
Obesity and Dyslipidemia Synergistically Exacerbate Psoriatic Skin Inflammation

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
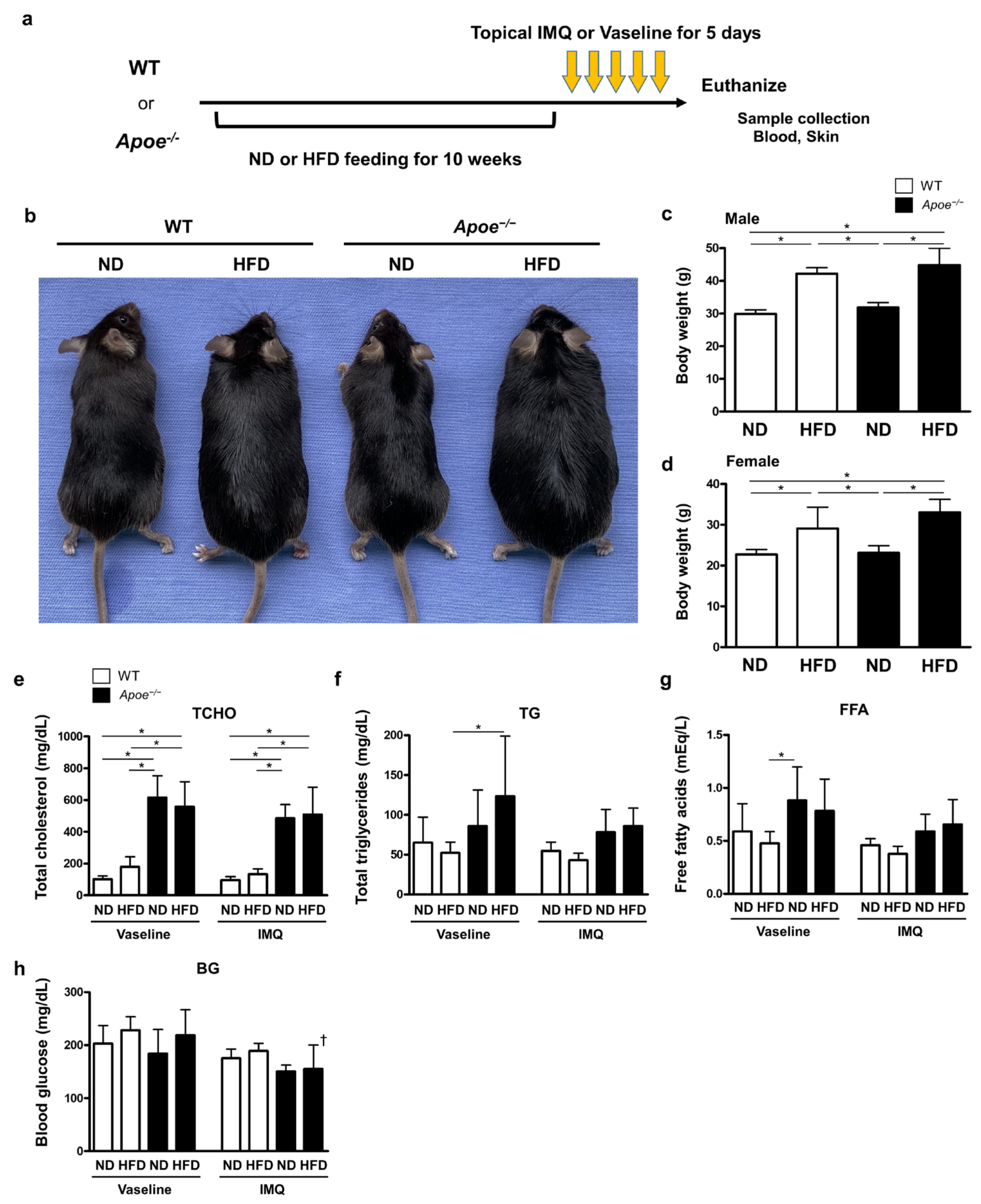
2.1. High-Fat-Diet-Induced Obesity and Apoe-Deficiency-Induced Dyslipidemia Synergistically Augment Psoriatic Skin Inflammation
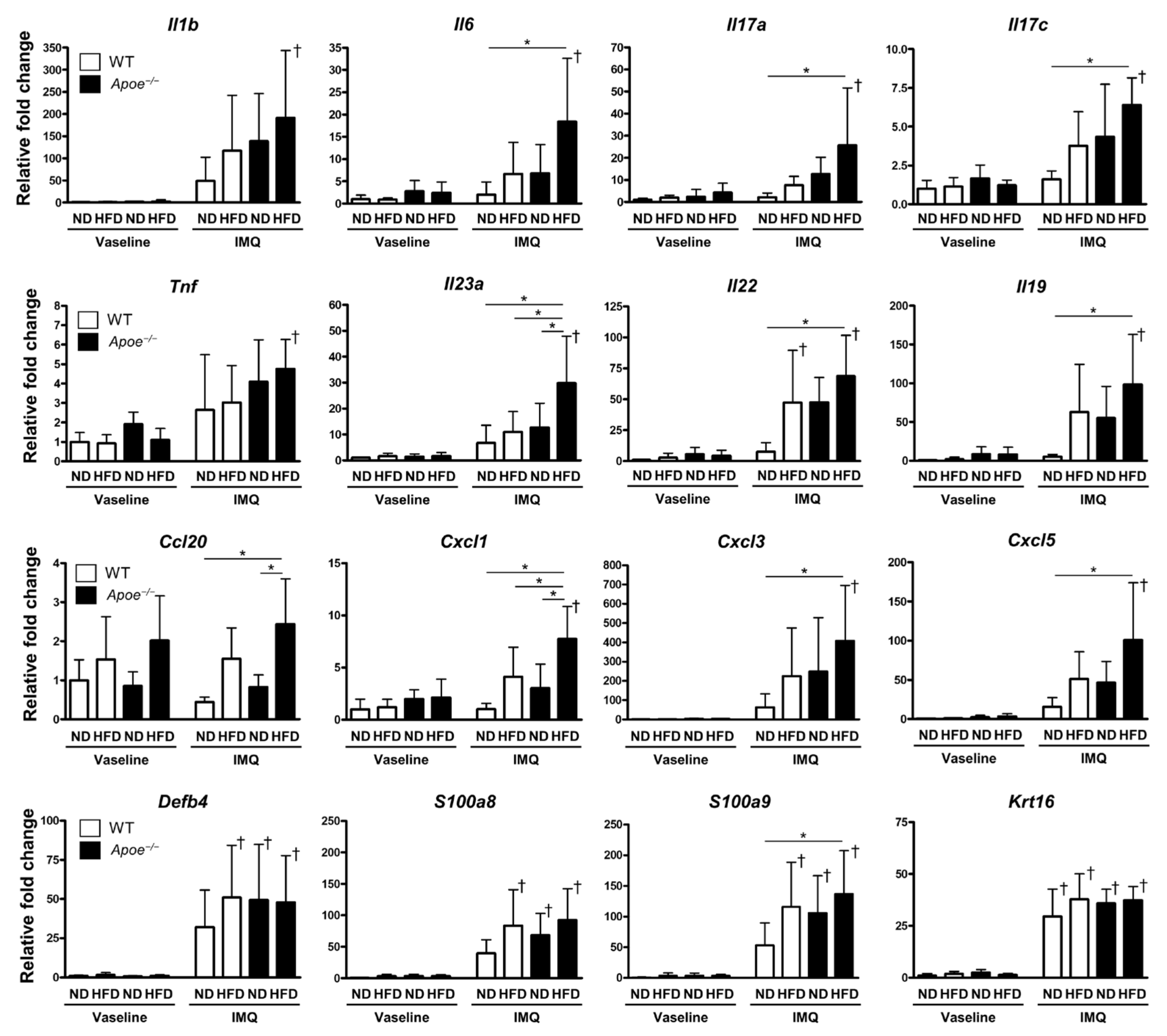
2.2. Coexistence of Obesity and Dyslipidemia Enhances Inflammatory Gene Expression in Psoriatic Lesions
2.3. Increased Serum Leptin Levels in HFD-Fed Mice
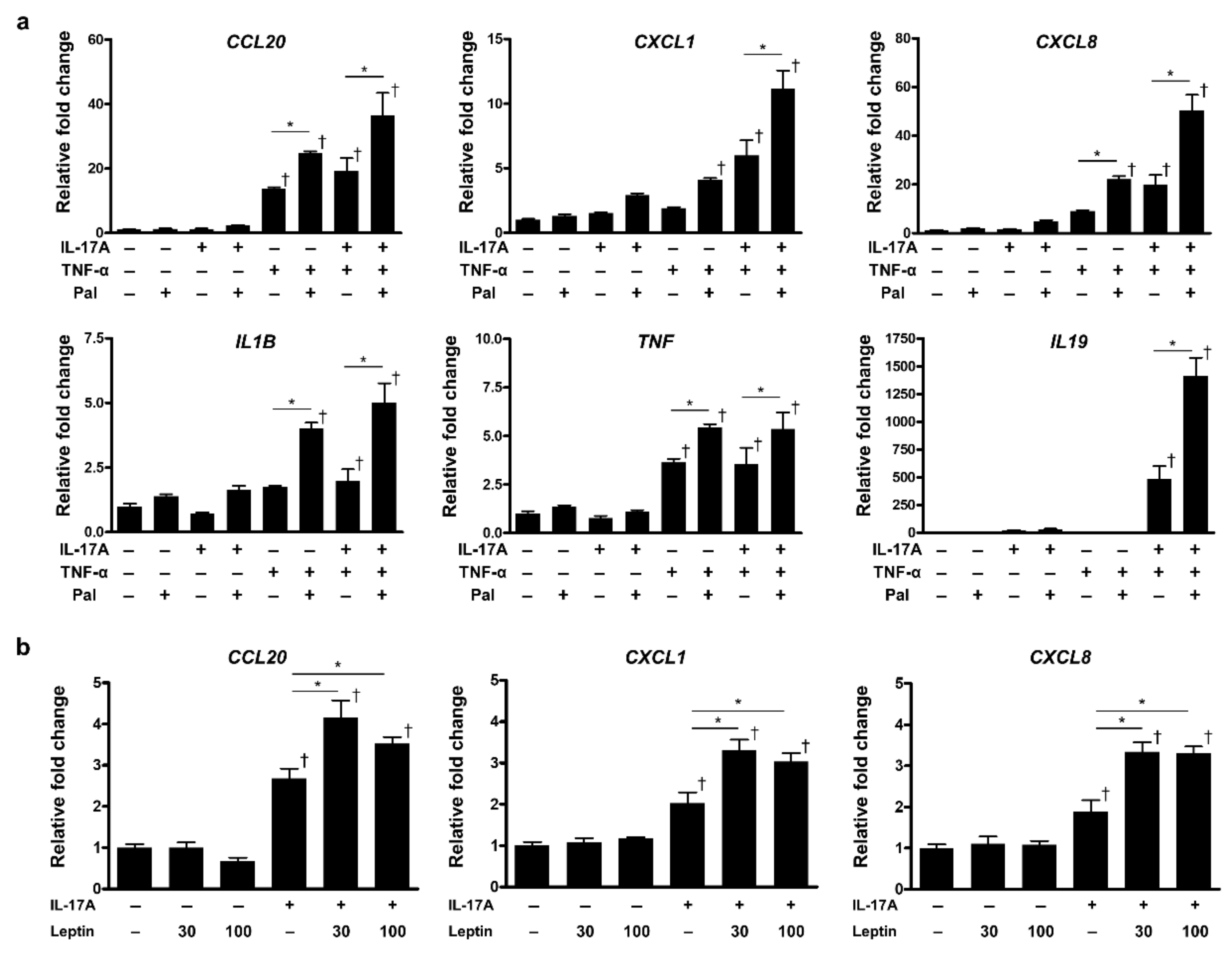
2.4. Palmitic Acid and Leptin Regulate Inflammatory Responses of Epidermal Keratinocytes in the Presence of Inflammatory Cytokines
2.5. Palmitic Acid and Leptin Activate the Phosphorylation of C-Jun N-Terminal Kinase Synergistically with IL-17A
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Mice and Diets
4.3. IMQ-Induced Psoriasis Model
4.4. Histological Analyses of the Ears
4.5. Immunohistochemistry
4.6. Measurement of Biochemical Parameters
4.7. Multiplex Analysis for Adipokines
4.8. ELISA for IL-17A
4.9. Real-Time Quantitative Polymerase Chain Reaction
4.10. Cell Culture and Stimuli
4.11. Western Blotting
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blauvelt, A.; Chiricozzi, A. The Immunologic Role of IL-17 in Psoriasis and Psoriatic Arthritis Pathogenesis. Clin. Rev. Allergy Immunol. 2018, 55, 379–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshita, J.; Grewal, S.; Langan, S.M.; Mehta, N.N.; Ogdie, A.; Van Voorhees, A.S.; Gelfand, J.M. Psoriasis and comorbid diseases. J. Am. Acad. Dermatol. 2017, 76, 377–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, A.W.; Harskamp, C.T.; Armstrong, E.J. The association between psoriasis and obesity: A systematic review and meta-analysis of observational studies. Nutr. Diabetes 2012, 2, e54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.; Harskamp, C.T.; Armstrong, E.J.; Armstrong, A.W. The association between psoriasis and dyslipidaemia: A systematic review. Br. J. Dermatol. 2013, 168, 486–495. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Zorena, K.; Jachimowicz-Duda, O.; Ślęzak, D.; Robakowska, M.; Mrugacz, M. Adipokines and Obesity. Potential Link to Metabolic Disorders and Chronic Complications. Int. J. Mol. Sci. 2020, 21, 3570. [Google Scholar] [CrossRef]
- Ravaut, G.; Légiot, A.; Bergeron, K.-F.; Mounier, C. Monounsaturated Fatty Acids in Obesity-Related Inflammation. Int. J. Mol. Sci. 2020, 22, 330. [Google Scholar] [CrossRef]
- Rogero, M.; Calder, P. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. [Google Scholar] [CrossRef] [Green Version]
- Haczeyni, F.; Bell-Anderson, K.S.; Farrell, G.C. Causes and mechanisms of adipocyte enlargement and adipose expansion. Obes. Rev. 2018, 19, 406–420. [Google Scholar] [CrossRef]
- Guo, Z.; Yang, Y.; Liao, Y.; Shi, Y.; Zhang, L.-J. Emerging Roles of Adipose Tissue in the Pathogenesis of Psoriasis and Atopic Dermatitis in Obesity. JID Innov. 2022, 2, 100064. [Google Scholar] [CrossRef]
- Kyriakou, A.; Patsatsi, A.; Sotiriadis, D.; Goulis, D.G. Serum Leptin, Resistin, and Adiponectin Concentrations in Psoriasis: A Meta-Analysis of Observational Studies. Dermatology 2017, 233, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Kanemaru, K.; Matsuyuki, A.; Nakamura, Y.; Fukami, K. Obesity exacerbates imiquimod-induced psoriasis-like epidermal hyperplasia and interleukin-17 and interleukin-22 production in mice. Exp. Dermatol. 2015, 24, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Nakamizo, S.; Honda, T.; Adachi, A.; Nagatake, T.; Kunisawa, J.; Kitoh, A.; Otsuka, A.; Dainichi, T.; Nomura, T.; Ginhoux, F.; et al. High fat diet exacerbates murine psoriatic dermatitis by increasing the number of IL-17-producing γδ T cells. Sci. Rep. 2017, 7, 14076. [Google Scholar] [CrossRef] [Green Version]
- Herbert, D.; Franz, S.; Popkova, Y.; Anderegg, U.; Schiller, J.; Schwede, K.; Lorz, A.; Simon, J.C.; Saalbach, A. High-fat diet exacerbates early psoriatic skin inflammation independent of obesity: Saturated fatty acids as key players. J. Investig. Dermatol. 2018, 138, 1999–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Li, W.-Q.; Han, J.; Sun, Q.; Qureshi, A.A. Hypercholesterolemia and Risk of Incident Psoriasis and Psoriatic Arthritis in US Women. Arthritis Rheumatol. 2014, 66, 304–310. [Google Scholar] [CrossRef]
- Rucević, I.; Perl, A.; Barisić-Drusko, V.; Adam-Perl, M. The role of the low energy diet in psoriasis vulgaris treatment. Coll. Antropol. 2003, 27 (Suppl. 1), 41–48. [Google Scholar] [PubMed]
- Xie, X.; Zhang, L.; Lin, Y.; Wang, Y.; Liu, W.; Li, X.; Li, P. Imiquimod induced ApoE-deficient mice might be a composite animal model for the study of psoriasis and dyslipideamia comorbidity. J. Dermatol. Sci. 2017, 88, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Snekvik, I.; Nilsen, T.I.L.; Romundstad, P.R.; Saunes, M. Metabolic syndrome and risk of incident psoriasis: Prospective data from the HUNT Study, Norway. Br. J. Dermatol. 2019, 180, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, K.; Tang, N.; Urakami, H.; Kajita, A.; Kobashi, M.; Nomura, H.; Sasakura, M.; Sugihara, S.; Jiang, F.; Tomonobu, N.; et al. Multifaceted Analysis of IL-23A- and/or EBI3-Including Cytokines Produced by Psoriatic Keratinocytes. Int. J. Mol. Sci. 2021, 22, 12659. [Google Scholar] [CrossRef]
- Sano, S.; Chan, K.S.; Carbajal, S.; Clifford, J.; Peavey, M.; Kiguchi, K.; Itami, S.; Nickoloff, B.J.; Digiovanni, J. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat. Med. 2005, 11, 43–49. [Google Scholar] [CrossRef]
- Hawkes, J.E.; Chan, T.C.; Krueger, J.G. Psoriasis pathogenesis and the development of novel targeted immune therapies. J. Allergy Clin. Immunol. 2017, 140, 645–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauffer, F.; Jargosch, M.; Baghin, V.; Krause, L.; Kempf, W.; Absmaier-Kijak, M.; Morelli, M.; Madonna, S.; Marsais, F.; Lepescheux, L.; et al. IL-17C amplifies epithelial inflammation in human psoriasis and atopic eczema. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Furue, K.; Ito, T.; Tsuji, G.; Nakahara, T.; Furue, M. The CCL20 and CCR6 axis in psoriasis. Scand. J. Immunol. 2020, 91, e12846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unamuno, X.; Gómez-Ambrosi, J.; Rodríguez, A.; Becerril, S.; Frühbeck, G.; Catalán, V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur. J. Clin. Investig. 2018, 48, e12997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.E.; Kim, J.M.; Joung, K.H.; Lee, J.H.; You, B.R.; Choi, M.J.; Ryu, M.J.; Ko, Y.B.; Lee, M.A.; Lee, J.; et al. The Roles of Adipokines, Proinflammatory Cytokines, and Adipose Tissue Macrophages in Obesity-Associated Insulin Resistance in Modest Obesity and Early Metabolic Dysfunction. PLoS ONE 2016, 11, e0154003. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.; Pessin, J.E. Adipokines mediate inflammation and insulin resistance. Front. Endocrinol. 2013, 4, 71. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Thomas, T.C.; Storlien, L.H.; Huang, X.F. Development of high fat diet-induced obesity and leptin resistance in C57Bl/6J mice. Int. J. Obes. Relat. Metab. Disord. 2000, 24, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Wang, Q.; Yu, Y.; Zhao, F.; Huang, P.; Zeng, R.; Qi, R.Z.; Li, W.; Liu, Y. Leptin Contributes to the Adaptive Responses of Mice to High-Fat Diet Intake through Suppressing the Lipogenic Pathway. PLoS ONE 2009, 4, e6884. [Google Scholar] [CrossRef] [Green Version]
- Albanesi, C.; Madonna, S.; Gisondi, P.; Girolomoni, G. The Interplay between Keratinocytes and Immune Cells in the Pathogenesis of Psoriasis. Front. Immunol. 2018, 9, 1549. [Google Scholar] [CrossRef] [Green Version]
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic Acid: Physiological Role, Metabolism and Nutritional Implications. Front. Physiol. 2017, 8, 902. [Google Scholar] [CrossRef] [Green Version]
- Morizane, S.; Nomura, H.; Tachibana, K.; Nakagawa, Y.; Iwatsuki, K. The synergistic activities of the combination of tumour necrosis factor-α, interleukin-17A and interferon-γ in epidermal keratinocytes. Br. J. Dermatol. 2018, 179, 496–498. [Google Scholar] [CrossRef] [PubMed]
- Pandit, R.; Beerens, S.; Adan, R.A.H. Role of leptin in energy expenditure: The hypothalamic perspective. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2017, 312, R938–R947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Candia, P.; Prattichizzo, F.; Garavelli, S.; Alviggi, C.; La Cava, A.; Matarese, G. The pleiotropic roles of leptin in metabolism, immunity, and cancer. J. Exp. Med. 2021, 218, e20191593. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Lee, E.; Jin, S.H.; Ahn, S.; Kim, S.O.; Kim, J.; Choi, D.; Lim, K.-M.; Lee, S.-T.; Noh, M. Leptin regulates the pro-inflammatory response in human epidermal keratinocytes. Arch. Dermatol. Res. 2018, 310, 351–362. [Google Scholar] [CrossRef]
- Stjernholm, T.; Ommen, P.; Langkilde, A.; Johansen, C.; Iversen, L.; Rosada, C.; Stenderup, K. Leptin deficiency in mice counteracts imiquimod (IMQ)-induced psoriasis-like skin inflammation while leptin stimulation induces inflammation in human keratinocytes. Exp. Dermatol. 2017, 26, 338–345. [Google Scholar] [CrossRef]
- Shih, C.-M.; Huang, C.-Y.; Wang, K.-H.; Huang, C.-Y.; Wei, P.-L.; Chang, Y.-J.; Hsieh, C.-K.; Liu, K.-T.; Lee, A.-W. Oxidized Low-Density Lipoprotein-Deteriorated Psoriasis Is Associated with the Upregulation of Lox-1 Receptor and Il-23 Expression In Vivo and In Vitro. Int. J. Mol. Sci. 2018, 19, 2610. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, P. The role of adipokines in chronic inflammation. ImmunoTargets Ther. 2016, 5, 47. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Zhang, S.; Wu, R.; Su, X.; Peng, D.; Zhao, M.; Su, Y. New insights into different adipokines in linking the pathophysiology of obesity and psoriasis. Lipids Health Dis. 2019, 18, 171. [Google Scholar] [CrossRef] [Green Version]
- Scotece, M.; Conde, J.; López, V.; Lago, F.; Pino, J.; Gómez-Reino, J.J.; Gualillo, O. Adiponectin and Leptin: New Targets in Inflammation. Basic Clin. Pharmacol. Toxicol. 2014, 114, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Xue, K.; Liu, H.; Jian, Q.; Liu, B.; Zhu, D.; Zhang, M.; Gao, L.; Li, C. Leptin induces secretion of pro-inflammatory cytokines by human keratinocytes in vitro—A possible reason for increased severity of psoriasis in patients with a high body mass index. Exp. Dermatol. 2013, 22, 406–410. [Google Scholar] [CrossRef]
- Visioli, F.; Poli, A. Fatty Acids and Cardiovascular Risk. Evidence, Lack of Evidence, and Diligence. Nutrients 2020, 12, 3782. [Google Scholar] [CrossRef] [PubMed]
- Nakamizo, S.; Honda, T.; Sato, T.; Al Mamun, M.; Chow, Z.; Duan, K.; Lum, J.; Tan, K.J.; Tomari, K.; Sato, R.; et al. High-fat diet induces a predisposition to follicular hyperkeratosis and neutrophilic folliculitis in mice. J. Allergy Clin. Immunol. 2021, 148, 473–485.e410. [Google Scholar] [CrossRef] [PubMed]
- Myśliwiec, H.; Baran, A.; Harasim-Symbor, E.; Myśliwiec, P.; Milewska, A.J.; Chabowski, A.; Flisiak, I. Serum fatty acid profile in psoriasis and its comorbidity. Arch. Dermatol. Res. 2017, 309, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Baumer, Y.; Ng, Q.; Sanda, G.E.; Dey, A.K.; Teague, H.L.; Sorokin, A.V.; Dagur, P.K.; Silverman, J.I.; Harrington, C.L.; Rodante, J.A.; et al. Chronic skin inflammation accelerates macrophage cholesterol crystal formation and atherosclerosis. JCI Insight 2018, 3, e97179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbecki, J.; Bajdak-Rusinek, K. The effect of palmitic acid on inflammatory response in macrophages: An overview of molecular mechanisms. Inflamm. Res. 2019, 68, 915–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Urso, C.J.; Jadeja, V. Saturated Fatty Acids in Obesity-Associated Inflammation. J. Inflamm. Res. 2020, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.H.; Dong, C. Signaling of interleukin-17 family cytokines in immunity and inflammation. Cell. Signal. 2011, 23, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Frühbeck, G. Intracellular signalling pathways activated by leptin. Biochem. J. 2006, 393, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Kant, S.; Standen, C.L.; Morel, C.; Jung, D.Y.; Kim, J.K.; Swat, W.; Flavell, R.A.; Davis, R.J. A Protein Scaffold Coordinates SRC-Mediated JNK Activation in Response to Metabolic Stress. Cell Rep. 2017, 20, 2775–2783. [Google Scholar] [CrossRef] [Green Version]
- Billi, A.C.; Ludwig, J.E.; Fritz, Y.; Rozic, R.; Swindell, W.R.; Tsoi, L.C.; Gruzska, D.; Abdollahi-Roodsaz, S.; Xing, X.; Diaconu, D.; et al. KLK6 expression in skin induces PAR1-mediated psoriasiform dermatitis and inflammatory joint disease. J. Clin. Investig. 2020, 130, 3151–3157. [Google Scholar] [CrossRef]
- Nakajima, K.; Kanda, T.; Takaishi, M.; Shiga, T.; Miyoshi, K.; Nakajima, H.; Kamijima, R.; Tarutani, M.; Benson, J.M.; Elloso, M.M.; et al. Distinct Roles of IL-23 and IL-17 in the Development of Psoriasis-Like Lesions in a Mouse Model. J. Immunol. 2011, 186, 4481–4489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witte, E.; Kokolakis, G.; Witte, K.; Philipp, S.; Doecke, W.-D.; Babel, N.; Wittig, B.M.; Warszawska, K.; Kurek, A.; Erdmann-Keding, M.; et al. IL-19 Is a Component of the Pathogenetic IL-23/IL-17 Cascade in Psoriasis. J. Investig. Dermatol. 2014, 134, 2757–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Wu, X.; Zhou, Y.; Sheng, L.; Jena, P.K.; Han, D.; Wan, Y.-J.Y.; Hwang, S.T. A Western Diet, but Not a High-Fat and Low-Sugar Diet, Predisposes Mice to Enhanced Susceptibility to Imiquimod-Induced Psoriasiform Dermatitis. J. Investig. Dermatol. 2019, 139, 1404–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, Y.; Yamakuchi, M.; Fukushige, T.; Ibusuki, A.; Hashiguchi, T.; Kanekura, T. High-fat diet exacerbates imiquimod-induced psoriasis-like dermatitis in mice. Exp. Dermatol. 2018, 27, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J.; Esser, N.; Paquot, N. Antidiabetic agents: Potential anti-inflammatory activity beyond glucose control. Diabetes Metab. 2015, 41, 183–194. [Google Scholar] [CrossRef]
- Fuhriman, J.M.; Winge, M.C.G.; Haberstock-Debic, H.; Funk, J.O.; Bradshaw, J.M.; Marinkovich, M.P. ITK and RLK Inhibitor PRN694 Improves Skin Disease in Two Mouse Models of Psoriasis. J. Investig. Dermatol. 2018, 138, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, S.; Matsuzaki, H.; Iseki, M.; Nagasu, A.; Hirano, H.; Ishihara, K.; Ueda, N.; Honda, Y.; Horiuchi, T.; Nishikomori, R.; et al. Functional analysis of a novel G87V TNFRSF1A mutation in patients with TNF receptor-associated periodic syndrome. Clin. Exp. Immunol. 2019, 198, 416–429. [Google Scholar] [CrossRef]
- Mukai, T.; Ishida, S.; Ishikawa, R.; Yoshitaka, T.; Kittaka, M.; Gallant, R.; Lin, Y.-L.; Rottapel, R.; Brotto, M.; Reichenberger, E.J.; et al. SH3BP2 Cherubism Mutation Potentiates TNF-α-Induced Osteoclastogenesis via NFATc1 and TNF-α-Mediated Inflammatory Bone Loss. J. Bone Miner. Res. 2014, 29, 2618–2635. [Google Scholar] [CrossRef] [Green Version]
- Mukai, T.; Gallant, R.; Ishida, S.; Kittaka, M.; Yoshitaka, T.; Fox, D.A.; Morita, Y.; Nishida, K.; Rottapel, R.; Ueki, Y. Loss of SH3 Domain-Binding Protein 2 Function Suppresses Bone Destruction in Tumor Necrosis Factor-Driven and Collagen-Induced Arthritis in Mice. Arthritis Rheumatol. 2015, 67, 656–667. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikeda, K.; Morizane, S.; Akagi, T.; Hiramatsu-Asano, S.; Tachibana, K.; Yahagi, A.; Iseki, M.; Kaneto, H.; Wada, J.; Ishihara, K.; et al. Obesity and Dyslipidemia Synergistically Exacerbate Psoriatic Skin Inflammation. Int. J. Mol. Sci. 2022, 23, 4312. https://doi.org/10.3390/ijms23084312
Ikeda K, Morizane S, Akagi T, Hiramatsu-Asano S, Tachibana K, Yahagi A, Iseki M, Kaneto H, Wada J, Ishihara K, et al. Obesity and Dyslipidemia Synergistically Exacerbate Psoriatic Skin Inflammation. International Journal of Molecular Sciences. 2022; 23(8):4312. https://doi.org/10.3390/ijms23084312
Chicago/Turabian StyleIkeda, Kenta, Shin Morizane, Takahiko Akagi, Sumie Hiramatsu-Asano, Kota Tachibana, Ayano Yahagi, Masanori Iseki, Hideaki Kaneto, Jun Wada, Katsuhiko Ishihara, and et al. 2022. "Obesity and Dyslipidemia Synergistically Exacerbate Psoriatic Skin Inflammation" International Journal of Molecular Sciences 23, no. 8: 4312. https://doi.org/10.3390/ijms23084312
APA StyleIkeda, K., Morizane, S., Akagi, T., Hiramatsu-Asano, S., Tachibana, K., Yahagi, A., Iseki, M., Kaneto, H., Wada, J., Ishihara, K., Morita, Y., & Mukai, T. (2022). Obesity and Dyslipidemia Synergistically Exacerbate Psoriatic Skin Inflammation. International Journal of Molecular Sciences, 23(8), 4312. https://doi.org/10.3390/ijms23084312