
The Role of the Neutrophilic Network in the Pathogenesis of Psoriasis



Abstract
:1. Neutrophils
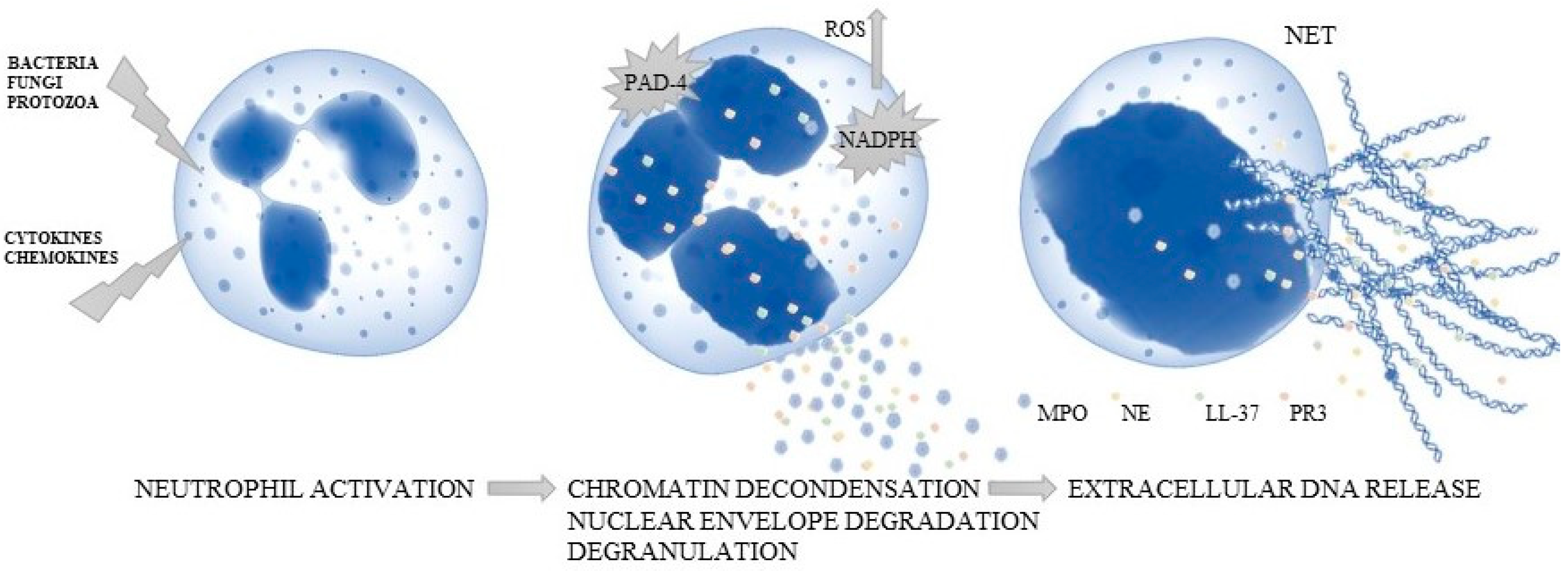
2. Mechanism of Neutrophil Extracellular Traps (NETs)
3. Psoriasis
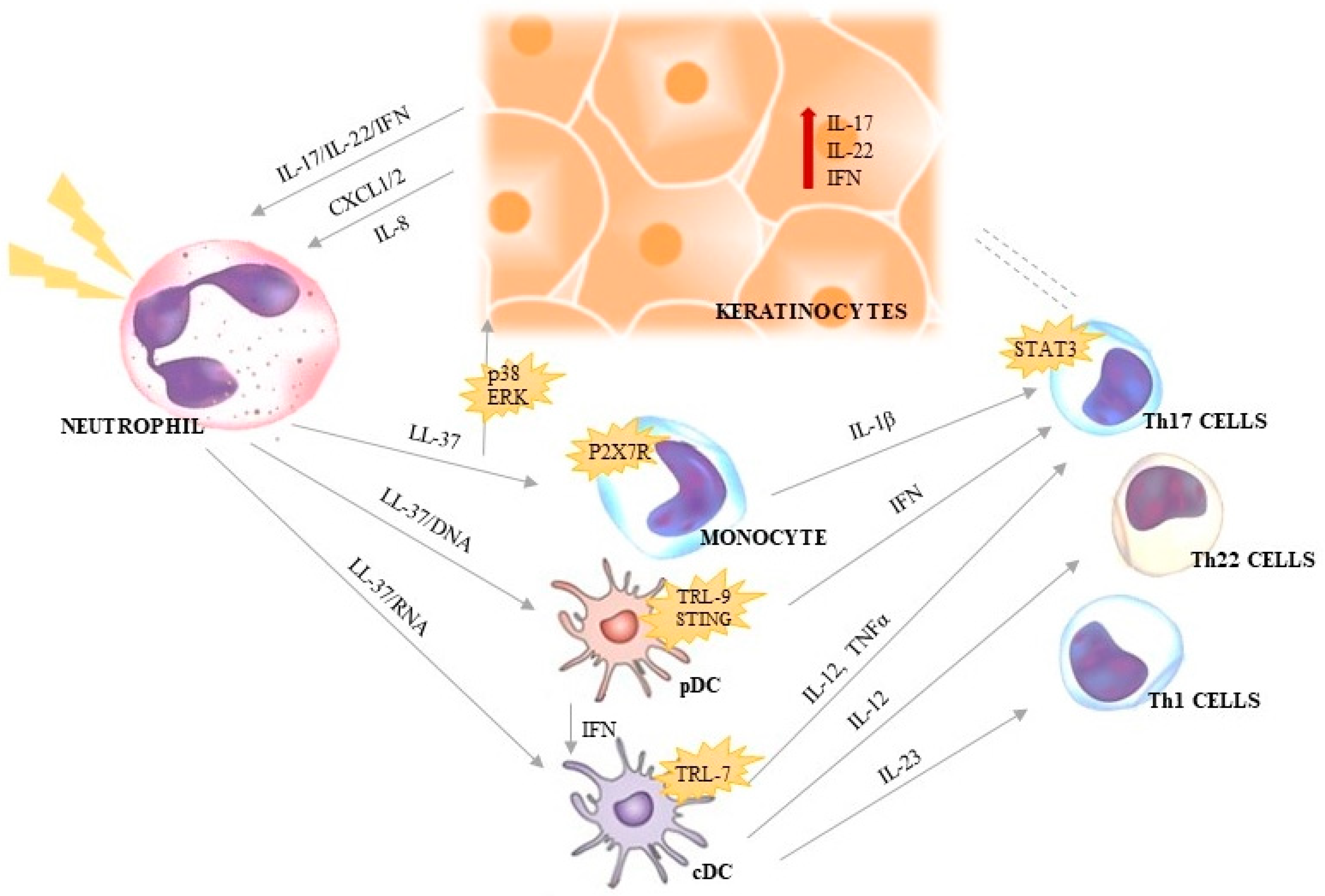
4. Neutrophils and NETs in Psoriasis
4.1. LL-37
4.2. Serine Proteases
4.3. Peptidyl Arginine Deiminase 4 (PAD-4)
4.4. Additional Triggers of NETs
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Carmona-Rivera, C.; Kaplan, M.J. Low-density granulocytes: A distinct class of neutrophils in systemic autoimmunity. Semin Immunopathol. 2013, 35, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Sheshachalam, A.; Srivastava, N.; Mitchell, T.; Lacy, P.; Eitzen, G. Granule protein processing and regulated secretion in neutrophils. Front Immunol. 2014, 19, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacy, P. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin. Immunol. 2006, 2, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Kahlenberg, J.M.; Kaplan, M.J. Little peptide, big effects: The role of LL-37 in inflammation and autoimmune disease. J Immunol. 2013, 191, 4895–4901. [Google Scholar] [CrossRef] [Green Version]
- Schon, M.P.; Broekaert, S.M.; Erpenbeck, L. Sexy again: The renaissance of neutrophils in psoriasis. Exp. Dermatol. 2017, 26, 305–311. [Google Scholar] [CrossRef] [Green Version]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef] [PubMed]
- Cowland, J.B.; Borregaard, N. Granulopoiesis and granules of human neutrophils. Immunol. Rev. 2016, 273, 11–28. [Google Scholar] [CrossRef]
- Yin, C.; Heit, B. Armed for destruction: Formation, function and trafficking of neutrophil granules. Cell Tissue Res. 2018, 371, 455–471. [Google Scholar] [CrossRef]
- Ueki, S.; Konno, Y.; Takeda, M.; Moritoki, Y.; Hirokawa, M.; Matsuwaki, Y.; Honda, K.; Ohta, N.; Yamamoto, S.; Takagi, Y.; et al. Eosinophil extracellular trap cell deathderived DNA traps: Their presence in secretions and functional attributes. J. Allergy Clin. Immunol. 2016, 137, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, S.; Morshed, M.; Amini, P.; Stojkov, D.; Simon, D.; von Gunten, S.; Kaufmann, T.; Simon, H.U. Basophils exhibit antibacterial activity through extracellular trap formation. Allergy 2015, 70, 1184–1188. [Google Scholar] [CrossRef]
- Daniel, C.; Leppkes, M.; Muñoz, L.E.; Schley, G.; Schett, G.; Herrmann, M. Extracellular DNA traps in inflammation, injury and healing. Nat. Rev. Nephrol. 2019, 15, 559–575. [Google Scholar] [CrossRef]
- Von Kockritz-Blickwede, M.; Goldmann, O.; Thulin, P.; Heinemann, K.; Norrby-Teglund, A.; Rohde, M.; Medina, E. Phagocytosisindependent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 2008, 111, 3070–3080. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, X.; Li, G.; Gong, P.; Zhang, X.; Yang, Z.; Yang, J.; Li, J. Mouse macrophages capture and kill Giardia lamblia by means of releasing extracellular trap. Dev. Comp. Immunol. 2018, 88, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Rawat, S.; Vrati, S.; Banerjee, A. Neutrophils at the crossroads of acute viral infections and severity. Mol. Aspects Med. 2021, 81, 100996. [Google Scholar] [CrossRef] [PubMed]
- Vorobjeva, N.V.; Pinegin, B.V. Neutrophil extracellular traps: Mechanisms of formation and role in health and disease. Biochemistry 2014, 79, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Vorobjeva, N.V.; Chernyak, B.V. NETosis: Molecular Mechanisms, Role in Physiology and Pathology. Biochemistry 2020, 85, 1178–1190. [Google Scholar] [CrossRef]
- Ravindran, M.; Khan, M.A.; Palaniyar, N. Neutrophil extracellular trap formation: Physiology, pathology and pharmacology. Biomolecules 2019, 9, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, S.; Urosevic, M.; Daryadel, A.; Oberholzer, P.A.; Baumann, C.; Fey, M.F.; Dummer, R.; Simon, H.; Yousefi, S. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J. Biol. Chem. 2004, 279, 44123–44132. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.K.; Hasler, P.; Holzgreve, W.; Gebhardt, S.; Hahn, S. Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum. Immunol. 2005, 66, 1146–1154. [Google Scholar] [CrossRef]
- Munoz, L.E.; Bilyy, R.; Biermann, M.H.C.; Kienhöfer, D.; Maueröder, C.; Hahn, J.; Brauner, J.M.; Weidner, D.; Chen, J.; Scharin-Mehlmann, M.; et al. Nanoparticles size-dependently initiate self-limiting NETosis-driven inflammation. Proc. Natl. Acad. Sci. USA 2016, 113, E5856–E5865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, E.F.; Herzig, A.; Kruger, R.; Muth, A.; Mondal, S.; Thompson, P.R. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife 2017, 6, e24437. [Google Scholar] [CrossRef] [PubMed]
- Cooper, P.R.; Palmer, L.J.; Chapple, I.L.C. Neutrophil extracellular traps as a new paradigm in innate immunity: Friend or foe? Perioodontology 2000 2013, 63, 165–197. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [Green Version]
- Kienhofer, D.; Hahn, J.; Stoof, J.; Csepregi, J.Z.; Reinwald, C.; Urbonaviciute, V.; Johnsson, C.; Maueröder, C.; Podolska, M.J.; Biermann, M.H.; et al. Experimental lupus is aggravated in mouse strains with impaired induction of neutrophil extracellular traps. JCI Insight 2017, 2, 92920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Alcázar, M.; Napirei, M.; Panda, R.; Köhler, E.C.; Kremer Hovinga, J.A.; Mannherz, H.G.; Peine, S.; Renné, T.; Lämmle, B.; Fuchs, T.A. Impaired DNase1-mediated degradation of neutrophil extracellular traps is associated with acute thrombotic microangiopathies. J. Thromb. Haemost. 2015, 13, 732–742. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Alcazar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, J.H.O.; Enk, A.H. Neutrophil extracellular traps in dermatology: Caught in the NET. J. Dermatol. Sci. 2016, 84, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.C.; Cheng, W.J.; Korinek, M.; Lin, C.Y.; Hwang, T.L. Neutrophils in Psoriasis. Front Immunol. 2019, 9, 2376. [Google Scholar] [CrossRef]
- Greb, J.E.; Goldminz, A.M.; Elder, J.T.; Lebwohl, M.G.; Gladman, D.D.; Wu, J.J. Psoriasis. Nat. Rev. Dis. Primers 2016, 2, 16082. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.C.; Yu, H.S.; Yen, F.L.; Lin, C.L.; Chen, G.S.; Lan, C.C. Neutrophil extracellular trap formation is increased in psoriasis and induces human beta-defensin-2 production in epidermal keratinocytes. Sci Rep. 2016, 6, 31119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sticherling, M. Psoriasis and autoimmunity. Autoimmun Rev. 2016, 15, 1167–1170. [Google Scholar] [CrossRef] [PubMed]
- Prinz, J.C. Autoimmune aspects of psoriasis: Heritability and autoantigens. Autoimmun. Rev. 2017, 16, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, R.; Altomare, G.; Marchesoni, A.; Balato, N.; Matucci Cerinic, M.; Lotti, T.; Olivieri, I.; Vena, G.A.; Salvarani, C.; Valesini, G.; et al. Psoriatic disease: Concepts and implications. J. Eur. Acad. Dermatol. Venereol. 2010, 24, 627–630. [Google Scholar] [CrossRef]
- Ganguly, D.; Chamilos, G.; Lande, R.; Gregorio, J.; Meller, S.; Facchinetti, V. Self-RNAantimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J. Exp. Med. 2009, 206, 1983–1994. [Google Scholar] [CrossRef] [PubMed]
- Zabieglo, K.; Majewski, P.; Majchrzak-Gorecka, M.; Wlodarczyk, A.; Grygier, B.; Zegar, A. The inhibitory effect of secretory leukocyte protease inhibitor (SLPI) on formation of neutrophil extracellular traps. J. Leukoc. Biol. 2015, 98, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Skrzeczynska-Moncznik, J.; Zabieglo, K.; Bossowski, J.P.; Osiecka, O.; Wlodarczyk, A.; Kapinska-Mrowiecka, M.; Kwitniewski, M.; Majewski, P.; Dubin, A.; Cichy, J. Eosinophils Regulate Interferon Alpha Production in Plasmacytoid Dendritic Cells Stimulated with Components of Neutrophil Extracellular Traps. J. Interferon Cytokine Res. 2017, 37, 119–128. [Google Scholar] [CrossRef]
- Bergen, L.L.; Petrovic, A.; Aarebrot, A.K.; Appel, S. The TNF/IL-23/IL-17 axis-Head-to-head trials comparing different biologics in psoriasis treatment. Scand. J. Immunol. 2020, 92, e12946. [Google Scholar] [CrossRef]
- Toichi, E.; Tachibana, T.; Furukawa, F. Rapid improvement of psoriasis vulgaris during drug-induced agranulocytosis. J. Am. Acad. Dermatol. 2000, 43, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Naik, H.B.; Natarajan, B.; Stansky, E.; Ahlman, M.A.; Teague, H.; Salahuddin, T.; Ng, Q.; Joshi, A.A.; Krishnamoorthy, P.; Dave, J.; et al. Severity of psoriasis associates with aortic vascular inflammation detected by FDG PET/CT and neutrophil activation in a prospective observational study. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2667–2676. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Umezawa, Y.; Yamagiwa, A.; Saeki, H.; Kondo, M.; Gabazza, E.C. Biologic therapy improves psoriasis by decreasing the activity of monocytes and neutrophils. J. Dermatol. 2014, 41, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Lu, J.; Allan, B.W.; Tang, Y.; Tetreault, J.; Chow, C.; Barmettler, B.; Nelson, J.; Bina, H.; Huang, L.; et al. Generation and characterization of ixekizumab, a humanized monoclonal antibody that neutralizes interleukin-17A. J. Inflamm. Res. 2016, 9, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Teague, H.L.; Varghese, N.J.; Tsoi, L.C.; Dey, A.K.; Garshick, M.S.; Silverman, J.I.; Baumer, Y.; Harrington, C.L.; Stempinski, E.; Elnabawi, Y.A.; et al. Neutrophil Subsets, Platelets, and Vascular Disease in Psoriasis. JACC Basic Transl. Sci. 2019, 4, 1–14. [Google Scholar] [CrossRef]
- Denny, M.F.; Yalavarthi, S.; Zhao, W.; Thacker, S.G.; Anderson, M.; Sandy, A.R.; McCune, W.J.; Kaplan, M.J. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J. Immunol. 2010, 184, 3284–3297. [Google Scholar] [CrossRef] [Green Version]
- Skrzeczynska-Moncznik, J.; Zabieglo, K.; Osiecka, O.; Morytko, A.; Brzoza, P.; Drozdz, L.; Kapinska-Mrowiecka, M.; Korkmaz, B.; Pastuszczak, M.; Kosalka-Wegiel, J.; et al. Differences in Staining for Neutrophil Elastase and its Controlling Inhibitor SLPI Reveal Heterogeneity among Neutrophils in Psoriasis. J. Investig. Dermatol. 2020, 140, 1371–1378.e3. [Google Scholar] [CrossRef] [PubMed]
- Ong, P.Y.; Ohtake, T.; Brandt, C.; Strickland, I.; Boguniewicz, M.; Ganz, T.; Gallo, R.L.; Leung, D.Y.M. Endogenous Antimicrobial Peptides and Skin Infections in Atopic Dermatitis. N. Engl. J. Med. 2002, 347, 1151–1160. [Google Scholar] [CrossRef] [Green Version]
- Reinholz, M.; Ruzicka, T.; Schauber, J. Cathelicidin LL-37: An Antimicrobial Peptide with a Role in Inflammatory Skin Disease. Ann. Dermatol. 2012, 24, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Nestle, F.O.; Conrad, C.; Tun-Kyi, A.; Homey, B.; Gombert, M.; Boyman, O.; Burg, G.; Liu, J.; Gilliet, M. Plasmacytoid predendritic cells initiate psoriasis through interferon-α production. J. Exp. Med. 2005, 202, 135–143. [Google Scholar] [CrossRef]
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.; Homey, B.; Cao, W.; Wang, Y.; Su, B.; Nestle, F.O.; et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 2007, 449, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Botti, E.; Jandus, C.; Dojcinovic, D.; Fanelli, G.; Conrad, C.; Chamilos, G.; Feldmeyer, L.; Marinari, B.; Chon, S.; et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat. Commun. 2014, 3, 5621. [Google Scholar] [CrossRef]
- Chamilos, G.; Gregorio, J.; Meller, S.; Lande, R.; Kontoyiannis, D.P.; Modlin, R.L.; Gilliet, M. Cytosolic sensing of extracellular self-DNA transported into monocytes by the antimicrobial peptide LL37. Blood 2012, 120, 3699–3707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, G.; Chen, M.; Su, Y.; Xu, L.X.; Zhao, M.H.; Li, K.S. Serum B-cell activating factor in myeloperoxiase-antineutrophil cytoplasmic antibodies-associated vasculitis. Am. J. Med. Sci. 2014, 348, 25–29. [Google Scholar] [CrossRef]
- Kryczek, I.; Bruce, A.T.; Gudjonsson, J.E.; Johnston, A.; Aphale, A.; Vatan, L.; Szeliga, W.; Wang, Y.; Liu, Y.; Welling, T.H.; et al. Induction of IL-17+ T cell trafficking and development by IFN-gamma: Mechanism and pathological relevance in psoriasis. J Immunol. 2008, 181, 4733–4741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowes, M.A.; Kikuchi, T.; Fuentes-Duculan, J.; Cardinale, I.; Zaba, L.C.; Haider, A.S.; Bowman, E.P.; Krueger, J.G. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J. Investig. Dermatol. 2008, 128, 1207–1211. [Google Scholar] [CrossRef]
- Katayama, H. Development of psoriasis by continuous neutrophil infiltration into the epidermis. Exp. Dermatol. 2018, 27, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- Langley, R.G.; Elewski, B.E.; Lebwohl, M.; Reich, K.; Griffiths, C.E.M.; Papp, K.; Puig, L.; Nakagawa, H.; Spelman, L.; Sigurgeirsson, B.; et al. ERASURE Study Group; FIXTURE Study Group. Secukinumab in plaque psoriasis--results of two phase 3 trials. N. Engl. J. Med. 2014, 371, 326–338. [Google Scholar] [CrossRef] [Green Version]
- Papp, K.A.; Menter, A.; Strober, B.; Langley, R.G.; Buonanno, M.; Wolk, R.; Gupta, P.; Krishnaswami, S.; Tan, H.; Harness, J.A. Efficacy and safety of tofacitinib, an oral Janus kinase inhibitor, in the treatment of psoriasis: A Phase 2b randomized placebo-controlled dose-ranging study. Br. J. Dermatol. 2012, 167, 668–677. [Google Scholar] [CrossRef]
- Zheng, Y.; Niyonsaba, F.; Ushio, H.; Nagaoka, I.; Ikeda, S.; Okumura, K.; Ogawa, H. Cathelicidin LL-37 induces the generation of reactive oxygen species and release of human alpha-defensins from neutrophils. Br. J. Dermatol. 2007, 157, 1124–1131. [Google Scholar] [CrossRef]
- Chamorro, C.I.; Weber, G.; Gronberg, A.; Pivarcsi, A.; Stahle, M. The Human Antimicrobial Peptide LL-37 Suppresses Apoptosis in Keratinocytes. J. Investig. Dermatol. 2008, 129, 937–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Domizio, J.; Gilliet, M. Psoriasis Caught in the NET. J. Investig. Dermatol. 2019, 139, 1426–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombrowski, Y.; Peric, M.; Koglin, S.; Kammerbauer, C.; Göβ, C.; Anz, D.; Simanski, M.; Gläser, R.; Harder, J.; Hornung, V.; et al. Cytosolic DNA Triggers Inflammasome Activation in Keratinocytes in Psoriatic Lesions. Sci. Transl. Med. 2011, 3, 82ra38. [Google Scholar] [CrossRef] [Green Version]
- Herster, F.; Bittner, Z.; Archer, N.K.; Dickhöfer, S.; Eisel, D.; Eigenbrod, T.; Knorpp, T.; Schneiderhan-Marra, N.; Löffler, M.W.; Kalbacher, H.; et al. Neutrophil extracellular trap-associated RNA and LL37 enable self-amplifying inflammation in psoriasis. Nat. Commun. 2020, 11, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, R.; Xian, D.; Xiong, X.; Yang, L.; Song, J.; Zhong, J. Proanthocyanidins: Novel treatment for psoriasis that reduces oxidative stress and modulates Th17 and Treg cells. Redox Rep. 2018, 23, 130–135. [Google Scholar] [CrossRef] [Green Version]
- Min, M.S.; Wu, J.; He, H.; Sanz-Cabanillas, J.L.; Del Duca, E.; Zhang, N.; Renert-Yuval, Y.; Pavel, A.B.; Lebwohl, M.; Guttman-Yassky, E. Granuloma annulare skin profile shows activation of T-helper cell type 1, T-helper cell type 2, and Janus kinase pathways. J. Am. Acad. Dermatol. 2020, 83, 63–70. [Google Scholar] [CrossRef]
- Liang, Y.; Xing, X.; Beamer, M.A.; Swindell, W.R.; Sarkar, M.K.; Roberts, L.W.; Voorhees, J.J.; Kahlenberg, J.M.; Harms, P.W.; Johnston, A.; et al. Six-transmembrane epithelial antigens of the prostate comprise a novel inflammatory nexus in patients with pustular skin disorders. J Allergy Clin. Immunol. 2017, 139, 1217–1227. [Google Scholar] [CrossRef]
- Henry, A.L.; Kyle, S.D.; Bhandari, S.; Chisholm, A.; Griffiths, C.E.; Bundy, C. Measurement, Classification and Evaluation of Sleep Disturbance in Psoriasis: A Systematic Review. PLoS ONE 2016, 11, e0157843. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.Y.; Soler, D.C.; Debanne, S.M.; Grozdev, I.; Rodriguez, M.E.; Feig, R.L.; Carman, T.L.; Gilkeson, R.C.; Orringer, C.E.; Kern, E.; et al. Psoriasis and cardiovascular risk factors: Increased serum myeloperoxidase and corresponding immunocellular overexpression by Cd11b+ CD68+ macrophages in skin lesions. Am. J. Transl. Res. 2014, 6, 16–27. [Google Scholar]
- Vergnano, M.; Mockenhaupt, M.; Benzian-Olsson, N.; Paulmann, M.; Grys, K.; Mahil, S.K.; Chaloner, C.; Barbosa, I.A.; August, S.; Burden, A.D.; et al. Loss-of-Function Myeloperoxidase Mutations Are Associated with Increased Neutrophil Counts and Pustular Skin Disease. Am. J. Hum. Genet. 2020, 107, 539–543. [Google Scholar] [CrossRef]
- Haskamp, S.; Bruns, H.; Hahn, M.; Hoffmann, M.; Gregor, A.; Löhr, S.; Hahn, J.; Schauer, C.; Ringer, M.; Flamann, C.; et al. Myeloperoxidase Modulates Inflammation in Generalized Pustular Psoriasis and Additional Rare Pustular Skin Diseases. Am. J. Hum. Genet. 2020, 107, 527–538. [Google Scholar] [CrossRef]
- Gordon, R.A.; Herter, J.A.; Rosetti, F.; Campbell, A.M.; Nishi, H.; Kashgarian, M.; Bastacky, S.I.; Marinov, A.; Nickerson, K.M.; Mayadas, T.N.; et al. Lupus and proliferative nephritis are PAD4 independent in murine models. JCI Insight 2017, 2, e92926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrbach, A.S.; Hemmers, S.; Arandjelovic, S.; Corr, M.; Mowen, K.A. PAD4 is not essential for disease in the K/BxN murine autoantibody-mediated model of arthritis. Arthritis Res. Ther. 2012, 14, R104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef]
- Hemmers, S.; Teijaro, J.R.; Arandjelovic, S.; Mowen, K.A. PAD4- mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS ONE. 2011, 6, e22043. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Iwata, Y.; Fukushima, H.; Saito, K.; Tanaka, Y.; Hasegawa, Y.; Akiyama, M.; Sugiura, K. Neutrophil extracellular traps are induced in a psoriasis model of interleukin-36 receptor antagonist-deficient mice. Sci. Rep. 2020, 10, 20149. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Kronbichler, A.; Park, D.D.; Park, Y.; Moon, H.; Kim, H.; Choi, J.H.; Choi, J.; Shim, S.; Lyu, I.S.; et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: A comprehensive review. Autoimmun. Rev. 2017, 16, 1160–1173. [Google Scholar] [CrossRef]
- Skrzeczynska-Moncznik, J.; Wlodarczyk, A.; Zabieglo, K.; Kapinska-Mrowiecka, K.; Marewicz, E.; Dubin, A.; Potempa, J.; Cichy, J. Secretory leukocyte proteinase inhibitor-competent DNA deposits are potent stimulators of plasmacytoid dendritic cells: Implication for psoriasis. J. Immunol. 2012, 189, 1611–1617. [Google Scholar] [CrossRef] [Green Version]
- Wolk, K.; Frambach, Y.; Jacobi, A.; Wilsmann-Theis, D.; Phillipp, S.; WitteHandel, E.; Wenzel, J.; Mössner, R.; Sabat, R. Increased levels of lipocalin 2 in palmoplantar pustular psoriasis. J. Dermatol. Sci. 2018, 90, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Kolls, J.K. Interluekin-17A (IL17A). Gene 2017, 614, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.; Hambro, C.A.; Johnston, A.; Stuart, P.E.; Tsoi, L.C.; Nair, R.P.; Elder, J.T. Neutrophil Extracellular Traps Induce Human Th17 Cells: Effect of Psoriasis-Associated TRAF3IP2 Genotype. J. Investig. Dermatol. 2019, 139, 1245–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Wang, N.; Xu, Y.; Tan, H.; Li, S.; Feng, Y. Molecular Mechanisms Involved in Oxidative Stress-Associated Liver Injury Induced by Chinese Herbal Medicine: An Experimental Evidence-Based Literature Review and Network Pharmacology Study. Int. J. Mol. Sci. 2018, 19, 2745. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Type of Granules | Other Name | Type of Proteins | Function of Proteins |
|---|---|---|---|
| Azurophilic | Peroxidase-positive or primary | myeloperoxidase, defensins, lysozyme, alkaline phosphatase, hydrolases, phospholipases (A2,C,D), bacterial permeability increasing protein, peroxidase 3, elastase, cathepsin G | antimicrobial activity |
| Specific | secondary | lactoferrin, lysozyme, alkaline phosphatase, NADPH oxidase, cathelicidin, collagenase | migration, antimicrobial activity |
| Gelatinase | tertiary | cathepsin, gelatinase, lipocalin, collagenase, cytochrome b558, ficolin | exocytosis, extracellular matrix degradation |
| Secretory | - | alkaline phosphatase | neutrophil recruitment (early stages of inflammatory response) |
| Ficolin-1 rich (gelatinase poor) | - | ficolin | lectin-initiated complement pathway |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czerwińska, J.; Owczarczyk-Saczonek, A. The Role of the Neutrophilic Network in the Pathogenesis of Psoriasis. Int. J. Mol. Sci. 2022, 23, 1840. https://doi.org/10.3390/ijms23031840
Czerwińska J, Owczarczyk-Saczonek A. The Role of the Neutrophilic Network in the Pathogenesis of Psoriasis. International Journal of Molecular Sciences. 2022; 23(3):1840. https://doi.org/10.3390/ijms23031840
Chicago/Turabian StyleCzerwińska, Joanna, and Agnieszka Owczarczyk-Saczonek. 2022. "The Role of the Neutrophilic Network in the Pathogenesis of Psoriasis" International Journal of Molecular Sciences 23, no. 3: 1840. https://doi.org/10.3390/ijms23031840
APA StyleCzerwińska, J., & Owczarczyk-Saczonek, A. (2022). The Role of the Neutrophilic Network in the Pathogenesis of Psoriasis. International Journal of Molecular Sciences, 23(3), 1840. https://doi.org/10.3390/ijms23031840