
Cognate DNA Recognition by Engrailed Homeodomain Involves a Conformational Change Controlled via an Electrostatic-Spring-Loaded Latch

 , , ,
, , , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
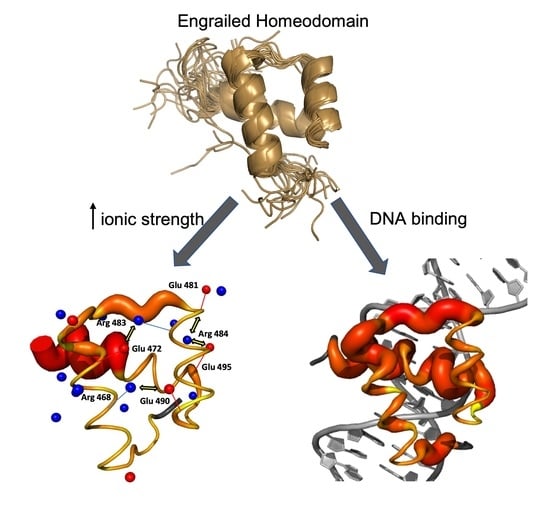
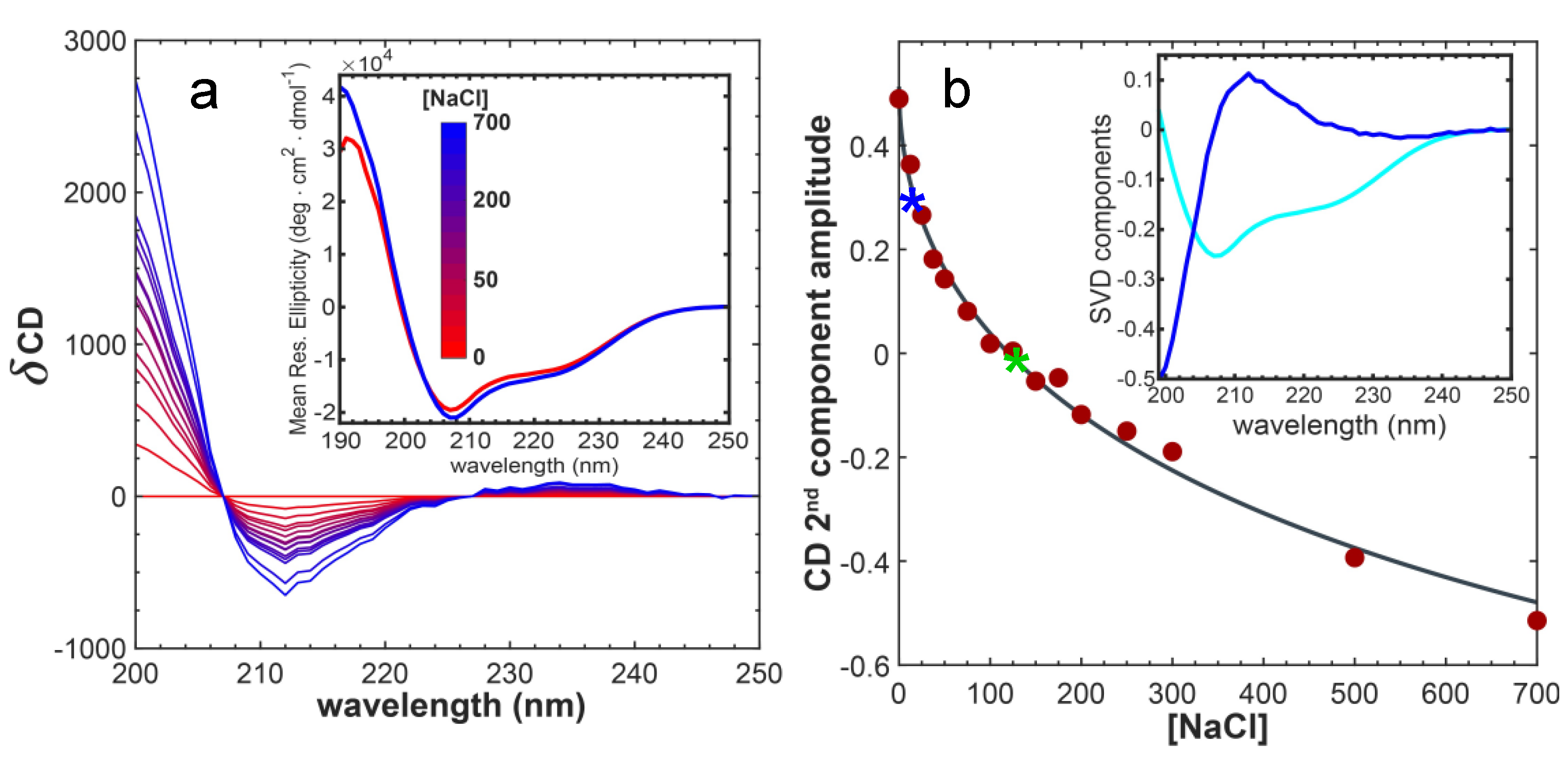
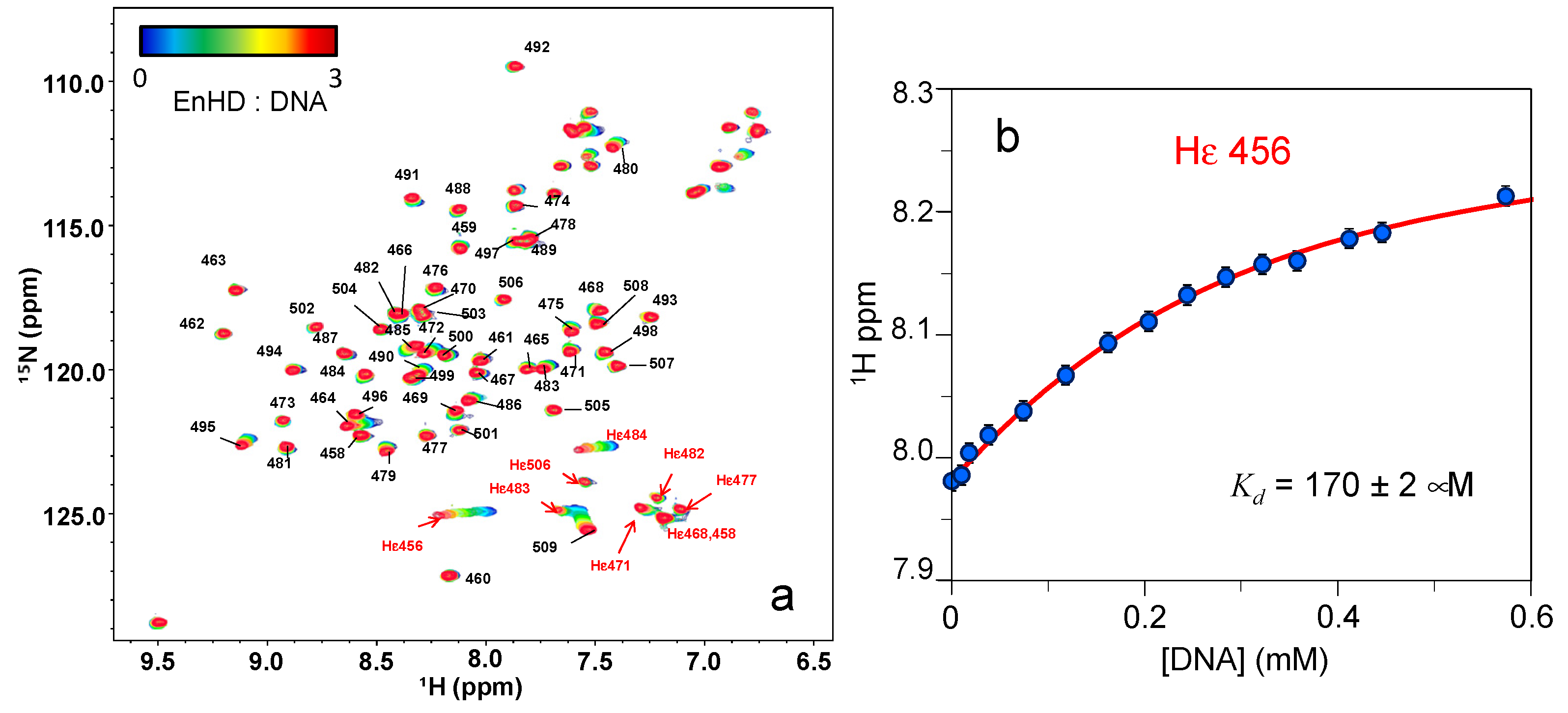
2.1. The Conformational Ensemble of EnHD Is Modulated by Ionic Strength
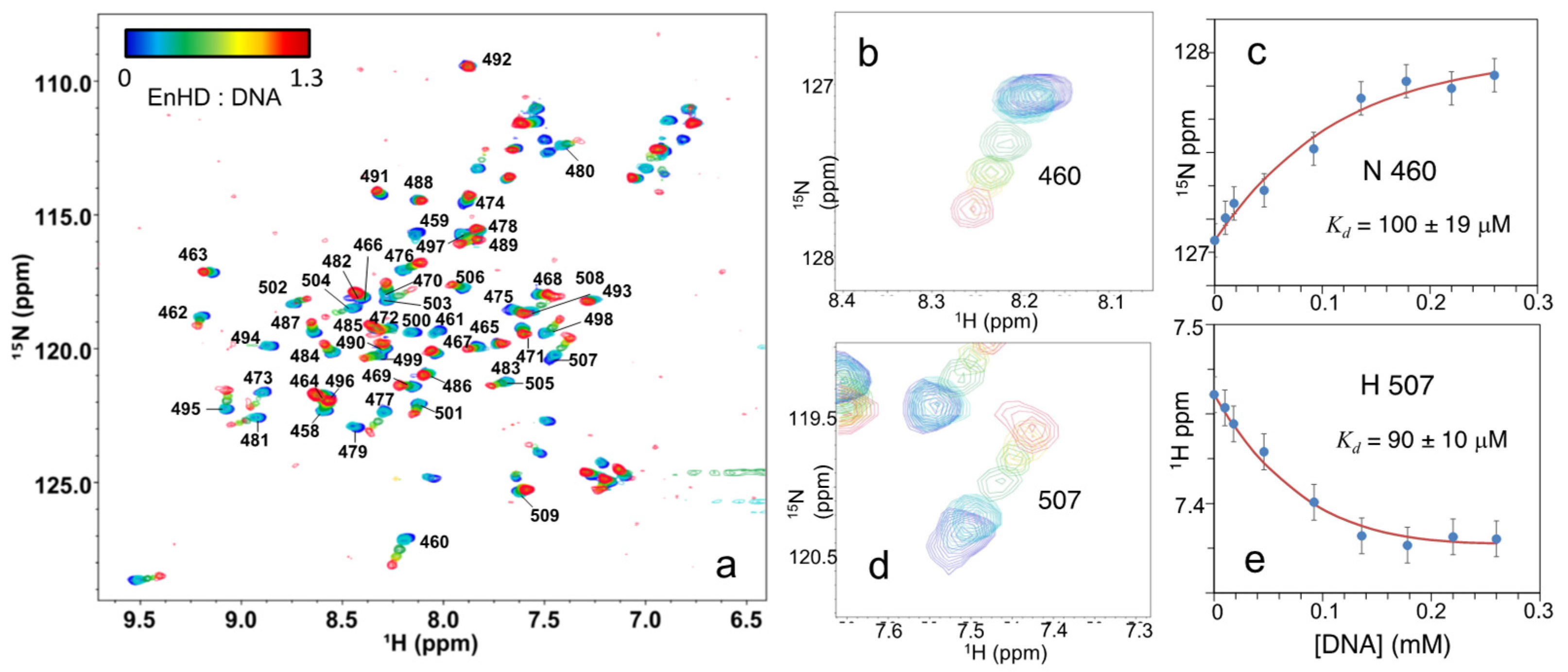
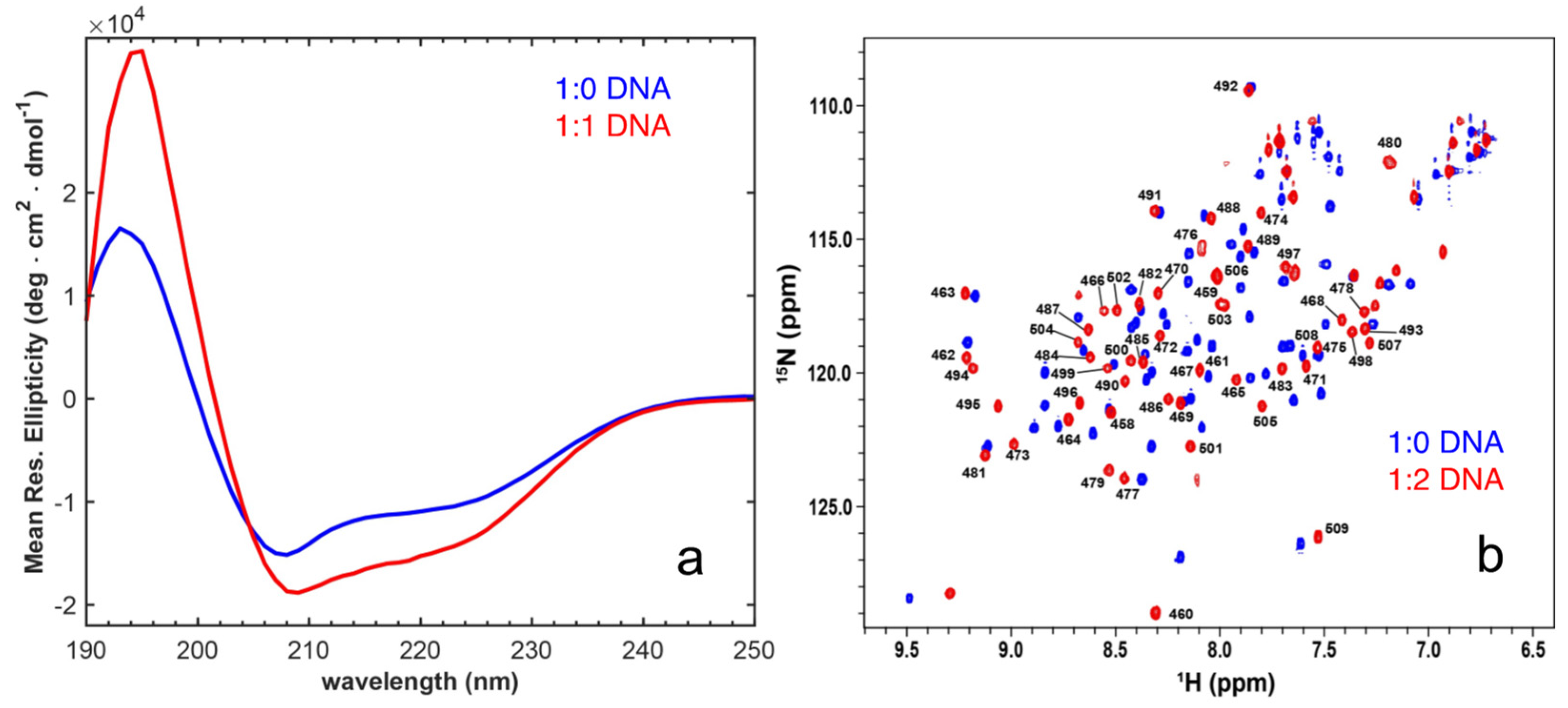
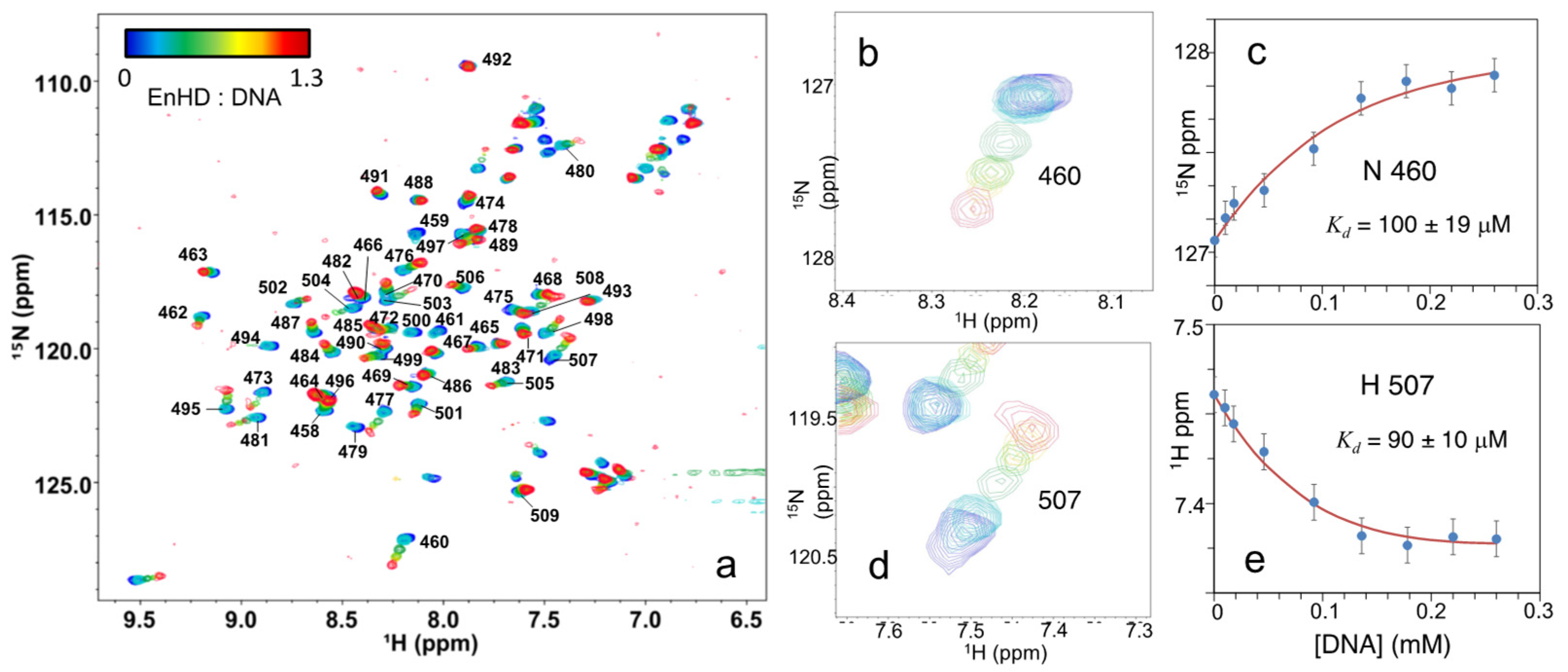
2.2. Binding to Cognate DNA Induces a Conformational Change on EnHD
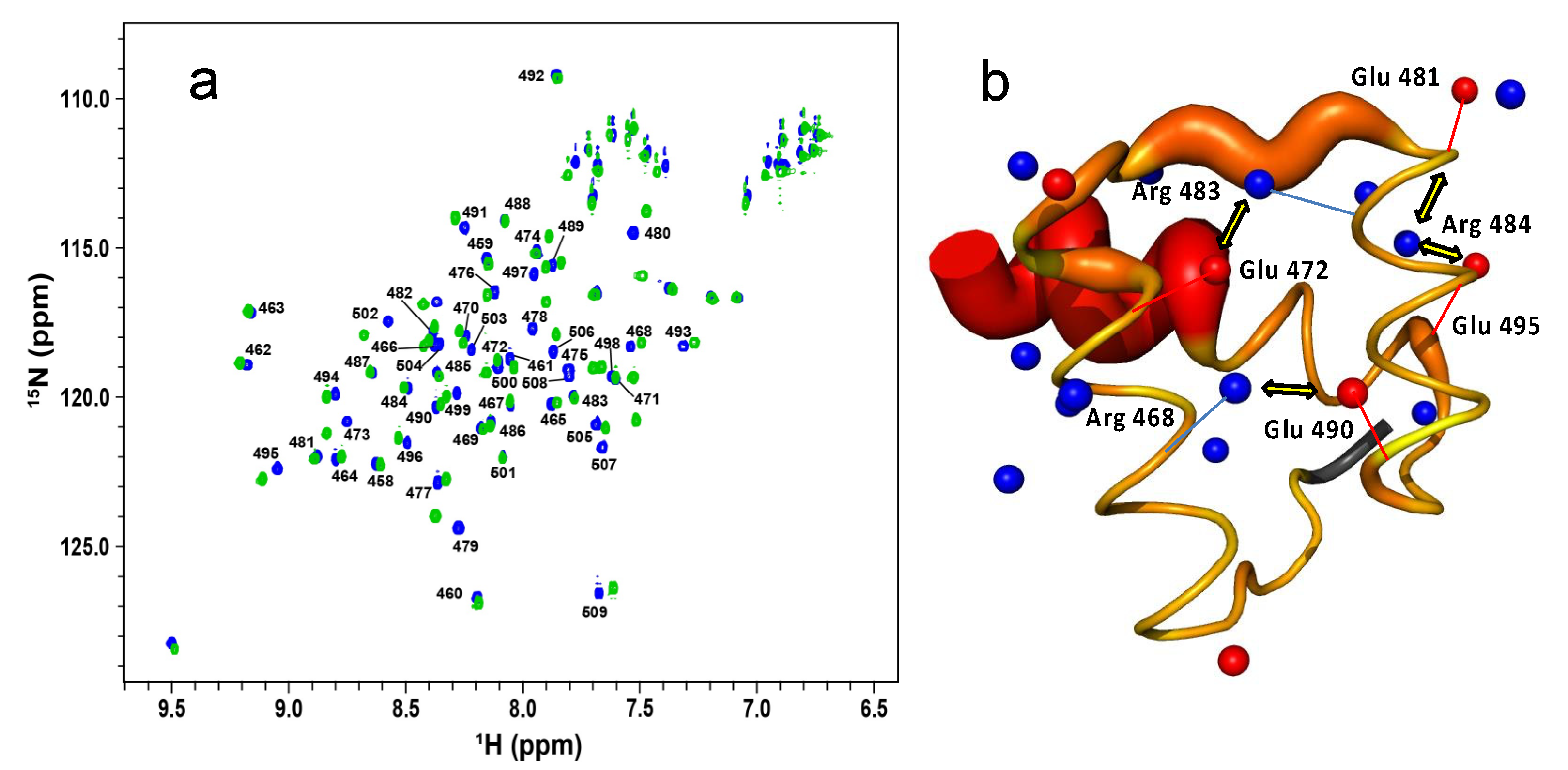
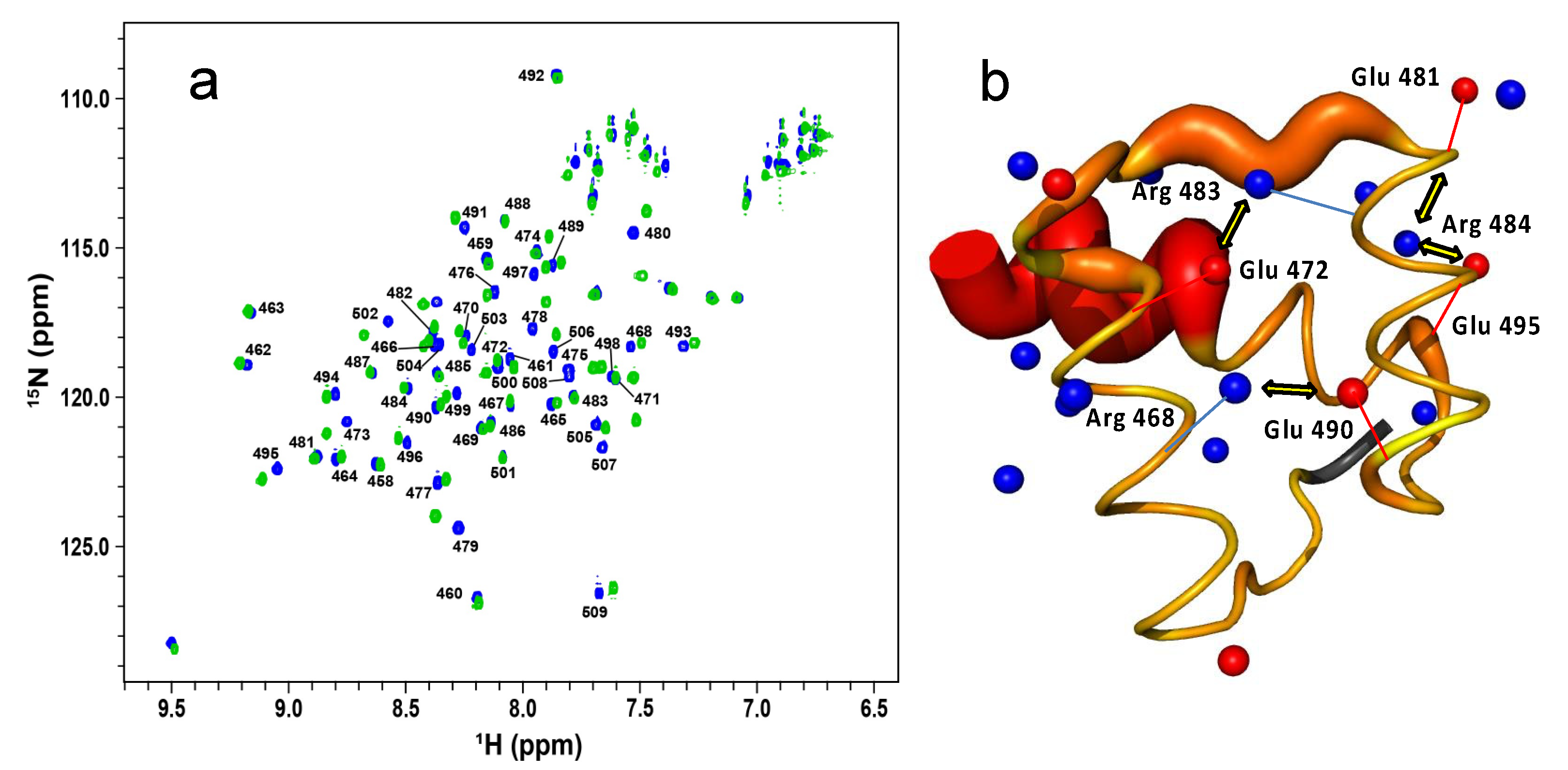
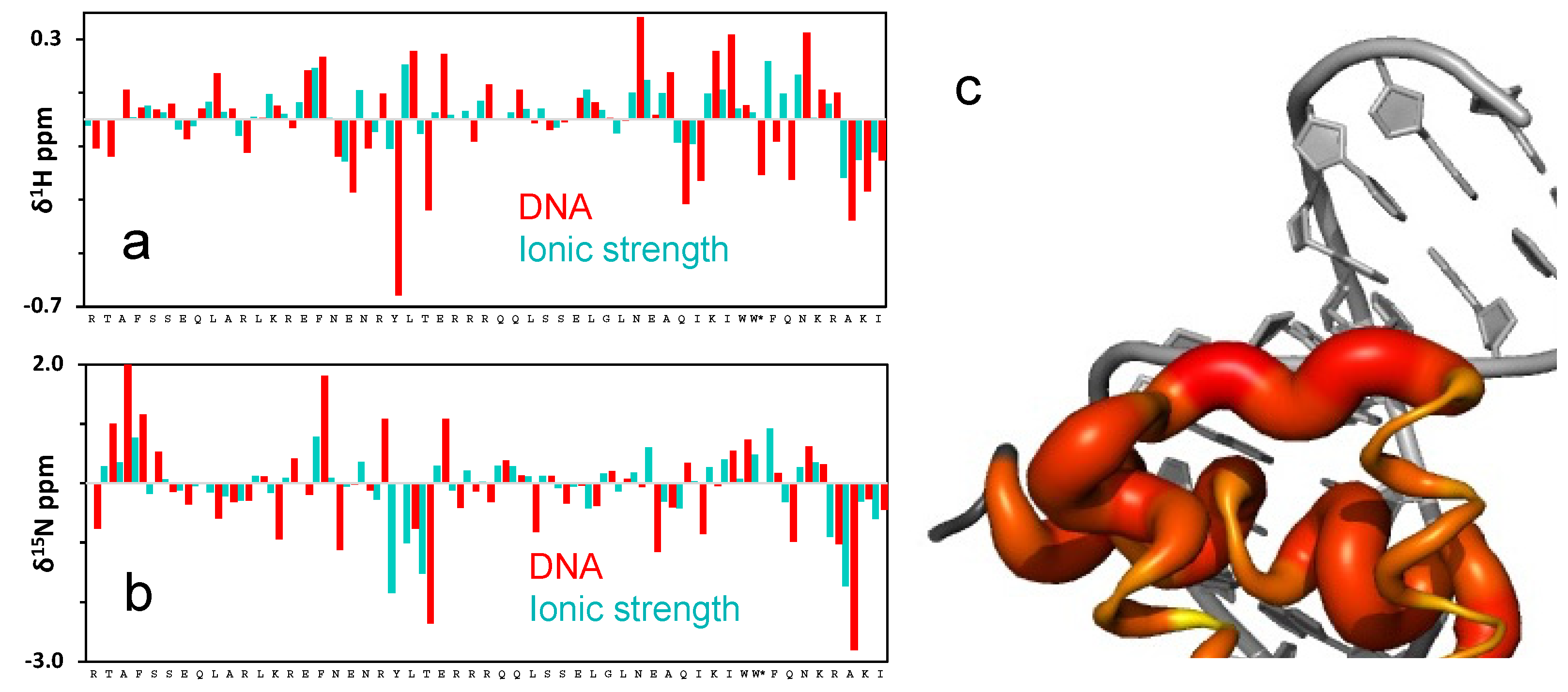
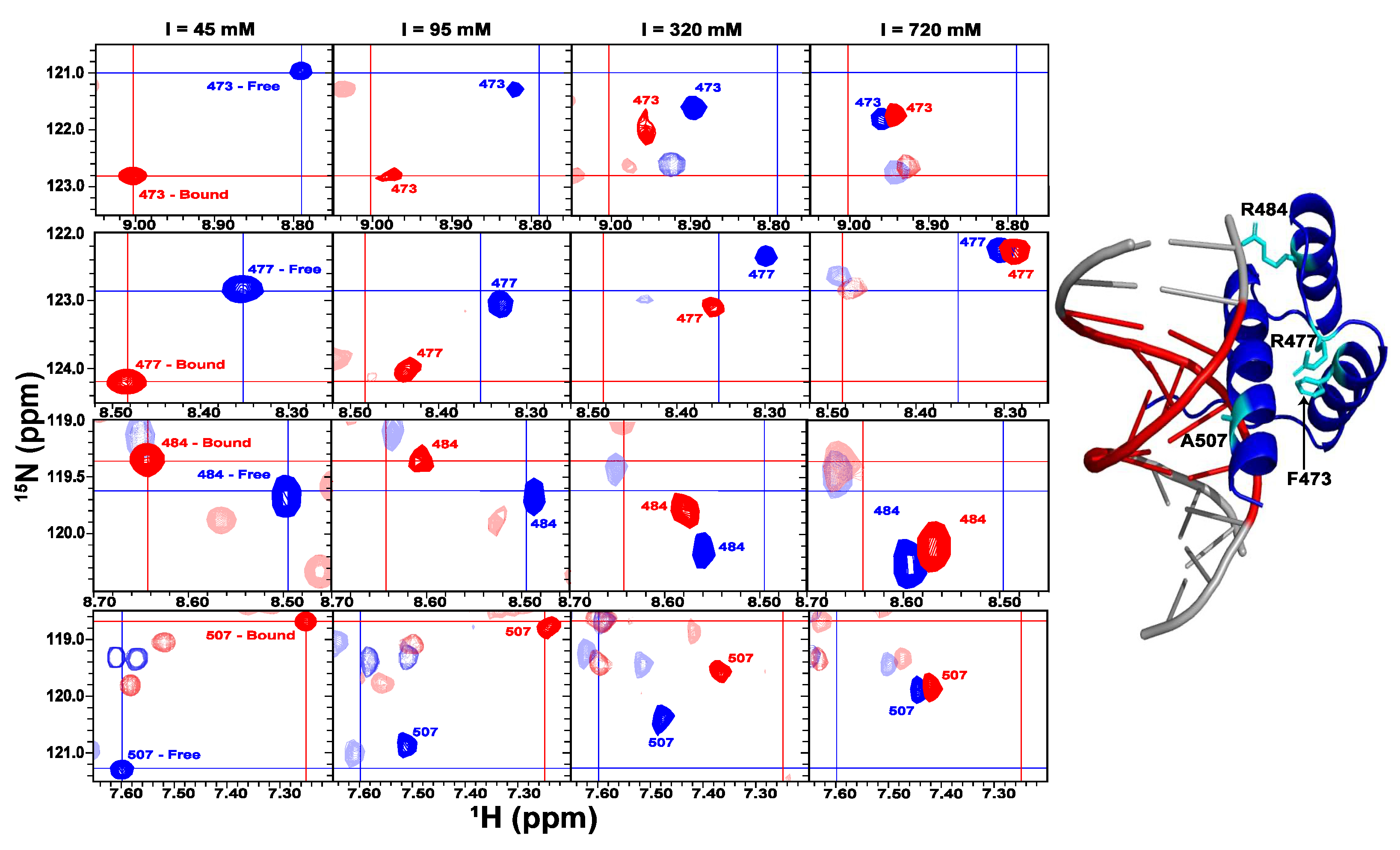
2.3. The Role of Electrostatics in EnHD’s Binding to Cognate DNA
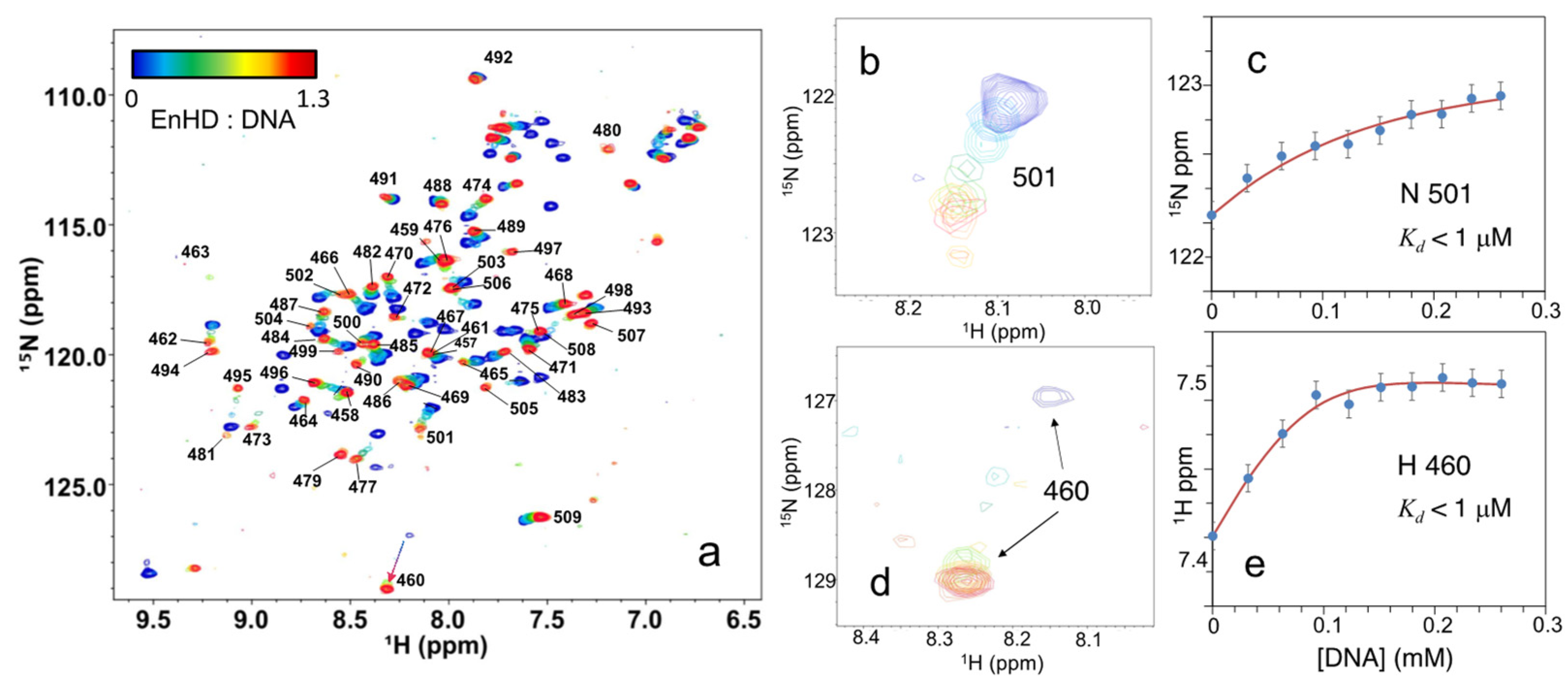
2.4. Electrostatic Shielding Blocks EnHD Conformation Leading to Lock-and-Key Binding to Cognate DNA
2.5. Interplay between EnHD Conformational Dynamics and Cognate DNA Binding
2.6. Cognate DNA Recognition near Physiological Conditions
2.7. Multilevel Interplay between DNA Interaction Energetics and EnHD Conformation
3. Discussion
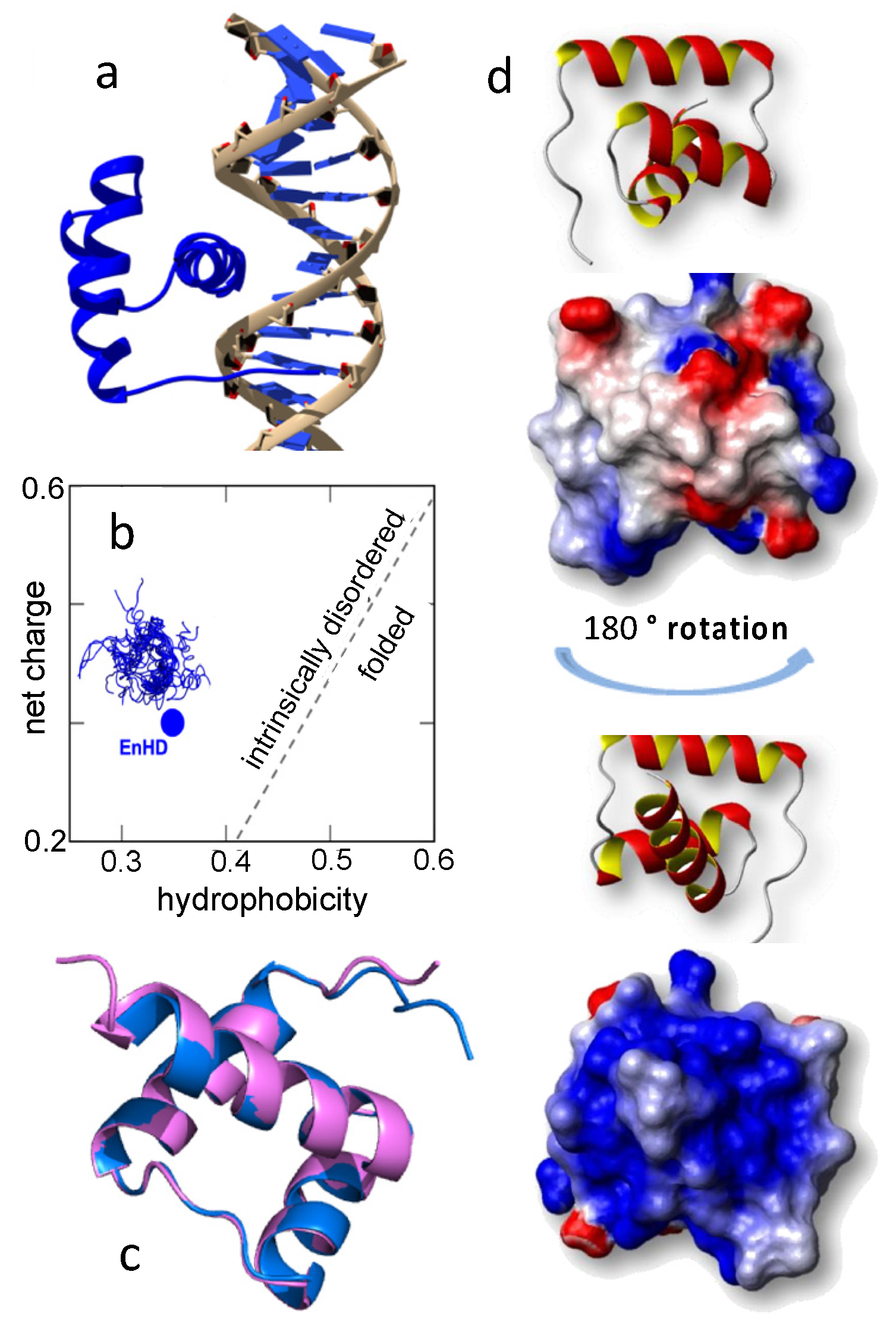
3.1. A Conformational Ensemble with a Builtin Electrostatic-Spring-Loaded Latch
3.2. Conformation versus Cognate DNA Recognition in EnHD
3.3. Functional Implications for an Interplay between EnHD Conformation and Interaction Energetics
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Cognate DNA Molecule
4.3. Far UV Circular Dichroism (CD) Spectroscopy
4.4. Nuclear Magnetic Resonance
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bintu, L.; Buchler, N.E.; Garcia, H.G.; Gerland, U.; Hwa, T.; Kondev, J.; Phillips, R. Transcriptional regulation by the numbers: Models. Curr. Opin. Gen. Dev. 2005, 15, 116–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milo, R.; Jorgensen, P.; Moran, U.; Weber, G.; Springer, M. BioNumbers—The database of key numbers in molecular and cell biology. Nucleic Acids Res 2010, 38 (Suppl. 1), D750–D753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hippel, P.H.; Berg, O.G. Facilitated target location in biological systems. J. Biol. Chem. 1989, 264, 675–678. [Google Scholar] [CrossRef]
- Mirny, L.; Slutsky, M.; Wunderlich, Z.; Tafvizi, A.; Leith, J.; Kosmrlj, A. How a protein searches for its site on DNA: The mechanism of facilitated diffusion. J. Phys. A Math. Theor. 2009, 42, 434013. [Google Scholar] [CrossRef] [Green Version]
- Kolomeisky, A.B. Physics of protein–DNA interactions: Mechanisms of facilitated target search. Phys. Chem. Chem. Phys. 2011, 13, 2088–2095. [Google Scholar] [CrossRef] [PubMed]
- Bhattacherjee, A.; Krepel, D.; Levy, Y. Coarse-grained models for studying protein diffusion along DNA. WIREs Comput. Mol. Sci. 2016, 6, 515–531. [Google Scholar] [CrossRef]
- Liu, L.; Cherstvy, A.G.; Metzler, R. Facilitated diffusion of transcription factor proteins with anomalous bulk diffusion. J. Phys. Chem. B 2017, 121, 1284–1289. [Google Scholar] [CrossRef]
- Esadze, A.; Stivers, J.T. Facilitated diffusion mechanisms in DNA base excision repair and transcriptional activation. Chem. Rev. 2018, 118, 11298–11323. [Google Scholar] [CrossRef]
- Iwahara, J.; Zandarashvili, L.; Kemme, C.A.; Esadze, A. NMR-based investigations into target DNA search processes of proteins. Methods 2018, 148, 57–66. [Google Scholar] [CrossRef]
- Monico, C.; Capitanio, M.; Belcastro, G.; Vanzi, F.; Pavone, F.S. Optical methods to study protein-DNA interactions in vitro and in living cells at the single-molecule level. Int. J. Mol. Sci. 2013, 14, 3961–3992. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Tjian, R. Visualizing transcription factor dynamics in living cells. J. Cell Biol. 2018, 217, 1181–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elf, J.; Li, G.W.; Xie, X.S. Probing transcription factor dynamics at the single-molecule level in a living cell. Science 2007, 316, 1191–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenfeld, G. Chromatin unfolds. Cell 1996, 86, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef]
- Bryne, J.C.; Valen, E.; Tang, M.H.; Marstrand, T.; Winther, O.; da Piedade, I.; Krogh, A.; Lenhard, B.; Sandelin, A. JASPAR, the open access database of transcription factor-binding profiles: New content and tools in the 2008 update. Nuceic Acids Res. 2008, 36, D102–D106. [Google Scholar] [CrossRef] [Green Version]
- Wunderlich, Z.; Mirny, L.A. Different gene regulation strategies revealed by analysis of binding motifs. Trends Genet. 2009, 25, 434–440. [Google Scholar] [CrossRef] [Green Version]
- Castellanos, M.; Mothi, N.; Muñoz, V. Eukaryotic transcription factors can track and control their target genes using DNA antennas. Nat. Commun. 2020, 11, 540. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Fuxreiter, M.; Tompa, P.; Simon, I.; Uversky, V.N.; Hansen, J.C.; Asturias, F.J. Malleable machines take shape in eukaryotic transcriptional regulation. Nat. Chem. Biol. 2008, 4, 728–737. [Google Scholar] [CrossRef] [Green Version]
- Vuzman, D.; Azia, A.; Levy, Y. Searching DNA via a “Monkey Bar” mechanism: The significance of disordered tails. J. Mol. Biol. 2010, 396, 674–684. [Google Scholar] [CrossRef]
- Vuzman, D.; Levy, Y. The “Monkey-Bar” Mechanism for Searching for the DNA Target Site: The Molecular Determinants. Isr. J. Chem. 2014, 54, 1374–1381. [Google Scholar] [CrossRef]
- Chu, X.; Liu, F.; Maxwell, B.A.; Wang, Y.; Suo, Z.; Wang, H.; Han, W.; Wang, J. Dynamic Conformational Change Regulates the Protein-DNA Recognition: An Investigation on Binding of a Y-Family Polymerase to Its Target DNA. PLoS Comput. Biol. 2014, 10, e1003804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tafvizi, A.; Huang, F.; Fersht, A.R.; Mirny, L.A.; van Oijen, A.M. A single-molecule characterization of p53 search on DNA. Proc. Natl. Acad. Sci. USA 2011, 108, 563–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marklund, E.; van Oosten, B.; Mao, G.; Amselem, E.; Kipper, K.; Sabantsev, A.; Emmerich, A.; Globisch, D.; Zheng, X.; Lehmann, L.C. DNA surface exploration and operator bypassing during target search. Nature 2020, 583, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Zandarashvili, L.; Esadze, A.; Vuzman, D.; Kemme, C.A.; Levy, Y.; Iwahara, J. Balancing between affinity and speed in target DNA search by zinc-finger proteins via modulation of dynamic conformational ensemble. Proc. Natl. Acad. Sci. USA 2015, 112, E5142–E5149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, N.D.; Kissinger, C.R.; Desjarlais, J.; Gilliland, G.L.; Pabo, C.O. Structural studies of the engrailed homeodomain. Protein Sci. 1994, 3, 1779–1787. [Google Scholar] [CrossRef]
- Kissinger, C.R.; Liu, B.S.; Martin-Blanco, E.; Kornberg, T.B.; Pabo, C.O. Crystal structure of an engrailed homeodomain-DNA complex at 2.8 A resolution: A framework for understanding homeodomain-DNA interactions. Cell 1990, 63, 579–590. [Google Scholar] [CrossRef]
- Fraenkel, E.; Rould, M.A.; Chambers, K.A.; Pabo, C.O. Engrailed homeodomain-DNA complex at 2.2 Å resolution: A detailed view of the interface and comparison with other engrailed structures. J. Mol. Biol. 1998, 284, 351–361. [Google Scholar] [CrossRef]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Dragan, A.I.; Li, Z.; Makeyeva, E.N.; Milgotina, E.I.; Liu, Y.; Crane-Robinson, C.; Privalov, P.L. Forces driving the binding of homeodomains to DNA. Biochemistry 2006, 45, 141–151. [Google Scholar] [CrossRef]
- Privalov, P.L.; Dragan, A.I. Microcalorimetry of biological macromolecules. Biophys. Chem. 2007, 126, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Billeter, M.; Qian, Y.Q.; Otting, G.; Müller, M.; Gehring, W.; Wüthrich, K. Determination of the nuclear magnetic resonance solution structure of an Antennapedia homeodomain-DNA complex. J. Mol. Biol. 1993, 234, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Tsao, D.H.; Gruschus, J.M.; Wang, L.-H.; Nirenberg, M.; Ferretti, J.A. The Three-dimensional Solution Structure of the NK-2 Homeodomain fromDrosophila. J. Mol. Biol. 1995, 251, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Ippel, H.; Larsson, G.; Behravan, G.; Zdunek, J.; Lundqvist, M.; Schleucher, J.; Lycksell, P.-O.; Wijmenga, S. The solution structure of the homeodomain of the rat insulin-gene enhancer protein isl-1. Comparison with other homeodomains. J. Mol. Biol. 1999, 288, 689–703. [Google Scholar] [CrossRef]
- Bürglin, T.R.; Affolter, M. Homeodomain proteins: An update. Chromosoma 2016, 125, 497–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGinnis, W.; Krumlauf, R. Homeobox genes and axial patterning. Cell 1992, 68, 283–302. [Google Scholar] [CrossRef]
- Lifanov, A.P.; Makeev, V.J.; Nazina, A.G.; Papatsenko, D.A. Homotypic regulatory clusters in Drosophila. Genome Res. 2003, 13, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Ingham, P.; Martinez-Arias, A.; Lawrence, P.A.; Howard, K. Expression of engrailed in the parasegment of Drosophila. Nature 1985, 317, 634–636. [Google Scholar] [CrossRef]
- Eggert, T.; Hauck, B.; Hildebrandt, N.; Gehring, W.J.; Walldorf, U. Isolation of a Drosophila homolog of the vertebrate homeobox gene Rx and its possible role in brain and eye development. Proc. Natl. Acad. Sci. USA 1998, 95, 2343–2348. [Google Scholar] [CrossRef] [Green Version]
- Abate-Shen, C. Deregulated homeobox gene expression in cancer: Cause or consequence? Nat. Rev. Cancer 2002, 2, 777–785. [Google Scholar] [CrossRef]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.I.; Abecasis, G.R.; Cardon, L.R.; Goldstein, D.B.; Little, J.; Ioannidis, J.P.; Hirschhorn, J.N. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 2008, 9, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Ades, S.E.; Sauer, R.T. Specificity of minor-groove and major-groove interactions in a homeodomain-DNA complex. Biochemistry 1995, 34, 14601–14608. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Kissinger, C.R.; Pabo, C.O.; Martin-Blanco, E.; Kornberg, T.B. Crystallization and preliminary X-ray diffraction studies of the engrailed homeodomain and of an engrailed homeodomain/DNA complex. Biochem. Biophys. Res. Commun. 1990, 171, 257–259. [Google Scholar] [CrossRef]
- Mayor, U.; Johnson, C.M.; Daggett, V.; Fersht, A.R. Protein folding and unfolding in microseconds to nanoseconds by experiment and simulation. Proc. Natl. Acad. Sci. USA 2000, 97, 13518–13522. [Google Scholar] [CrossRef] [Green Version]
- Mayor, U.; Freund, S.M.; Fersht, A.R.; Stollar, E.J.; Lovell, S.C.; Federici, L.; Luisi, B.F. Crystal structures of engrailed homeodomain mutants: Implications for stability and dynamics. J. Biol. Chem. 2003, 278, 43699–43708. [Google Scholar]
- Omichinski, J.G.; Clore, G.M.; Schaad, O.; Felsenfeld, G.; Trainor, C.; Appella, E.; Stahl, S.J.; Gronenborn, A.M. NMR structure of a specific DNA complex of Zn-containing DNA binding domain of GATA-1. Science 1993, 261, 438–446. [Google Scholar] [CrossRef]
- Chuprina, V.P.; Rullmann, J.A.; Lamerichs, R.M.; van Boom, J.H.; Boelens, R.; Kaptein, R. Structure of the complex of lac repressor headpiece and an 11 base-pair half-operator determined by nuclear magnetic resonance spectroscopy and restrained molecular dynamics. J. Mol. Biol. 1993, 234, 446–462. [Google Scholar] [CrossRef]
- Huth, J.R.; Bewley, C.A.; Nissen, M.S.; Evans, J.N.S.; Reeves, R.; Gronenborn, A.M.; Marius Clore, G. The solution structure of an HMG-I(Y)–DNA complex defines a new architectural minor groove binding motif. Nat. Struct. Biol. 1997, 4, 657–665. [Google Scholar] [CrossRef]
- Foster, M.P.; Wuttke, D.S.; Radhakrishnan, I.; Case, D.A.; Gottesfeld, J.M.; Wright, P.E. Domain packing and dynamics in the DNA complex of the N-terminal zinc fingers of TFIIIA. Nat. Struct. Biol. 1997, 4, 605–608. [Google Scholar] [CrossRef]
- Koizumi, K.; Lintas, C.; Nirenberg, M.; Maeng, J.-S.; Ju, J.-H.; Mack, J.W.; Gruschus, J.M.; Odenwald, W.F.; Ferretti, J.A. Mutations that affect the ability of the vnd/NK-2 homeoprotein to regulate gene expression: Transgenic alterations and tertiary structure. Proc. Natl. Acad. Sci. USA 2003, 100, 3119–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird-Titus, J.M.; Thapa, M.; Doerdelmann, T.; Combs, K.A.; Rance, M. Lysine Side-Chain Dynamics in the Binding Site of Homeodomain/DNA Complexes As Observed by NMR Relaxation Experiments and Molecular Dynamics Simulations. Biochemistry 2018, 57, 2796–2813. [Google Scholar] [CrossRef] [PubMed]
- Iwahara, J.; Clore, G.M. Direct observation of enhanced translocation of a homeodomain between DNA cognate sites by NMR exchange spectroscopy. J. Am. Chem. Soc. 2006, 128, 404–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalodimos, C.G.; Biris, N.; Bonvin, A.M.; Levandoski, M.M.; Guennuegues, M.; Boelens, R.; Kaptein, R. Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science 2004, 305, 386–389. [Google Scholar] [CrossRef] [Green Version]
- Iwahara, J.; Zweckstetter, M.; Clore, G.M. NMR structural and kinetic characterization of a homeodomain diffusing and hopping on nonspecific DNA. Proc. Natl. Acad. Sci. USA 2006, 103, 15062–15067. [Google Scholar] [CrossRef] [Green Version]
- Thordarson, P. Determining association constants from titration experiments in supramolecular chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. [Google Scholar] [CrossRef]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef]
- Ades, S.E.; Sauer, R.T. Differential DNA-binding specificity of the engrailed homeodomain: The role of residue 50. Biochemistry 1994, 33, 9187–9194. [Google Scholar] [CrossRef]
- Hermann, P.; Fredericq, E. The role of the AT pairs in the acid denaturation of DNA. Nucleic Acids Res. 1977, 4, 2939–2947. [Google Scholar] [CrossRef] [Green Version]
- Garvie, C.W.; Wolberger, C. Recognition of specific DNA sequences. Mol. Cell 2001, 8, 937–946. [Google Scholar] [CrossRef]
- Gehring, W.J.; Affolter, M.; Bürglin, T. Homeodomain proteins. Annu. Rev. Biochem. 1994, 63, 487–526. [Google Scholar] [CrossRef]
- Bauer, M.; Metzler, R. Generalized facilitated diffusion model for DNA-binding proteins with search and recognition states. Biophys. J. 2012, 102, 2321–2330. [Google Scholar] [CrossRef] [Green Version]
- De Sancho, D.; Doshi, U.; Muñoz, V. Protein folding rates and stability: How much is there beyond size? J. Am. Chem. Soc. 2009, 131, 2074–2075. [Google Scholar] [CrossRef]
- Campos, L.A.; Sadqi, M.; Muñoz, V. Lessons about Protein Folding and Binding from Archetypal Folds. Acc. Chem. Res. 2020, 53, 2180–2188. [Google Scholar] [CrossRef]
- Stein, A.; Whitlock, J.P.; Bina, M. Acidic polypeptides can assemble both histones and chromatin in vitro at physiological ionic strength. Proc. Natl. Acad. Sci. USA 1979, 76, 5000–5004. [Google Scholar] [CrossRef] [Green Version]
- Maguire, M.E.; Cowan, J.A. Magnesium chemistry and biochemistry. Biometals 2002, 15, 203–210. [Google Scholar] [CrossRef]
- Liu, B.; Poolman, B.; Boersma, A.J. Ionic strength sensing in living cells. ACS Chem. Biol. 2017, 12, 2510–2514. [Google Scholar] [CrossRef] [Green Version]
- Moore, R.; Morrill, G. A possible mechanism for concentrating sodium and potassium in the cell nucleus. Biophys. J. 1976, 16, 527–533. [Google Scholar] [CrossRef] [Green Version]
- Chu, X.; Muñoz, V. Roles of conformational disorder and downhill folding in modulating protein-DNA recognition. Phys. Chem. Chem. Phys. 2017, 19, 28527–28539. [Google Scholar] [CrossRef] [Green Version]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Amelio, N.; Tanielian, B.; Sadqi, M.; López-Navajas, P.; Muñoz, V. Cognate DNA Recognition by Engrailed Homeodomain Involves a Conformational Change Controlled via an Electrostatic-Spring-Loaded Latch. Int. J. Mol. Sci. 2022, 23, 2412. https://doi.org/10.3390/ijms23052412
D’Amelio N, Tanielian B, Sadqi M, López-Navajas P, Muñoz V. Cognate DNA Recognition by Engrailed Homeodomain Involves a Conformational Change Controlled via an Electrostatic-Spring-Loaded Latch. International Journal of Molecular Sciences. 2022; 23(5):2412. https://doi.org/10.3390/ijms23052412
Chicago/Turabian StyleD’Amelio, Nicola, Benjamin Tanielian, Mourad Sadqi, Pilar López-Navajas, and Victor Muñoz. 2022. "Cognate DNA Recognition by Engrailed Homeodomain Involves a Conformational Change Controlled via an Electrostatic-Spring-Loaded Latch" International Journal of Molecular Sciences 23, no. 5: 2412. https://doi.org/10.3390/ijms23052412
APA StyleD’Amelio, N., Tanielian, B., Sadqi, M., López-Navajas, P., & Muñoz, V. (2022). Cognate DNA Recognition by Engrailed Homeodomain Involves a Conformational Change Controlled via an Electrostatic-Spring-Loaded Latch. International Journal of Molecular Sciences, 23(5), 2412. https://doi.org/10.3390/ijms23052412