, 279, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Anelli, T.; Bergamelli, L.; Margittai, E.; Rimessi, A.; Fagioli, C.; Malgaroli, A.; Pinton, P.; Ripamonti, M.; Rizzuto, R.; Sitia, R. Ero1alpha regulates Ca(2+) fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxid. Redox Signal. 2012, 16, 1077–1087. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Smirnova, O.A.; Petrushanko, I.Y.; Ivanova, O.N.; Karpenko, I.L.; Alekseeva, E.; Sominskaya, I.; Makarov, A.A.; Bartosch, B.; Kochetkov, S.N.; et al. HCV core protein uses multiple mechanisms to induce oxidative stress in human hepatoma Huh7 cells. Viruses 2015, 7, 2745–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.I.; Hwang, J.M.; Im, J.H.; Lee, Y.I.; Kim, N.S.; Kim, D.G.; Yu, D.Y.; Moon, H.B.; Park, S.K. Human hepatitis B virus-X protein alters mitochondrial function and physiology in human liver cells. J. Biol. Chem. 2004, 279, 15460–15471. [Google Scholar] [CrossRef] [Green Version]
- Rahmani, Z.; Huh, K.W.; Lasher, R.; Siddiqui, A. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J. Virol. 2000, 74, 2840–2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, L.Y.; Zheng, B.Y.; Fang, X.F.; Li, D.; Huang, Y.H.; Chen, Z.X.; Zhou, L.Y.; Wang, X.Z. HBx co-localizes with COXIII in HL-7702 cells to upregulate mitochondrial function and ROS generation. Oncol. Rep. 2015, 33, 2461–2467. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Ding, J.; Chen, Z.; Chen, Y.; Lin, N.; Chen, F.; Wang, X. Accurately mapping the location of the binding site for the interaction between hepatitis B virus X protein and cytochrome c oxidase III. Int. J. Mol. Med. 2015, 35, 319–324. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Kim, H.; Lee, S.A.; Won, Y.S.; Kim, H.I.; Inn, K.S.; Kim, B.J. Upregulation of endoplasmic reticulum stress and reactive oxygen species by naturally occurring mutations in hepatitis B virus core antigen. J. Gen. Virol. 2015, 96, 1850–1854. [Google Scholar] [CrossRef]
- Lee, I.K.; Lee, S.A.; Kim, H.; Won, Y.S.; Kim, B.J. Induction of endoplasmic reticulum-derived oxidative stress by an occult infection related S surface antigen variant. World J. Gastroenterol. 2015, 21, 6872–6883. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, O.A.; Ivanova, O.N.; Bartosch, B.; Valuev-Elliston, V.T.; Mukhtarov, F.; Kochetkov, S.N.; Ivanov, A.V. Hepatitis C Virus NS5A Protein Triggers Oxidative Stress by Inducing NADPH Oxidases 1 and 4 and Cytochrome P450 2E1. Oxid. Med. Cell. Longev. 2016, 2016, 8341937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, V.; Brichler, S.; Khan, E.; Chami, M.; Deny, P.; Kremsdorf, D.; Gordien, E. Large hepatitis delta antigen activates STAT-3 and NF-kappaB via oxidative stress. J. Viral Hepat. 2012, 19, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.Y.; Ahmad, I.M.; Spitz, D.R.; Schmidt, W.N.; Britigan, B.E. Hepatitis C virus-core and non structural proteins lead to different effects on cellular antioxidant defenses. J. Med. Virol. 2005, 76, 489–497. [Google Scholar] [CrossRef]
- Carvajal-Yepes, M.; Himmelsbach, K.; Schaedler, S.; Ploen, D.; Krause, J.; Ludwig, L.; Weiss, T.; Klingel, K.; Hildt, E. Hepatitis C virus impairs the induction of cytoprotective Nrf2 target genes by delocalization of small Maf proteins. J. Biol. Chem. 2011, 286, 8941–8951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, A.V.; Smirnova, O.A.; Ivanova, O.N.; Masalova, O.V.; Kochetkov, S.N.; Isaguliants, M.G. Hepatitis C virus proteins activate NRF2/ARE pathway by distinct ROS-dependent and independent mechanisms in HUH7 cells. PLoS ONE 2011, 6, e24957. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Bao, H.; Ge, Y.; Tang, W.; Cheng, D.; Luo, K.; Gong, G.; Gong, R. Therapeutic targeting of GSK3beta enhances the Nrf2 antioxidant response and confers hepatic cytoprotection in hepatitis C. Gut 2015, 64, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, O.A.; Ivanova, O.N.; Mukhtarov, F.S.; Tunitskaya, V.L.; Jansons, J.; Isaguliants, M.G.; Kochetkov, S.N.; Ivanov, A.V. Analysis of the Domains of Hepatitis C Virus Core and NS5A Proteins that Activate the Nrf2/ARE Cascade. Acta Nat. 2016, 8, 123–127. [Google Scholar] [CrossRef]
- Tang, W.; Lazaro, C.A.; Campbell, J.S.; Parks, W.T.; Katze, M.G.; Fausto, N. Responses of nontransformed human hepatocytes to conditional expression of full-length hepatitis C virus open reading frame. Am. J. Pathol. 2007, 171, 1831–1846. [Google Scholar] [CrossRef] [Green Version]
- Blackham, S.; Baillie, A.; Al-Hababi, F.; Remlinger, K.; You, S.; Hamatake, R.; McGarvey, M.J. Gene expression profiling indicates the roles of host oxidative stress, apoptosis, lipid metabolism, and intracellular transport genes in the replication of hepatitis C virus. J. Virol. 2010, 84, 5404–5414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, K.A.; Syder, A.J.; Lederer, S.L.; Diamond, D.L.; Paeper, B.; Rice, C.M.; Katze, M.G. Genomic analysis reveals a potential role for cell cycle perturbation in HCV-mediated apoptosis of cultured hepatocytes. PLoS Pathog. 2009, 5, e1000269. [Google Scholar] [CrossRef] [PubMed]
- Schaedler, S.; Krause, J.; Himmelsbach, K.; Carvajal-Yepes, M.; Lieder, F.; Klingel, K.; Nassal, M.; Weiss, T.S.; Werner, S.; Hildt, E. Hepatitis B virus induces expression of antioxidant response element-regulated genes by activation of Nrf2. J. Biol. Chem. 2010, 285, 41074–41086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Fang, M.; He, Z.; Cui, D.; Jia, S.; Lin, X.; Xu, X.; Zhou, T.; Liu, W. Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis. 2015, 6, e1980. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, Y.; Lin, Z.; Han, M.; Cheng, H. Altered serum copper homeostasis suggests higher oxidative stress and lower antioxidant capability in patients with chronic hepatitis B. Medicine 2018, 97, e11137. [Google Scholar] [CrossRef]
- Nakashima, T.; Sumida, Y.; Yoh, T.; Kakisaka, Y.; Nakajima, Y.; Ishikawa, H.; Mitsuyoshi, H.; Kashima, K.; Nakamura, H.; Yodoi, J. Thioredoxin levels in the sera of untreated viral hepatitis patients and those treated with glycyrrhizin or ursodeoxycholic acid. Antioxid. Redox Signal. 2000, 2, 687–694. [Google Scholar] [CrossRef]
- Wang, Q.; Na, B.; Ou, J.H.; Pulliam, L.; Yen, T.S. Hepatitis B virus alters the antioxidant system in transgenic mice and sensitizes hepatocytes to Fas signaling. PLoS ONE 2012, 7, e36818. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Zou, Z.Q.; Wang, L.Y.; Gao, S.; Fan, Y.C.; Long, B.; Guo, Y.M.; Xu, A.L.; Han, J.; Li, T.; et al. Methylation of the glutathione-S-transferase M3 gene promoter is associated with oxidative stress in acute-on-chronic hepatitis B liver failure. Tohoku J. Exp. Med. 2012, 228, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Niu, D.; Zhang, J.; Ren, Y.; Feng, H.; Chen, W.N. HBx genotype D represses GSTP1 expression and increases the oxidative level and apoptosis in HepG2 cells. Mol. Oncol. 2009, 3, 67–76. [Google Scholar] [CrossRef]
- Cho, I.J.; Ki, S.H.; Brooks, C., 3rd; Kim, S.G. Role of hepatitis B virus X repression of C/EBPbeta activity in the down-regulation of glutathione S-transferase A2 gene: Implications in other phase II detoxifying enzyme expression. Xenobiotica 2009, 39, 182–192. [Google Scholar] [CrossRef]
- Ding, C.; Wei, H.; Sun, R.; Zhang, J.; Tian, Z. Hepatocytes proteomic alteration and seroproteome analysis of HBV-transgenic mice. Proteomics 2009, 9, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.S.; Park, S.G.; Byeon, S.M.; Kwon, Y.G.; Jung, G. Hepatitis B virus X protein induces TNF-alpha expression via down-regulation of selenoprotein P in human hepatoma cell line, HepG2. Biochim. Biophys. Acta 2003, 1638, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Du, D.; Zheng, W.; Liao, M.; Zhang, L.; Liang, G.; Gong, M. Small hepatitis delta antigen selectively binds to target mRNA in hepatic cells: A potential mechanism by which hepatitis D virus downregulates glutathione S-transferase P1 and induces liver injury and hepatocarcinogenesis. Biochem. Cell Biol. 2019, 97, 130–139. [Google Scholar] [CrossRef]
- Jaeschke, H. Reactive oxygen and mechanisms of inflammatory liver injury. J. Gastroenterol. Hepatol. 2000, 15, 718–724. [Google Scholar] [CrossRef]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc. Natl. Acad. Sci. USA 2001, 98, 9599–9604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Tsai, W.L.; Shao, R.X.; Wu, G.; Peng, L.F.; Barlow, L.L.; Chung, W.J.; Zhang, L.; Zhao, H.; Jang, J.Y.; et al. Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology 2010, 138, 2509–2518.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bataller, R.; Lemon, S.M. Fueling fibrosis in chronic hepatitis C. Proc. Natl. Acad. Sci. USA 2012, 109, 14293–14294. [Google Scholar] [CrossRef] [Green Version]
- Presser, L.D.; Haskett, A.; Waris, G. Hepatitis C virus-induced furin and thrombospondin-1 activate TGF-beta1: Role of TGF-beta1 in HCV replication. Virology 2011, 412, 284–296. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Tovar, E.; Muriel, P. Free radicals, antioxidants, nuclear factor-E2-related factor-2 and liver damage. J. Appl. Toxicol. 2020, 40, 151–168. [Google Scholar] [CrossRef]
- Higgs, M.R.; Lerat, H.; Pawlotsky, J.M. Hepatitis C virus-induced activation of beta-catenin promotes c-Myc expression and a cascade of pro-carcinogenetic events. Oncogene 2013, 32, 4683–4693. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, T.; Kohara, M.; Izumi, K.; Kasama, Y.; Hirata, Y.; Huang, Y.; Shuda, M.; Mukaidani, C.; Takano, T.; Tokunaga, Y.; et al. Hepatitis C virus impairs p53 via persistent overexpression of 3beta-hydroxysterol Delta24-reductase. J. Biol. Chem. 2009, 284, 36442–36452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mankouri, J.; Dallas, M.L.; Hughes, M.E.; Griffin, S.D.; Macdonald, A.; Peers, C.; Harris, M. Suppression of a pro-apoptotic K+ channel as a mechanism for hepatitis C virus persistence. Proc. Natl. Acad. Sci. USA 2009, 106, 15903–15908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Moriya, K.; Kiyosawa, K.; Koike, K.; Gonzalez, F.J.; Aoyama, T. PPARalpha activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J. Clin. Investig. 2008, 118, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.L.; Heo, S.; Jang, K.L. Hepatitis C virus core protein overcomes H2O2-induced apoptosis by downregulating p14 expression via DNA methylation. J. Gen. Virol. 2015, 96, 822–832. [Google Scholar] [CrossRef]
- O’Reilly, M.A. Redox activation of p21Cip1/WAF1/Sdi1: A multifunctional regulator of cell survival and death. Antioxid. Redox Signal. 2005, 7, 108–118. [Google Scholar] [CrossRef]
- Inoue, T.; Kato, K.; Kato, H.; Asanoma, K.; Kuboyama, A.; Ueoka, Y.; Yamaguchi, S.; Ohgami, T.; Wake, N. Level of reactive oxygen species induced by p21Waf1/CIP1 is critical for the determination of cell fate. Cancer Sci. 2009, 100, 1275–1283. [Google Scholar] [CrossRef]
- Villeneuve, N.F.; Sun, Z.; Chen, W.; Zhang, D.D. Nrf2 and p21 regulate the fine balance between life and death by controlling ROS levels. Cell Cycle 2009, 8, 3255–3256. [Google Scholar] [CrossRef] [Green Version]
- Majumder, M.; Ghosh, A.K.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J. Virol. 2001, 75, 1401–1407. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Yoshida, I.; Takamatsu, M.; Ishido, S.; Fujita, T.; Oka, K.; Hotta, H. Complex formation between hepatitis C virus core protein and p21Waf1/Cip1/Sdi1. Biochem. Biophys. Res. Commun. 2000, 273, 479–484. [Google Scholar] [CrossRef]
- Elpek, G.O. Molecular pathways in viral hepatitis-associated liver carcinogenesis: An update. World J. Clin. Cases 2021, 9, 4890–4917. [Google Scholar] [CrossRef]
- Park, C.Y.; Oh, S.H.; Kang, S.M.; Lim, Y.S.; Hwang, S.B. Hepatitis delta virus large antigen sensitizes to TNF-alpha-induced NF-kappaB signaling. Mol. Cells 2009, 28, 49–55. [Google Scholar] [CrossRef]
- Williams, V.; Brichler, S.; Radjef, N.; Lebon, P.; Goffard, A.; Hober, D.; Fagard, R.; Kremsdorf, D.; Deny, P.; Gordien, E. Hepatitis delta virus proteins repress hepatitis B virus enhancers and activate the alpha/beta interferon-inducible MxA gene. J. Gen. Virol. 2009, 90, 2759–2767. [Google Scholar] [CrossRef]
- Zampino, R.; Marrone, A.; Restivo, L.; Guerrera, B.; Sellitto, A.; Rinaldi, L.; Romano, C.; Adinolfi, L.E. Chronic HCV infection and inflammation: Clinical impact on hepatic and extra-hepatic manifestations. World J. Hepatol. 2013, 5, 528–540. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Brezgin, S.; Kostyusheva, A.; Bayurova, E.; Volchkova, E.; Gegechkori, V.; Gordeychuk, I.; Glebe, D.; Kostyushev, D.; Chulanov, V. Immunity and Viral Infections: Modulating Antiviral Response via CRISPR-Cas Systems. Viruses 2021, 13, 1373. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.H.; Thimme, R. Innate and adaptive immune responses in HCV infections. J. Hepatol. 2014, 61, S14–S25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.; Altstetter, S.M.; Protzer, U. Innate immune recognition and modulation in hepatitis D virus infection. World J. Gastroenterol. 2020, 26, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Pugnale, P.; Pazienza, V.; Guilloux, K.; Negro, F. Hepatitis delta virus inhibits alpha interferon signaling. Hepatology 2009, 49, 398–406. [Google Scholar] [CrossRef]
- Lupberger, J.; Duong, F.H.; Fofana, I.; Zona, L.; Xiao, F.; Thumann, C.; Durand, S.C.; Pessaux, P.; Zeisel, M.B.; Heim, M.H.; et al. Epidermal growth factor receptor signaling impairs the antiviral activity of interferon-alpha. Hepatology 2013, 58, 1225–1235. [Google Scholar] [CrossRef]
- Hosel, M.; Quasdorff, M.; Ringelhan, M.; Kashkar, H.; Debey-Pascher, S.; Sprinzl, M.F.; Bockmann, J.H.; Arzberger, S.; Webb, D.; von Olshausen, G.; et al. Hepatitis B Virus Activates Signal Transducer and Activator of Transcription 3 Supporting Hepatocyte Survival and Virus Replication. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 339–363. [Google Scholar] [CrossRef]
- Yang, Y.; Zheng, B.; Han, Q.; Zhang, C.; Tian, Z.; Zhang, J. Targeting blockage of STAT3 inhibits hepatitis B virus-related hepatocellular carcinoma. Cancer Biol. Ther. 2016, 17, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Saso, W.; Sugiyama, R.; Ishii, K.; Ohki, M.; Nagamori, S.; Suzuki, R.; Aizaki, H.; Ryo, A.; Yun, J.H.; et al. Epidermal growth factor receptor is a host-entry cofactor triggering hepatitis B virus internalization. Proc. Natl. Acad. Sci. USA 2019, 116, 8487–8492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zona, L.; Lupberger, J.; Sidahmed-Adrar, N.; Thumann, C.; Harris, H.J.; Barnes, A.; Florentin, J.; Tawar, R.G.; Xiao, F.; Turek, M.; et al. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe 2013, 13, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Van Renne, N.; Roca Suarez, A.A.; Duong, F.H.T.; Gondeau, C.; Calabrese, D.; Fontaine, N.; Ababsa, A.; Bandiera, S.; Croonenborghs, T.; Pochet, N.; et al. miR-135a-5p-mediated downregulation of protein tyrosine phosphatase receptor delta is a candidate driver of HCV-associated hepatocarcinogenesis. Gut 2018, 67, 953–962. [Google Scholar] [CrossRef]
- Mori, R.; Wauman, J.; Icardi, L.; Van der Heyden, J.; De Cauwer, L.; Peelman, F.; De Bosscher, K.; Tavernier, J. TYK2-induced phosphorylation of Y640 suppresses STAT3 transcriptional activity. Sci. Rep. 2017, 7, 15919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.C.; Cheng, P.N.; Kao, J.H. Systematic review: Chronic viral hepatitis and metabolic derangement. Aliment. Pharmacol. Ther. 2020, 51, 216–230. [Google Scholar] [CrossRef]
- Hui, R.W.H.; Seto, W.K.; Cheung, K.S.; Mak, L.Y.; Liu, K.S.H.; Fung, J.; Wong, D.K.; Lai, C.L.; Yuen, M.F. Inverse relationship between hepatic steatosis and hepatitis B viremia: Results of a large case-control study. J. Viral Hepat. 2018, 25, 97–104. [Google Scholar] [CrossRef]
- Joo, E.J.; Chang, Y.; Yeom, J.S.; Ryu, S. Hepatitis B virus infection and decreased risk of nonalcoholic fatty liver disease: A cohort study. Hepatology 2017, 65, 828–835. [Google Scholar] [CrossRef] [Green Version]
- Seto, W.K. Chronic hepatitis B and metabolic risk factors: A call for rigorous longitudinal studies. World J. Gastroenterol. 2019, 25, 282–286. [Google Scholar] [CrossRef]
- Patel, A.; Harrison, S.A. Hepatitis C virus infection and nonalcoholic steatohepatitis. Gastroenterol. Hepatol. 2012, 8, 305–312. [Google Scholar]
- Wrensch, F.; Crouchet, E.; Ligat, G.; Zeisel, M.B.; Keck, Z.Y.; Foung, S.K.H.; Schuster, C.; Baumert, T.F. Hepatitis C Virus (HCV)-Apolipoprotein Interactions and Immune Evasion and Their Impact on HCV Vaccine Design. Front. Immunol. 2018, 9, 1436. [Google Scholar] [CrossRef]
- Benga, W.J.; Krieger, S.E.; Dimitrova, M.; Zeisel, M.B.; Parnot, M.; Lupberger, J.; Hildt, E.; Luo, G.; McLauchlan, J.; Baumert, T.F.; et al. Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology 2010, 51, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.S.; Jiang, J.; Cai, Z.; Luo, G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 2007, 81, 13783–13793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herker, E.; Harris, C.; Hernandez, C.; Carpentier, A.; Kaehlcke, K.; Rosenberg, A.R.; Farese, R.V., Jr.; Ott, M. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 2010, 16, 1295–1298. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhang, Y.Y.; Chiu, S.; Hu, Z.; Lan, K.H.; Cha, H.; Sodroski, C.; Zhang, F.; Hsu, C.S.; Thomas, E.; et al. Integrative functional genomics of hepatitis C virus infection identifies host dependencies in complete viral replication cycle. PLoS Pathog. 2014, 10, e1004163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, P.L.; Duponchel, S.; Eischeid, H.; Molle, J.; Michelet, M.; Diserens, G.; Vermathen, M.; Vermathen, P.; Dufour, J.F.; Dienes, H.P.; et al. Hepatitis C virus infection triggers a tumor-like glutamine metabolism. Hepatology 2017, 65, 789–803. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Karin, M. NF-kappaB and STAT3—Key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Chang, Z.; Wang, Y.; Zhou, X.; Long, J.E. STAT3 roles in viral infection: Antiviral or proviral? Future Virol. 2018, 13, 557–574. [Google Scholar] [CrossRef]
- Hiscott, J.; Kwon, H.; Genin, P. Hostile takeovers: Viral appropriation of the NF-kappaB pathway. J. Clin. Investig. 2001, 107, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Huh, K.W.; Siddiqui, A. Mitochondrially associated hepatitis B virus X protein constitutively activates transcription factors STAT-3 and NF-kappa B via oxidative stress. Mol. Cell. Biol. 2001, 21, 7721–7730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Hanada, T.; Tokuhisa, T.; Kosai, K.; Sata, M.; Kohara, M.; Yoshimura, A. Activation of STAT3 by the hepatitis C virus core protein leads to cellular transformation. J. Exp. Med. 2002, 196, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Ma, Y.; Liu, M.; Yan, L.; Tang, H. Peroxisome Proliferators Activated Receptor (PPAR) agonists activate hepatitis B virus replication in vivo. Virol J. 2017, 14, 96. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Wang, X.; Ding, X.; Li, Y.; Zhang, X.; Xie, P.; Yang, J.; Wang, S. MicroRNA-141 represses HBV replication by targeting PPARA. PLoS ONE 2012, 7, e34165. [Google Scholar] [CrossRef] [Green Version]
- Dubuquoy, L.; Louvet, A.; Hollebecque, A.; Mathurin, P.; Dharancy, S. Peroxisome proliferator-activated receptors in HBV-related infection. PPAR Res. 2009, 2009, 145124. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.D.; Wu, H.; Huang, S.; Zhang, H.L.; Qin, C.J.; Zhao, L.H.; Fu, G.B.; Zhou, X.; Wang, X.M.; Tang, L.; et al. HBx regulates fatty acid oxidation to promote hepatocellular carcinoma survival during metabolic stress. Oncotarget 2016, 7, 6711–6726. [Google Scholar] [CrossRef]
- Wu, Y.L.; Peng, X.E.; Zhu, Y.B.; Yan, X.L.; Chen, W.N.; Lin, X. Hepatitis B Virus X Protein Induces Hepatic Steatosis by Enhancing the Expression of Liver Fatty Acid Binding Protein. J. Virol. 2016, 90, 1729–1740. [Google Scholar] [CrossRef] [Green Version]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Haga, Y.; Kanda, T.; Sasaki, R.; Nakamura, M.; Nakamoto, S.; Yokosuka, O. Nonalcoholic fatty liver disease and hepatic cirrhosis: Comparison with viral hepatitis-associated steatosis. World J. Gastroenterol. 2015, 21, 12989–12995. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Yan, S.; He, Y.; Wang, F.; Song, S.; Guo, Y.; Zhou, Q.; Wang, Y.; Lin, Z.; Yang, Y.; et al. Expression of hepatitis B virus proteins in transgenic mice alters lipid metabolism and induces oxidative stress in the liver. J. Hepatol. 2008, 48, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Hajjou, M.; Norel, R.; Carver, R.; Marion, P.; Cullen, J.; Rogler, L.E.; Rogler, C.E. cDNA microarray analysis of HBV transgenic mouse liver identifies genes in lipid biosynthetic and growth control pathways affected by HBV. J. Med. Virol. 2005, 77, 57–65. [Google Scholar] [CrossRef]
- Hertzel, A.V.; Bernlohr, D.A. The mammalian fatty acid-binding protein multigene family: Molecular and genetic insights into function. Trends Endocrinol. Metab. 2000, 11, 175–180. [Google Scholar] [CrossRef]
- Liu, G.; Liu, G.; Cui, X.; Xu, Y. Transcriptomic Data Analyses Reveal a Reprogramed Lipid Metabolism in HCV-Derived Hepatocellular Cancer. Front. Cell Dev. Biol. 2020, 8, 581863. [Google Scholar] [CrossRef]
- Shieh, Y.S.; Chang, Y.S.; Hong, J.R.; Chen, L.J.; Jou, L.K.; Hsu, C.C.; Her, G.M. Increase of hepatic fat accumulation by liver specific expression of Hepatitis B virus X protein in zebrafish. Biochim. Biophys. Acta 2010, 1801, 721–730. [Google Scholar] [CrossRef]
- Na, T.Y.; Shin, Y.K.; Roh, K.J.; Kang, S.A.; Hong, I.; Oh, S.J.; Seong, J.K.; Park, C.K.; Choi, Y.L.; Lee, M.O. Liver X receptor mediates hepatitis B virus X protein-induced lipogenesis in hepatitis B virus-associated hepatocellular carcinoma. Hepatology 2009, 49, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kim, K.H.; Kim, H.H.; Cheong, J. Hepatitis B virus X protein induces lipogenic transcription factor SREBP1 and fatty acid synthase through the activation of nuclear receptor LXRalpha. Biochem. J. 2008, 416, 219–230. [Google Scholar] [CrossRef]
- Kim, K.H.; Shin, H.J.; Kim, K.; Choi, H.M.; Rhee, S.H.; Moon, H.B.; Kim, H.H.; Yang, U.S.; Yu, D.Y.; Cheong, J. Hepatitis B virus X protein induces hepatic steatosis via transcriptional activation of SREBP1 and PPARgamma. Gastroenterology 2007, 132, 1955–1967. [Google Scholar] [CrossRef]
- Mota, S.; Mendes, M.; Freitas, N.; Penque, D.; Coelho, A.V.; Cunha, C. Proteome analysis of a human liver carcinoma cell line stably expressing hepatitis delta virus ribonucleoproteins. J. Proteomics 2009, 72, 616–627. [Google Scholar] [CrossRef]
- Petta, S.; Camma, C.; Di Marco, V.; Macaluso, F.S.; Maida, M.; Pizzolanti, G.; Belmonte, B.; Cabibi, D.; Di Stefano, R.; Ferraro, D.; et al. Hepatic steatosis and insulin resistance are associated with severe fibrosis in patients with chronic hepatitis caused by HBV or HCV infection. Liver Int. 2011, 31, 507–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verrier, E.R.; Weiss, A.; Bach, C.; Heydmann, L.; Turon-Lagot, V.; Kopp, A.; El Saghire, H.; Crouchet, E.; Pessaux, P.; Garcia, T.; et al. Combined small molecule and loss-of-function screen uncovers estrogen receptor alpha and CAD as host factors for HDV infection and antiviral targets. Gut 2020, 69, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Hatting, M.; Tavares, C.D.J.; Sharabi, K.; Rines, A.K.; Puigserver, P. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 2018, 1411, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, Y.; Kemper, T.; Chen, J.; Yuan, Z.; Liu, S.; Zhu, Y.; Broering, R.; Lu, M. AMPK and Akt/mTOR signalling pathways participate in glucose-mediated regulation of hepatitis B virus replication and cellular autophagy. Cell. Microbiol. 2020, 22, e13131. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Saito, K.; Ait-Goughoulte, M.; Meyer, K.; Ray, R.B.; Ray, R. Hepatitis C virus core protein upregulates serine phosphorylation of insulin receptor substrate-1 and impairs the downstream akt/protein kinase B signaling pathway for insulin resistance. J. Virol. 2008, 82, 2606–2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.T.; Qin, Z.L.; Ren, H.; Zhao, P.; Qi, Z.T. Inhibition of IRS-1 by hepatitis C virus infection leads to insulin resistance in a PTEN-dependent manner. Virol. J. 2015, 12, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvaiz, F.; Manzoor, S.; Iqbal, J.; Sarkar-Dutta, M.; Imran, M.; Waris, G. Hepatitis C virus NS5A promotes insulin resistance through IRS-1 serine phosphorylation and increased gluconeogenesis. World J. Gastroenterol. 2015, 21, 12361–12369. [Google Scholar] [CrossRef]
- Arai, T.; Kano, F.; Murata, M. Translocation of forkhead box O1 to the nuclear periphery induces histone modifications that regulate transcriptional repression of PCK1 in HepG2 cells. Genes Cells 2015, 20, 340–357. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.K.; Shrivastava, S.; Meyer, K.; Ray, R.B.; Ray, R. Hepatitis C virus activates the mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. J. Virol. 2012, 86, 6315–6322. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Shoji, I.; Ogawa, W.; Kaneda, S.; Soga, T.; Jiang, D.P.; Ide, Y.H.; Hotta, H. Hepatitis C virus infection promotes hepatic gluconeogenesis through an NS5A-mediated, FoxO1-dependent pathway. J. Virol. 2011, 85, 8556–8568. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.J.; Park, Y.H.; Kim, S.U.; Moon, H.B.; Park, D.S.; Han, Y.H.; Lee, C.H.; Lee, D.S.; Song, I.S.; Lee, D.H.; et al. Hepatitis B virus X protein regulates hepatic glucose homeostasis via activation of inducible nitric oxide synthase. J. Biol. Chem. 2011, 286, 29872–29881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhu, W.; Zhang, L.; Lei, H.; Wu, X.; Guo, L.; Chen, X.; Wang, Y.; Tang, H. The metabolic responses to hepatitis B virus infection shed new light on pathogenesis and targets for treatment. Sci. Rep. 2015, 5, 8421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalopoulos, G.K.; Bhushan, B. Liver regeneration: Biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Tomas, A.; Futter, C.E.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Mailly, L.; Xiao, F.; Lupberger, J.; Wilson, G.K.; Aubert, P.; Duong, F.H.T.; Calabrese, D.; Leboeuf, C.; Fofana, I.; Thumann, C.; et al. Clearance of persistent hepatitis C virus infection in humanized mice using a claudin-1-targeting monoclonal antibody. Nat. Biotechnol. 2015, 33, 549–554. [Google Scholar] [CrossRef] [Green Version]
- Diao, J.; Pantua, H.; Ngu, H.; Komuves, L.; Diehl, L.; Schaefer, G.; Kapadia, S.B. Hepatitis C virus induces epidermal growth factor receptor activation via CD81 binding for viral internalization and entry. J. Virol. 2012, 86, 10935–10949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mankouri, J.; Griffin, S.; Harris, M. The hepatitis C virus non-structural protein NS5A alters the trafficking profile of the epidermal growth factor receptor. Traffic 2008, 9, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Igloi, Z.; Kazlauskas, A.; Saksela, K.; Macdonald, A.; Mankouri, J.; Harris, M. Hepatitis C virus NS5A protein blocks epidermal growth factor receptor degradation via a proline motif- dependent interaction. J. Gen. Virol. 2015, 96, 2133–2144. [Google Scholar] [CrossRef]
- Stindt, S.; Cebula, P.; Albrecht, U.; Keitel, V.; Schulte am Esch, J.; Knoefel, W.T.; Bartenschlager, R.; Haussinger, D.; Bode, J.G. Hepatitis C Virus Activates a Neuregulin-Driven Circuit to Modify Surface Expression of Growth Factor Receptors of the ErbB Family. PLoS ONE 2016, 11, e0148711. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, M.; Saso, W.; Nishioka, K.; Ohashi, H.; Sugiyama, R.; Ryo, A.; Ohki, M.; Yun, J.H.; Park, S.Y.; Ohshima, T.; et al. The machinery for endocytosis of epidermal growth factor receptor coordinates the transport of incoming hepatitis B virus to the endosomal network. J. Biol. Chem. 2020, 295, 800–807. [Google Scholar] [CrossRef]
- Burckstummer, T.; Kriegs, M.; Lupberger, J.; Pauli, E.K.; Schmittel, S.; Hildt, E. Raf-1 kinase associates with Hepatitis C virus NS5A and regulates viral replication. FEBS Lett. 2006, 580, 575–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, D.Y.; Sun, Y.; Cheng, R.X.; Ouyang, X.M.; Zheng, H. Effect of hepatitis C virus nonstructural protein NS3 on proliferation and MAPK phosphorylation of normal hepatocyte line. World J. Gastroenterol. 2005, 11, 2157–2161. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, J.; Aoki, H.; Kajino, K.; Moriyama, M.; Arakawa, Y.; Hino, O. Hepatitis C virus core protein activates the MAPK/ERK cascade synergistically with tumor promoter TPA, but not with epidermal growth factor or transforming growth factor alpha. Hepatology 2000, 32, 958–961. [Google Scholar] [CrossRef]
- Zhao, L.J.; Wang, L.; Ren, H.; Cao, J.; Li, L.; Ke, J.S.; Qi, Z.T. Hepatitis C virus E2 protein promotes human hepatoma cell proliferation through the MAPK/ERK signaling pathway via cellular receptors. Exp. Cell Res. 2005, 305, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Chen, C.Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. 2018, 9, 338. [Google Scholar] [CrossRef] [Green Version]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.; Zhang, L.; Yang, G.; Zhao, L.; Peng, F.; Tian, Y.; Xiao, X.; Chung, R.T.; Gong, G. Hepatitis C virus NS5A drives a PTEN-PI3K/Akt feedback loop to support cell survival. Liver Int. 2015, 35, 1682–1691. [Google Scholar] [CrossRef]
- Rawat, S.; Bouchard, M.J. The hepatitis B virus (HBV) HBx protein activates AKT to simultaneously regulate HBV replication and hepatocyte survival. J. Virol. 2015, 89, 999–1012. [Google Scholar] [CrossRef] [Green Version]
- Xiang, K.; Wang, B. Role of the PI3KAKTmTOR pathway in hepatitis B virus infection and replication. Mol. Med. Rep. 2018, 17, 4713–4719. [Google Scholar] [CrossRef]
- Sun, Y.; Yu, M.; Qu, M.; Ma, Y.; Zheng, D.; Yue, Y.; Guo, S.; Tang, L.; Li, G.; Zheng, W.; et al. Hepatitis B virus-triggered PTEN/beta-catenin/c-Myc signaling enhances PD-L1 expression to promote immune evasion. Am. J. Physiol. Gastrointest Liver Physiol. 2020, 318, G162–G173. [Google Scholar] [CrossRef] [PubMed]
- Perugorria, M.J.; Olaizola, P.; Labiano, I.; Esparza-Baquer, A.; Marzioni, M.; Marin, J.J.G.; Bujanda, L.; Banales, J.M. Wnt-beta-catenin signalling in liver development, health and disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Pan, Q.; Fuhler, G.M.; Smits, R.; Peppelenbosch, M.P. Action and function of Wnt/beta-catenin signaling in the progression from chronic hepatitis C to hepatocellular carcinoma. J. Gastroenterol. 2017, 52, 419–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Ding, X.; Tang, J.; Cao, Y.; Hu, P.; Zhou, F.; Shan, X.; Cai, X.; Chen, Q.; Ling, N.; et al. Enhancement of canonical Wnt/beta-catenin signaling activity by HCV core protein promotes cell growth of hepatocellular carcinoma cells. PLoS ONE 2011, 6, e27496. [Google Scholar] [CrossRef]
- Street, A.; Macdonald, A.; McCormick, C.; Harris, M. Hepatitis C virus NS5A-mediated activation of phosphoinositide 3-kinase results in stabilization of cellular beta-catenin and stimulation of beta-catenin-responsive transcription. J. Virol. 2005, 79, 5006–5016. [Google Scholar] [CrossRef] [Green Version]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef] [Green Version]
- Chusri, P.; Kumthip, K.; Hong, J.; Zhu, C.; Duan, X.; Jilg, N.; Fusco, D.N.; Brisac, C.; Schaefer, E.A.; Cai, D.; et al. HCV induces transforming growth factor beta1 through activation of endoplasmic reticulum stress and the unfolded protein response. Sci. Rep. 2016, 6, 22487. [Google Scholar] [CrossRef]
- Zou, L.L.; Li, J.R.; Li, H.; Tan, J.L.; Wang, M.X.; Liu, N.N.; Gao, R.M.; Yan, H.Y.; Wang, X.K.; Dong, B.; et al. TGF-beta isoforms inhibit hepatitis C virus propagation in transforming growth factor beta/SMAD protein signalling pathway dependent and independent manners. J. Cell. Mol. Med. 2021, 25, 3498–3510. [Google Scholar] [CrossRef]
- Choi, S.H.; Hwang, S.B. Modulation of the transforming growth factor-beta signal transduction pathway by hepatitis C virus nonstructural 5A protein. J. Biol. Chem. 2006, 281, 7468–7478. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Liu, G.; Kitamura, K.; Wang, Z.; Chowdhury, S.; Monjurul, A.M.; Wakae, K.; Koura, M.; Shimadu, M.; Kinoshita, K.; et al. TGF-beta suppression of HBV RNA through AID-dependent recruitment of an RNA exosome complex. PLoS Pathog. 2015, 11, e1004780. [Google Scholar] [CrossRef]
- Choi, S.H.; Jeong, S.H.; Hwang, S.B. Large hepatitis delta antigen modulates transforming growth factor-beta signaling cascades: Implication of hepatitis delta virus-induced liver fibrosis. Gastroenterology 2007, 132, 343–357. [Google Scholar] [CrossRef]
- Liang, Y.J.; Sun, C.P.; Hsu, Y.C.; Chen, Y.W.; Wang, I.A.; Su, C.W.; Tao, M.H.; Wu, J.C. Statin inhibits large hepatitis delta antigen-Smad3 -twist-mediated epithelial-to-mesenchymal transition and hepatitis D virus secretion. J. Biomed. Sci. 2020, 27, 65. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, H.; Manns, M.P. Epidemiology, pathogenesis and management of hepatitis D: Update and challenges ahead. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 31–40. [Google Scholar] [CrossRef]
- Klohn, M.; Schrader, J.A.; Bruggemann, Y.; Todt, D.; Steinmann, E. Beyond the Usual Suspects: Hepatitis E Virus and Its Implications in Hepatocellular Carcinoma. Cancers 2021, 13, 5867. [Google Scholar] [CrossRef] [PubMed]
- Khuroo, M.S.; Khuroo, M.S.; Khuroo, N.S. Hepatitis E: Discovery, global impact, control and cure. World J. Gastroenterol. 2016, 22, 7030–7045. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jerusalem, G.; Longuespee, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, B.C.; Hoshida, Y.; Fujii, T.; Wei, L.; Yamada, S.; Lauwers, G.Y.; McGinn, C.M.; DePeralta, D.K.; Chen, X.; Kuroda, T.; et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology 2014, 59, 1577–1590. [Google Scholar] [CrossRef] [Green Version]
- El-Far, Y.M.; Khodir, A.E.; Emarah, Z.A.; Ebrahim, M.A.; Al-Gayyar, M.M.H. Chemopreventive and hepatoprotective effects of genistein via inhibition of oxidative stress and the versican/PDGF/PKC signaling pathway in experimentally induced hepatocellular carcinoma in rats by thioacetamide. Redox Rep. 2022, 27, 9–20. [Google Scholar] [CrossRef]
- Faure-Dupuy, S.; Riedl, T.; Rolland, M.; Hizir, Z.; Reisinger, F.; Neuhaus, K.; Schuehle, S.; Remouchamps, C.; Gillet, N.; Schonung, M.; et al. Control of APOBEC3B induction and cccDNA decay by NF-kappaB and miR-138-5p. JHEP Rep. 2021, 3, 100354. [Google Scholar] [CrossRef]
- Zeisel, M.B.; Lupberger, J.; Fofana, I.; Baumert, T.F. Host-targeting agents for prevention and treatment of chronic hepatitis C—Perspectives and challenges. J. Hepatol. 2013, 58, 375–384. [Google Scholar] [CrossRef]
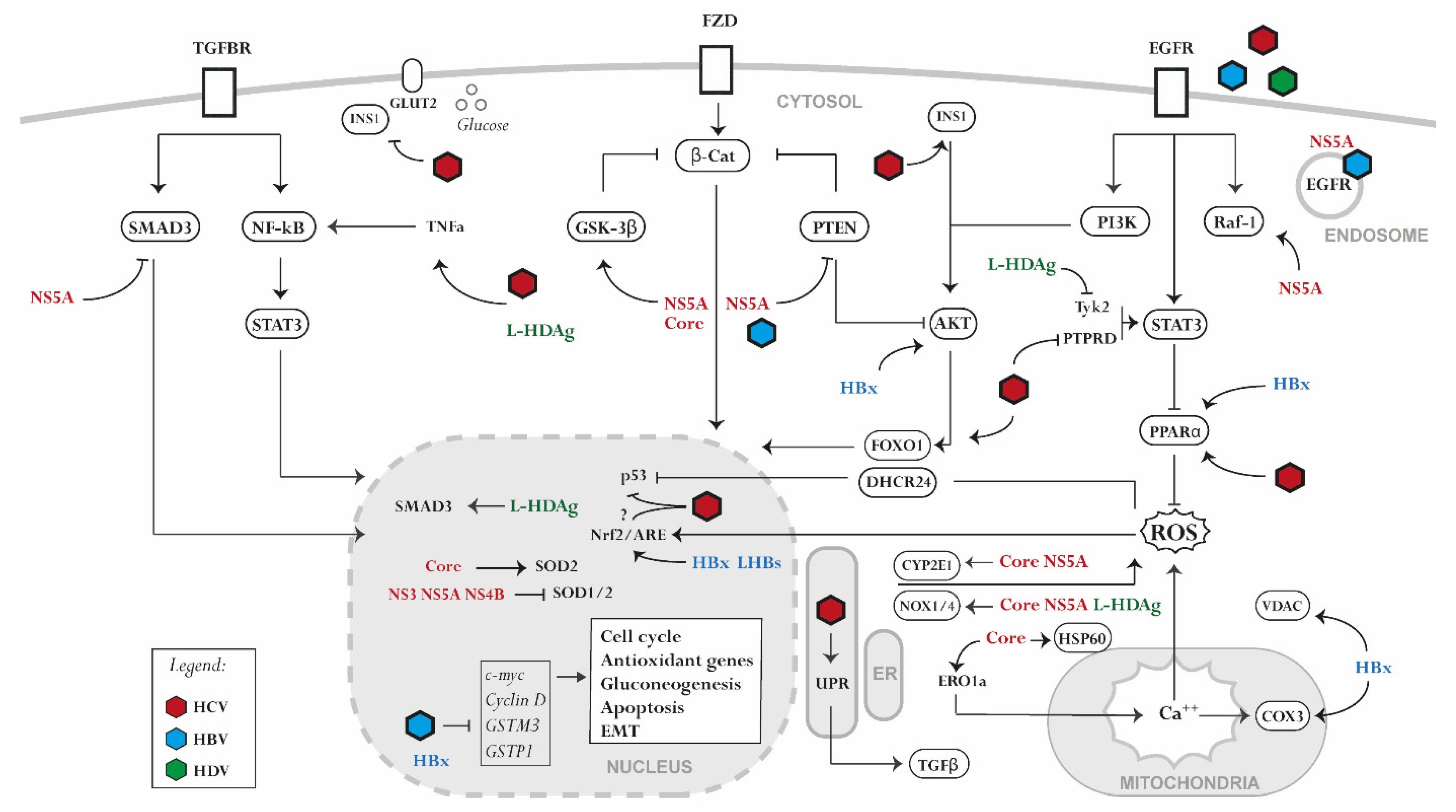


{kind=link}
{kind=link}
| Perturbed Signaling Pathway | Virus | References |
|---|---|---|
| IL-6/JAK/STAT3 | HBV, HCV, HDV | [45,83,89,90,91,92,96] |
| EGFR | HBV, HCV | [93,94,146,147,148,149,150,151] |
| TNF-α/NF-κB | HCV, HDV | [45,65,66,82] |
| Nrf2/ARE | HBV, HCV | [47,48,49,50,54,55,59,60,61] |
| PI3K/Akt | HBV, HCV | [48,160] |
| Ras/Raf | HCV | [152] |
| TGF-β/SMAD | HBV, HCV, HDV | [66,67,69,168,172,173] |
| Wnt/β-catenin | HBV, HCV | [71,162,164,165,166] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boulahtouf, Z.; Virzì, A.; Baumert, T.F.; Verrier, E.R.; Lupberger, J. Signaling Induced by Chronic Viral Hepatitis: Dependence and Consequences. Int. J. Mol. Sci. 2022, 23, 2787. https://doi.org/10.3390/ijms23052787
Boulahtouf Z, Virzì A, Baumert TF, Verrier ER, Lupberger J. Signaling Induced by Chronic Viral Hepatitis: Dependence and Consequences. International Journal of Molecular Sciences. 2022; 23(5):2787. https://doi.org/10.3390/ijms23052787
Chicago/Turabian StyleBoulahtouf, Zakaria, Alessia Virzì, Thomas F. Baumert, Eloi R. Verrier, and Joachim Lupberger. 2022. "Signaling Induced by Chronic Viral Hepatitis: Dependence and Consequences" International Journal of Molecular Sciences 23, no. 5: 2787. https://doi.org/10.3390/ijms23052787
APA StyleBoulahtouf, Z., Virzì, A., Baumert, T. F., Verrier, E. R., & Lupberger, J. (2022). Signaling Induced by Chronic Viral Hepatitis: Dependence and Consequences. International Journal of Molecular Sciences, 23(5), 2787. https://doi.org/10.3390/ijms23052787