
Allosteric Inter-Domain Contacts in Bacterial Hsp70 Are Located in Regions That Avoid Insertion and Deletion Events


Abstract
:1. Introduction
2. Results
2.1. Structural and Evolutionary Characterization of DnaK and Its Domains
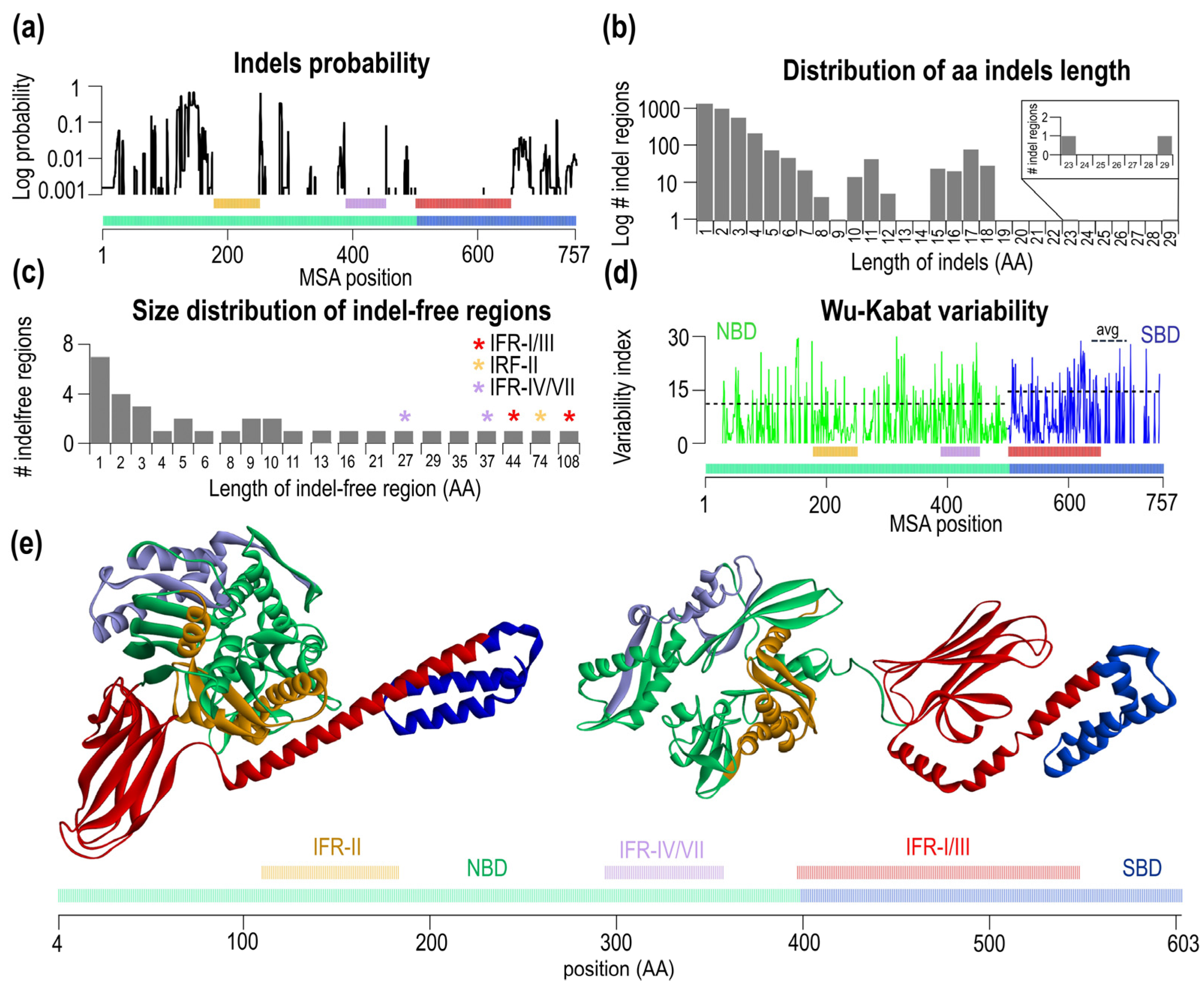
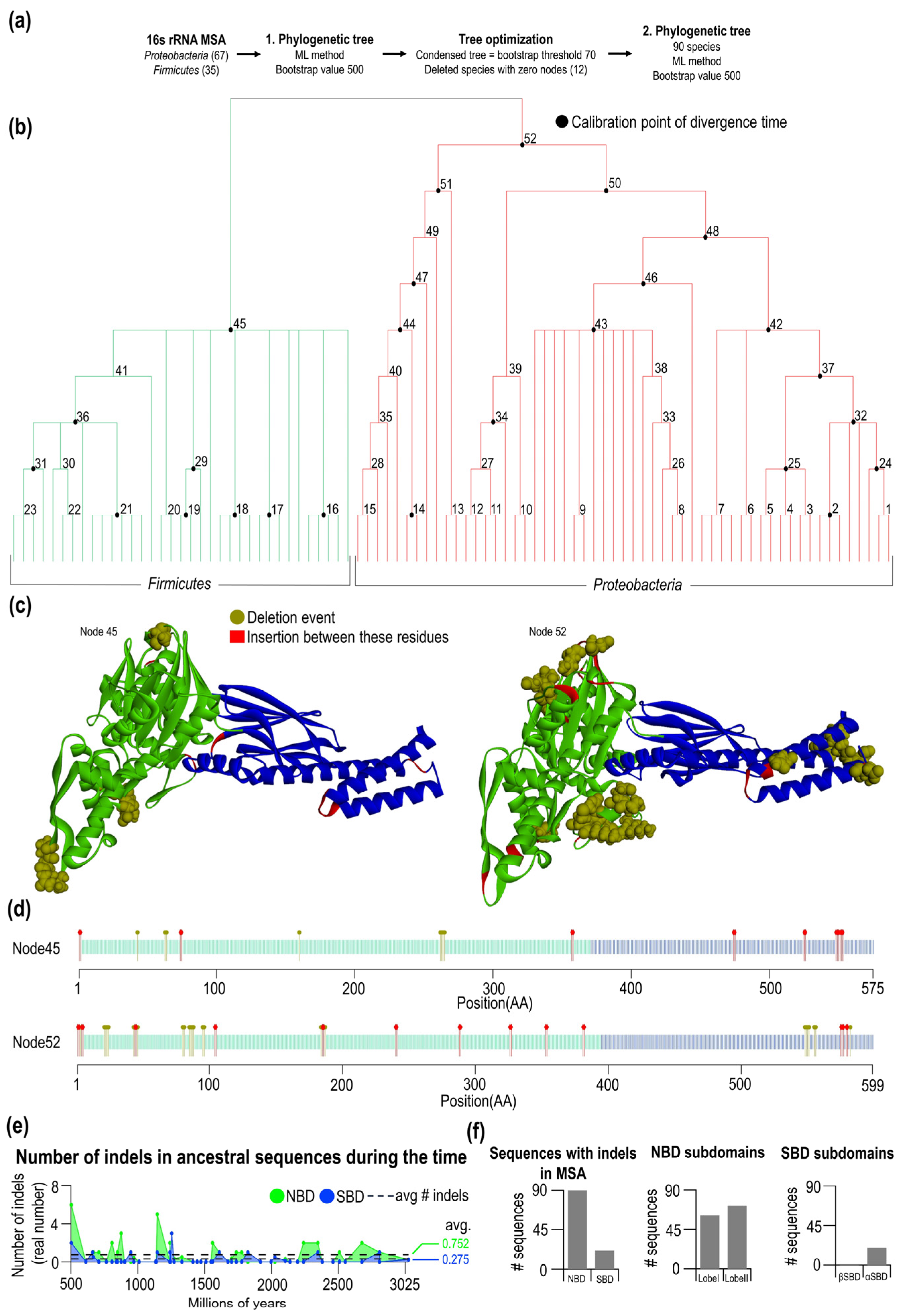
2.2. Indel Distribution from Multiple Sequence Alignment of Hsp70s
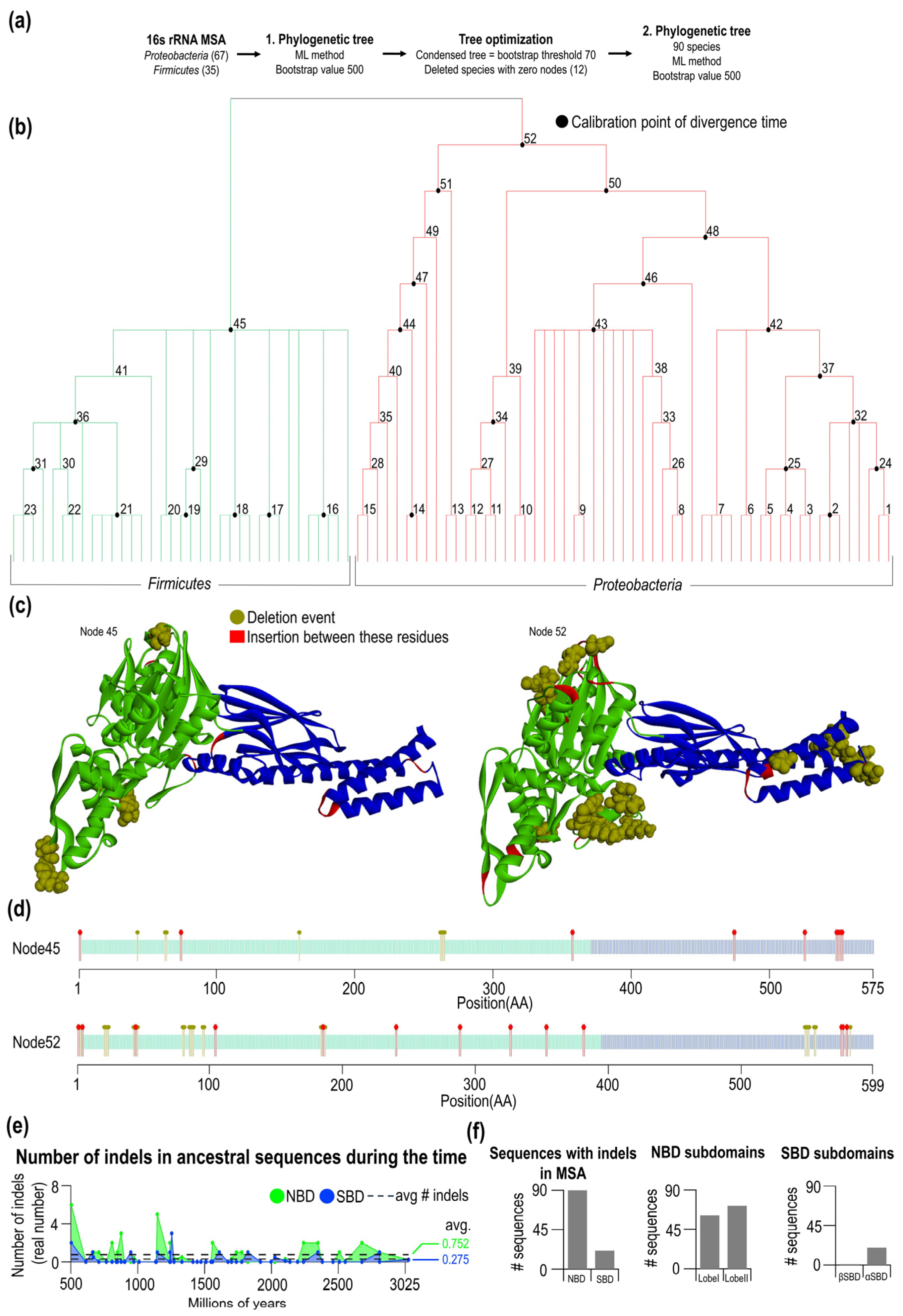
2.3. Phylogenesis of Hsp70 in Firmicutes and Proteobacteria
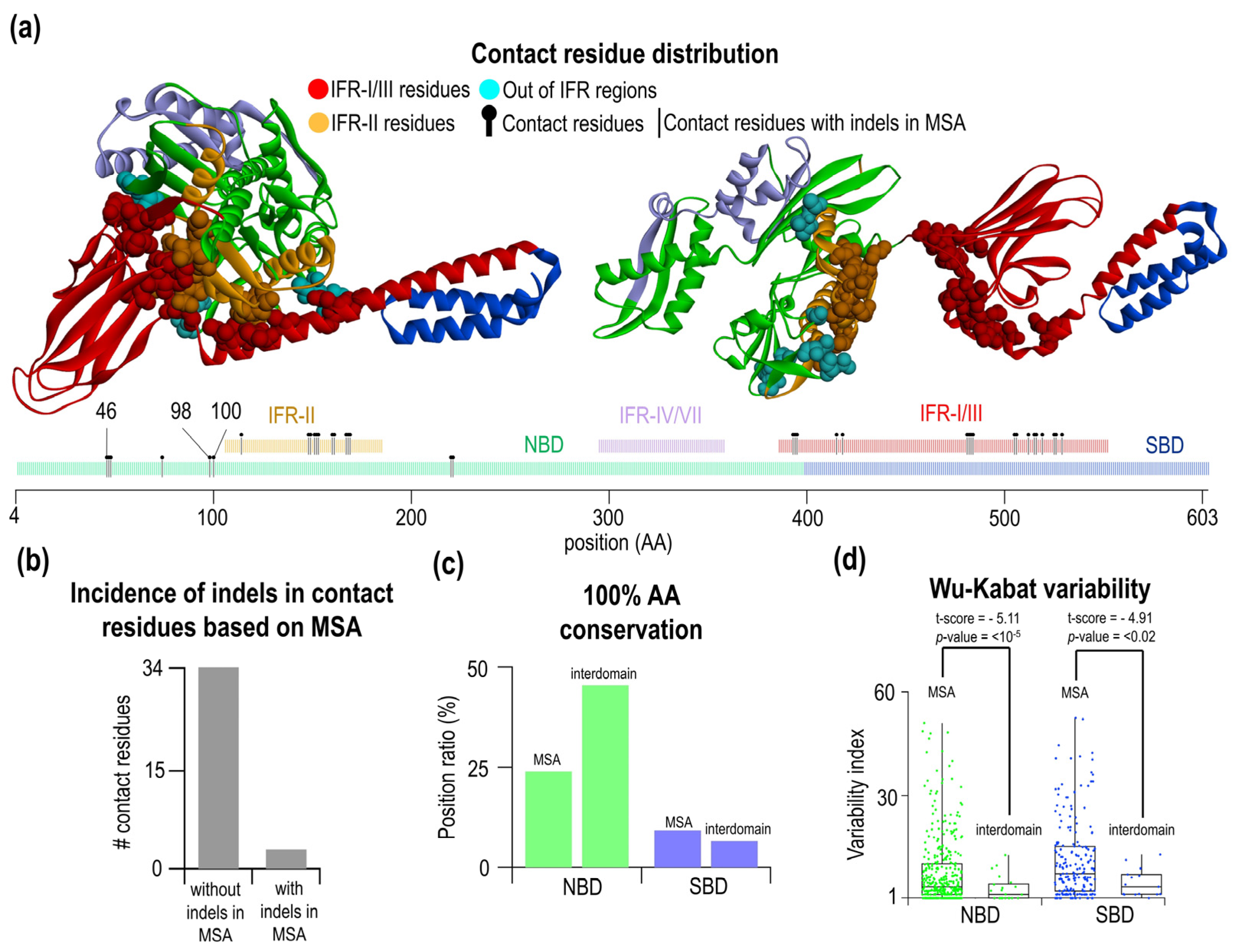
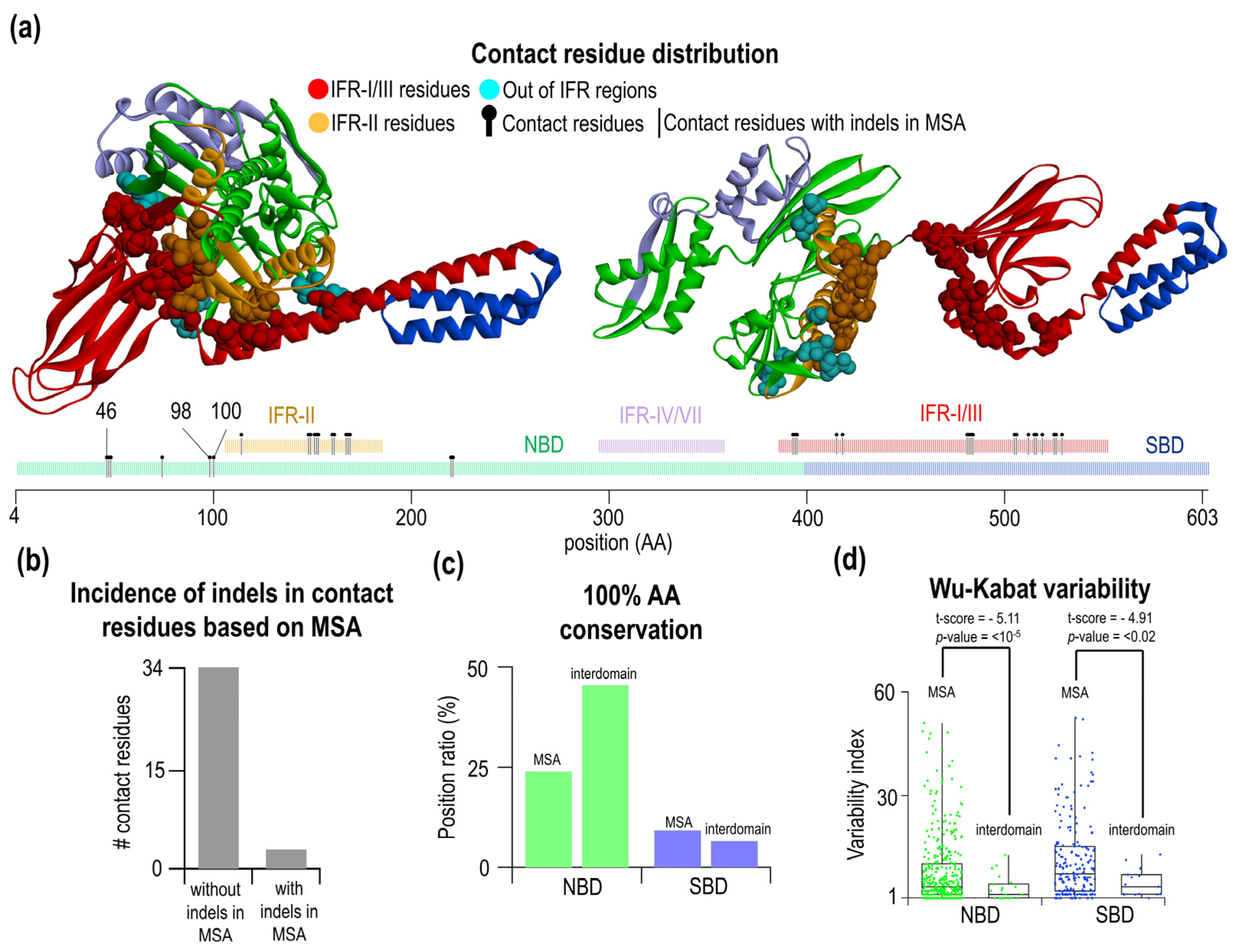
2.4. Amino Acid Residues Participating in Interdomain Contacts Are Frequently Located in Indel-Free Regions
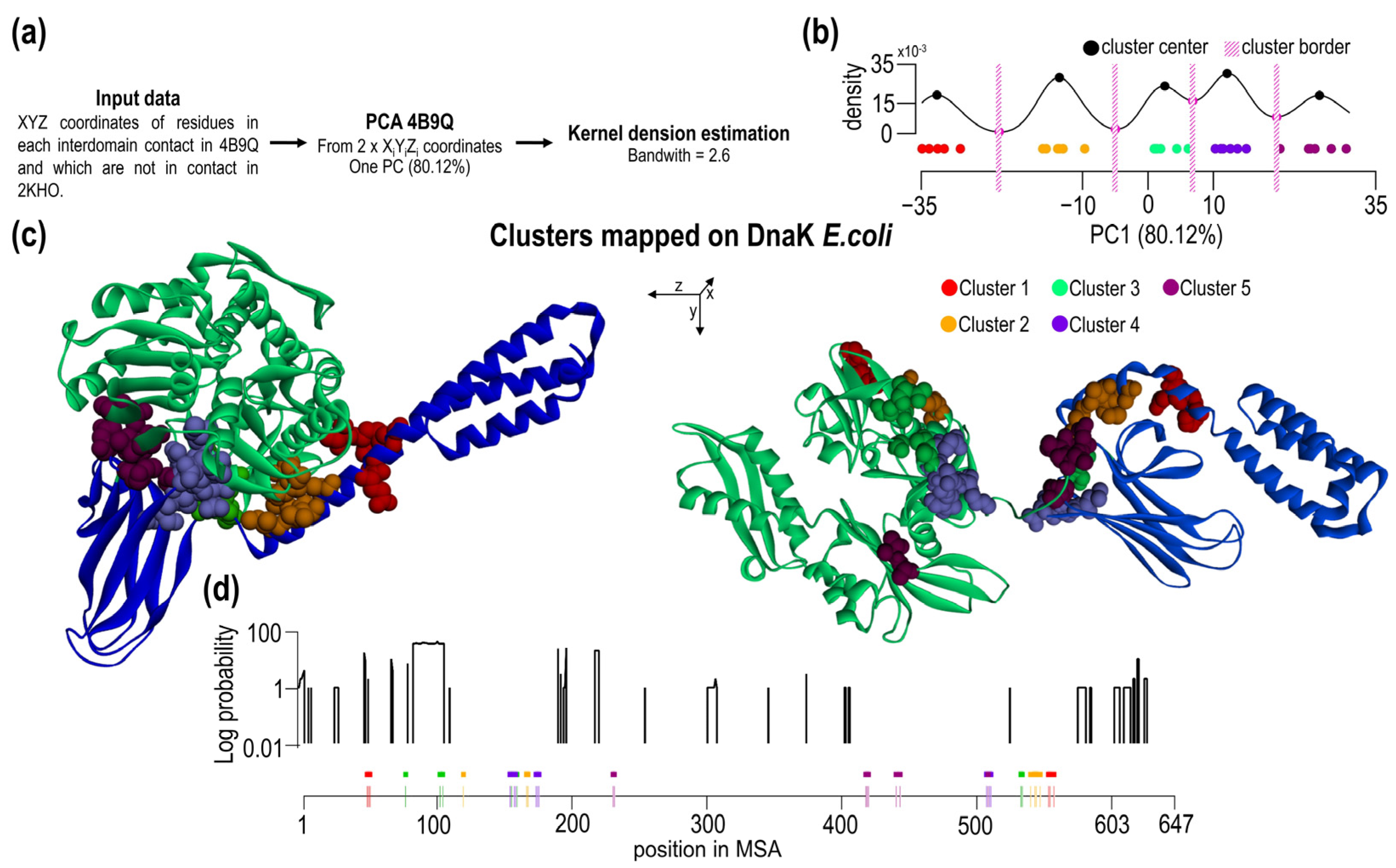
2.5. Clustering of Interdomain Residues Using Principal Component Analysis and Kernel Density Estimation
3. Discussion
3.1. The Evolutionary Rate of Structurally Distinct Subdomains of Hsp70 Are Considerably Different
3.2. Non-Random Distributions of Indel-Free Regions in Bacterial Hsp70s
3.3. Interdomain Contacts Tend to Be Located within Indel-Free Regions
3.4. Hsp70 Subfamily Member HscC Contains Indels in IFR-I/III
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Karlin, S.; Brocchieri, L. Heat Shock Protein 70 Family: Multiple Sequence Comparisons, Function, and Evolution. J. Mol. Evol. 1998, 47, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Genevaux, P.; Georgopoulos, C.; Kelley, W.L. The Hsp70 chaperone machines of Escherichia coli: A paradigm for the repartition of chaperone functions. Mol. Microbiol. 2007, 66, 840–857. [Google Scholar] [CrossRef] [PubMed]
- Rüdiger, S.; Buchberger, A.; Bukau, B. Interaction of Hsp70 chaperones with substrates. Nat. Struct. Mol. Biol. 1997, 4, 342–349. [Google Scholar] [CrossRef]
- Mayer, M.P.; Gierasch, L.M. Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J. Biol. Chem. 2019, 294, 2085–2097. [Google Scholar] [CrossRef] [Green Version]
- Bertelsena, E.B.; Chang, L.; Gestwicki, J.E.; Zuiderweg, E.R.P. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. USA 2009, 106, 8471–8476. [Google Scholar] [CrossRef] [Green Version]
- Gong, W.; Hu, W.; Xu, L.; Wu, H.; Wu, S.; Zhang, H.; Wang, J.; Jones, G.W.; Perrett, S. The C-terminal GGAP motif of Hsp70 mediates substrate recognition and stress response in yeast. J. Biol. Chem. 2018, 293, 17663–17675. [Google Scholar] [CrossRef] [Green Version]
- Smock, R.G.; Blackburn, M.E.; Gierasch, L.M. Correction: Conserved, disordered C terminus of DnaK enhances cellular survival upon stress and DnaK in vitro chaperone activity. J. Biol. Chem. 2018, 293, 14295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosendahl, S.; Ainelo, A.; Hõrak, R. The Disordered C-Terminus of the Chaperone DnaK Increases the Competitive Fitness of Pseudomonas putida and Facilitates the Toxicity of GraT. Microorganisms 2021, 9, 375. [Google Scholar] [CrossRef]
- Mayer, M.P.; Schröder, H.; Rüdiger, S.; Paal, K.; Laufen, T.; Bukau, B. Multistep mechanism of substrate binding determines chaperone activity of Hsp. Nat. Struct. Biol. 2000, 7, 586–593. [Google Scholar] [CrossRef]
- De Los Rios, P.; Barducci, A. Hsp70 chaperones are non-equilibrium machines that achieve ultra-affinity by energy consumption. eLife 2014, 3, e02218. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, B.; Hartich, D.; Seifert, U.; Rios, P.D.L. Thermodynamic Bounds on the Ultra- and Infra-affinity of Hsp70 for Its Substrates. Biophys. J. 2017, 113, 362–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.; Gupta, R.S. Conserved inserts in the Hsp60 (GroEL) and Hsp70 (DnaK) proteins are essential for cellular growth. Mol Genet. Genom. 2009, 281, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Golding, G.B. Evolution of HSP70 Gene and Its Implications Regarding Relationships Between Archaebacteria, Eubacteria, and Eukaryotes. J. Mol. Evol. 1993, 37, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Singh, B. Cloning of the HSP70 gene from Halobacterium marismortui: Relatedness of archaebacterial HSP70 to its eubacterial homologs and a model for the evolution of the HSP70 gene. J. Bacteriol. 1992, 174, 4594–4605. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.S.; Bustard, K.; Falah, M.; Singh, D. Sequencing of heat shock protein 70 (DnaK) homologs from Deinococcus proteolyticus and Thermomicrobium roseum and their integration in a protein-based phylogeny of prokaryotes. J. Bacteriol. 1997, 179, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Gribaldo, S.; Lumia, V.; Creti, R.; De Macario, E.C.; Sanangelantoni, A.; Cammarano, P. Discontinuous occurrence of the hsp70 (dnaK) gene among Archaea and sequence features of HSP70 suggest a novel outlook on phylogenies inferred from this protein. J. Bacteriol. 1999, 181, 434–443. [Google Scholar] [CrossRef] [Green Version]
- Rebeaud, M.E.; Mallik, S.; Goloubinoff, P.; Tawfik, D.S. On the evolution of chaperones and cochaperones and the expansion of proteomes across the Tree of Life. Proc. Natl. Acad. Sci. USA 2021, 118, e2020885118. [Google Scholar] [CrossRef]
- Liu, Y.; Gierasch, L.M.; Bahar, I. Role of Hsp70 ATPase Domain Intrinsic Dynamics and Sequence Evolution in Enabling its Functional Interactions with NEFs. PLoS Comput. Biol. 2010, 6, e1000931. [Google Scholar] [CrossRef] [Green Version]
- Meng, W.; Clerico, E.M.; McArthur, N.; Gierasch, L.M. Allosteric landscapes of eukaryotic cytoplasmic Hsp70s are shaped by evolutionary tuning of key interfaces. Proc. Natl. Acad. Sci. USA 2018, 115, 11970–11975. [Google Scholar] [CrossRef] [Green Version]
- Ang, D.; Georgopoulos, C. The heat-shock-regulated grpE gene of Escherichia coli is required for bacterial growth at all temperatures but is dispensable in certain mutant backgrounds. J. Bacteriol. 1989, 171, 2748–2755. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K.; Battistuzzi, F.U. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Kim, D.E.; Ovchinnikov, S.; Baker, D.; DiMaio, F. Automatic structure prediction of oligomeric assemblies using Robetta in CASP. Proteins Struct. Funct. Bioinform. 2018, 86, 283–291. [Google Scholar] [CrossRef]
- Yang, J.; Zong, Y.; Su, J.; Li, H.; Zhu, H.; Columbus, L.; Zhou, L.; Liu, Q. Conformation transitions of the polypeptide-binding pocket support an active substrate release from Hsp70s. Nat. Commun. 2017, 8, 1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, R.; Sarbeng, E.B.; Liu, Q.; Le, K.Q.; Xu, X.; Xu, H.; Yang, J.; Wong, J.L.; Vorvis, C.; Hendrickson, W.A.; et al. Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nat. Struct. Mol. Biol. 2013, 20, 900–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kityk, R.; Kopp, J.; Sinning, I.; Mayer, M.P. Structure and Dynamics of the ATP-Bound Open Conformation of Hsp70 Chaperones. Mol. Cell 2012, 48, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Rigo, M.M.; Borges, T.J.; Lang, B.J.; Murshid, A.; Nitika; Wolfgeher, D.; Calderwood, S.K.; Truman, A.W.; Bonorino, C. Host expression system modulates recombinant Hsp70 activity through post-translational modifications. FEBS J. 2020, 287, 4902–4916. [Google Scholar] [CrossRef]
- Calloni, G.; Chen, T.; Schermann, S.M.; Chang, H.-C.; Genevaux, P.; Agostini, F.; Tartaglia, G.G.; Hayer-Hartl, M.; Hartl, F.U. DnaK functions as a central hub in the E. coli chaperone network. Cell Rep. 2012, 1, 251–264. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.; Meinhold, S.; Jakob, R.P.; Stigler, J.; Merkel, U.; Maier, T.; Rief, M.; Žoldák, G. A folding nucleus and minimal ATP binding domain of Hsp70 identified by single-molecule force spectroscopy. Proc. Natl. Acad. Sci. USA 2018, 115, 4666–4671. [Google Scholar] [CrossRef] [Green Version]
- Tian, P.; Best, R.B. How Many Protein Sequences Fold to a Given Structure? A Coevolutionary Analysis. Biophys. J. 2017, 113, 1719–1730. [Google Scholar] [CrossRef] [Green Version]
- Mandal, S.S.; Merz, D.R.; Buchsteiner, M.; Dima, R.I.; Rief, M.; Žoldák, G. Nanomechanics of the substrate binding domain of Hsp70 determine its allosteric ATP-induced conformational change. Proc. Natl. Acad. Sci. USA 2017, 114, 6040–6045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vickery, L.E.; Cupp-Vickery, J.R. Molecular Chaperones HscA/Ssq1 and HscB/Jac1 and Their Roles in Iron-Sulfur Protein Maturation. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Kluck, C.J.; Patzelt, H.; Genevaux, P.; Brehmer, D.; Rist, W.; Schneider-Mergener, J.; Bukau, B.; Mayer, M.P. Structure-Function Analysis of HscC, theEscherichia coli Member of a Novel Subfamily of Specialized Hsp70 Chaperones. J. Biol. Chem. 2002, 277, 41060–41069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, M.P. The Hsp70-Chaperone Machines in Bacteria. Front. Mol. Biosci. 2021, 8, 512. [Google Scholar] [CrossRef]
- Hall, T. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Kabat, E.A.; Wu, T.T.; Bilofsky, H. Unusual distributions of amino acids in complementarity-determining (hypervariable) segments of heavy and light chains of immunoglobulins and their possible roles in specificity of antibody-combining sites. J. Biol. Chem. 1977, 252, 6609–6616. [Google Scholar] [CrossRef]
- Gala, M.; Žoldák, G. Classifying Residues in Mechanically Stable and Unstable Substructures Based on a Protein Sequence: The Case Study of the DnaK Hsp70 Chaperone. Nanomaterials 2021, 11, 2198. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M. Molecular structure, selective constraint and the rate of evolution. In The Neutral Theory of Molecular Evolution; Cambridge University Press: Cambridge, UK, 1983; Volume 5, pp. 149–193. [Google Scholar]
- Rice, P.; Longden, L.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Tamura, K.; Battistuzzi, F.U.; Billing-Ross, P.; Murillo, O.; Filipski, A.; Kumar, S. Estimating divergence times in large molecular phylogenies. Proc. Natl. Acad. Sci. USA 2012, 109, 19333–19338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Tao, Q.; Kumar, S. Theoretical Foundation of the RelTime Method for Estimating Divergence Times from Variable Evolutionary Rates. Mol. Biol. Evol. 2018, 35, 1770–1782. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Le, S.Q.; Gascuel, O. An Improved General Amino Acid Replacement Matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, H.; Shinomoto, S. Kernel bandwidth optimization in spike rate estimation. J. Comput. Neurosci. 2010, 29, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, B.L. The generalization of ‘student’s’ problem when several different population varlances are involved. Biometrika 1947, 34, 28–35. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster ID | The Number of Conserved Residues NBD + SBD | All in IFR | Contact Residues (E. coli Numbering) |
|---|---|---|---|
| 1 | 0 + 0 | No | G46-A525, E47-A525, E47-D526, E47-F529, T48-D526 |
| 2 | 1 + 0 | Yes | Q114-M515, * I160-I512, * I160-M515, * I160-V516, * I160-A519, A161-V516, A161-A519 |
| 3 | 2 + 0 | No | * G74-G506, N98-S505, D100-S505, 149-S505, A149-G506, * A153-G506 |
| 4 | 4 + 0 | Yes | * D148-I483, * D148-L484, * R151-I483, Q152-I483, * R167-D481, * I168-D481, I169-D481 |
| 5 | 3 + 1 | No | A220-N415, * T221-N415, * D393-I418, * V394-I418, T395-I418, T395-** G482 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gala, M.; Pristaš, P.; Žoldák, G. Allosteric Inter-Domain Contacts in Bacterial Hsp70 Are Located in Regions That Avoid Insertion and Deletion Events. Int. J. Mol. Sci. 2022, 23, 2788. https://doi.org/10.3390/ijms23052788
Gala M, Pristaš P, Žoldák G. Allosteric Inter-Domain Contacts in Bacterial Hsp70 Are Located in Regions That Avoid Insertion and Deletion Events. International Journal of Molecular Sciences. 2022; 23(5):2788. https://doi.org/10.3390/ijms23052788
Chicago/Turabian StyleGala, Michal, Peter Pristaš, and Gabriel Žoldák. 2022. "Allosteric Inter-Domain Contacts in Bacterial Hsp70 Are Located in Regions That Avoid Insertion and Deletion Events" International Journal of Molecular Sciences 23, no. 5: 2788. https://doi.org/10.3390/ijms23052788
APA StyleGala, M., Pristaš, P., & Žoldák, G. (2022). Allosteric Inter-Domain Contacts in Bacterial Hsp70 Are Located in Regions That Avoid Insertion and Deletion Events. International Journal of Molecular Sciences, 23(5), 2788. https://doi.org/10.3390/ijms23052788