
Multidimensional Functional Profiling of Human Neuropathogenic FOXG1 Alleles in Primary Cultures of Murine Pallial Precursors

 ,
,  and
and
Abstract
1. Introduction
2. Results
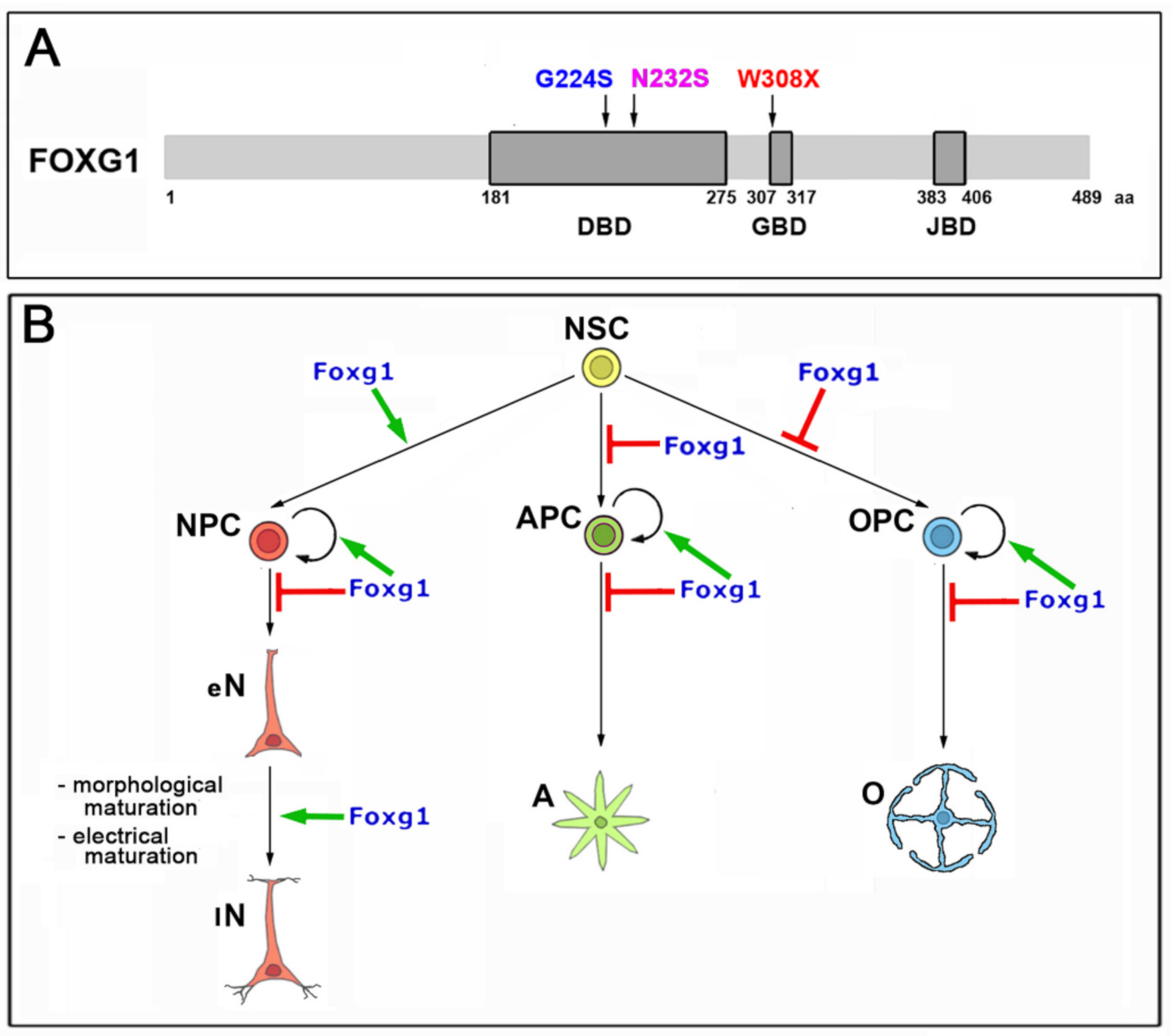
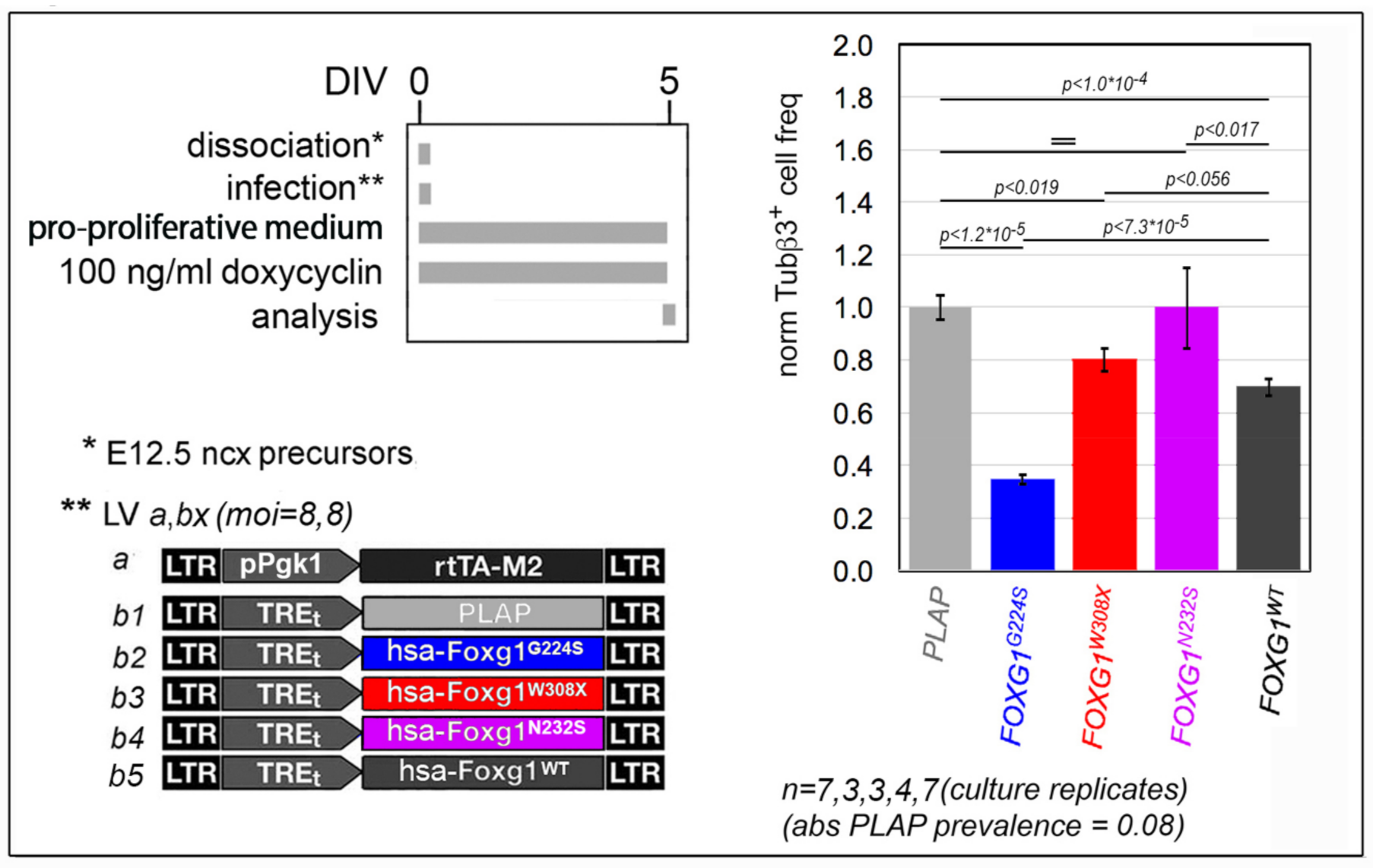
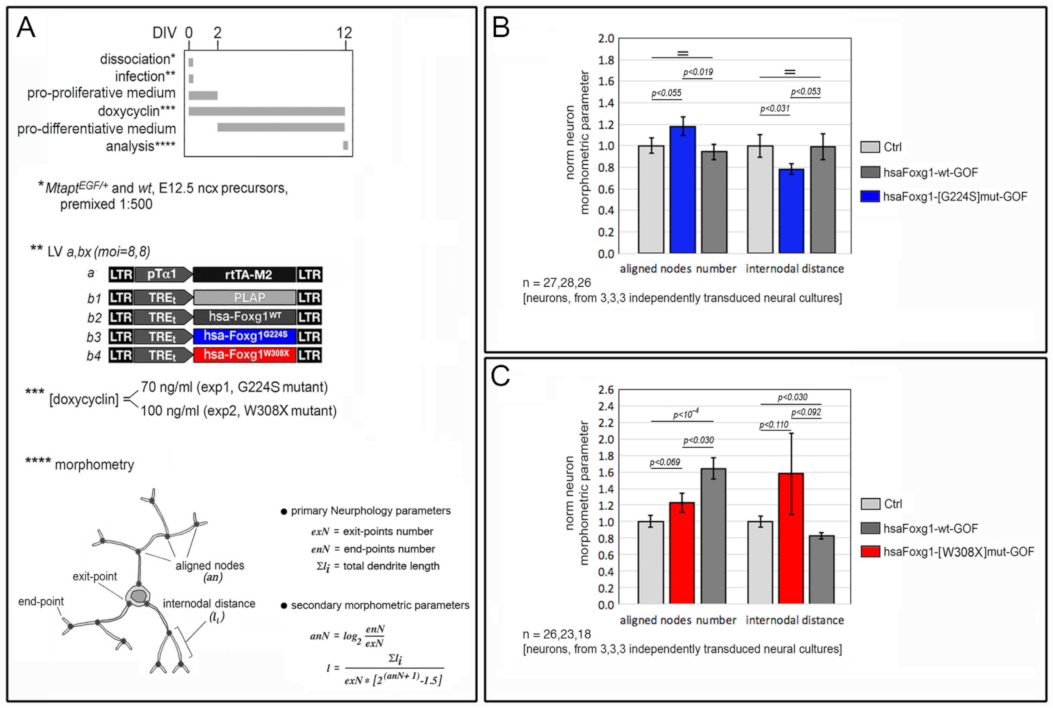
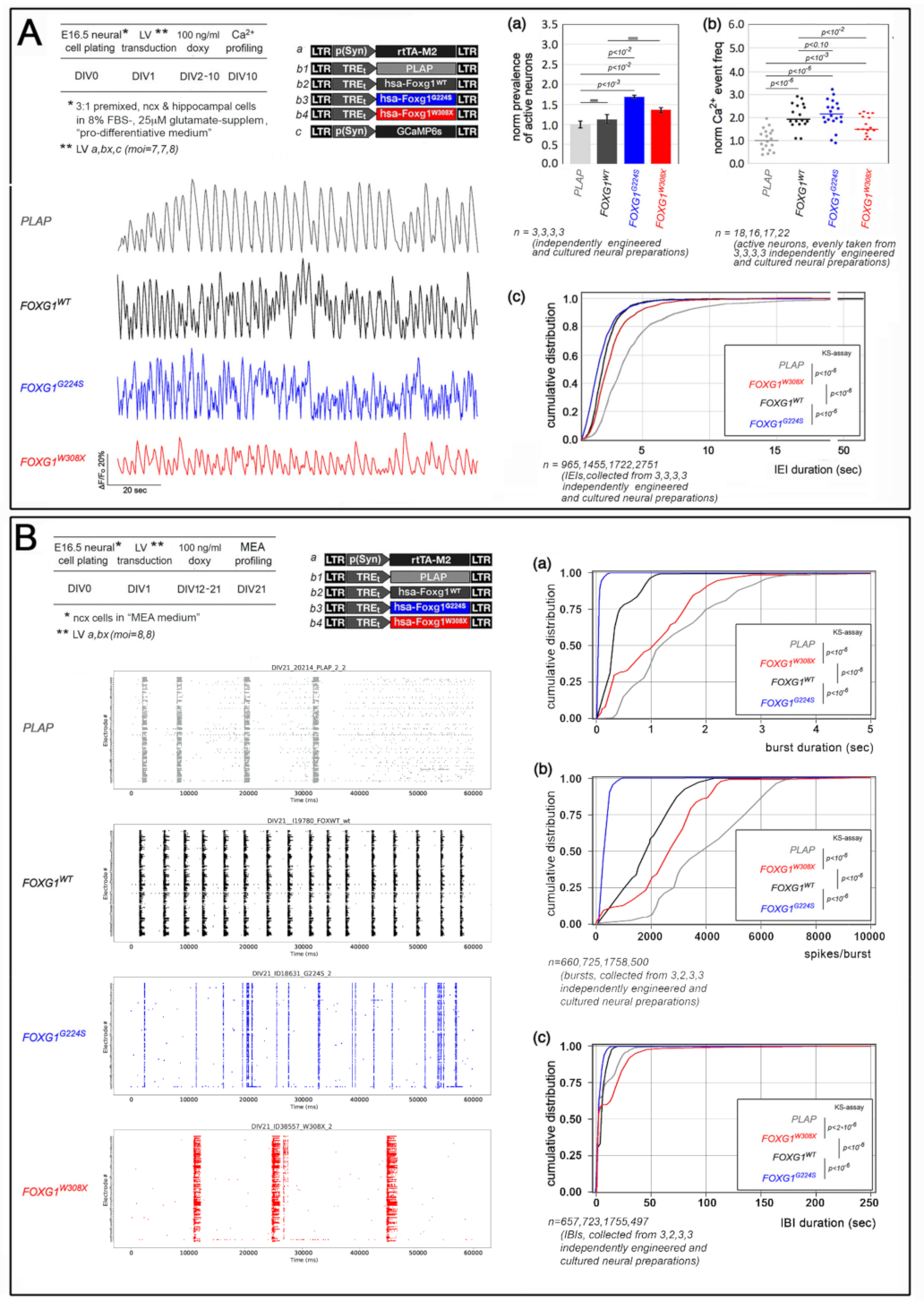
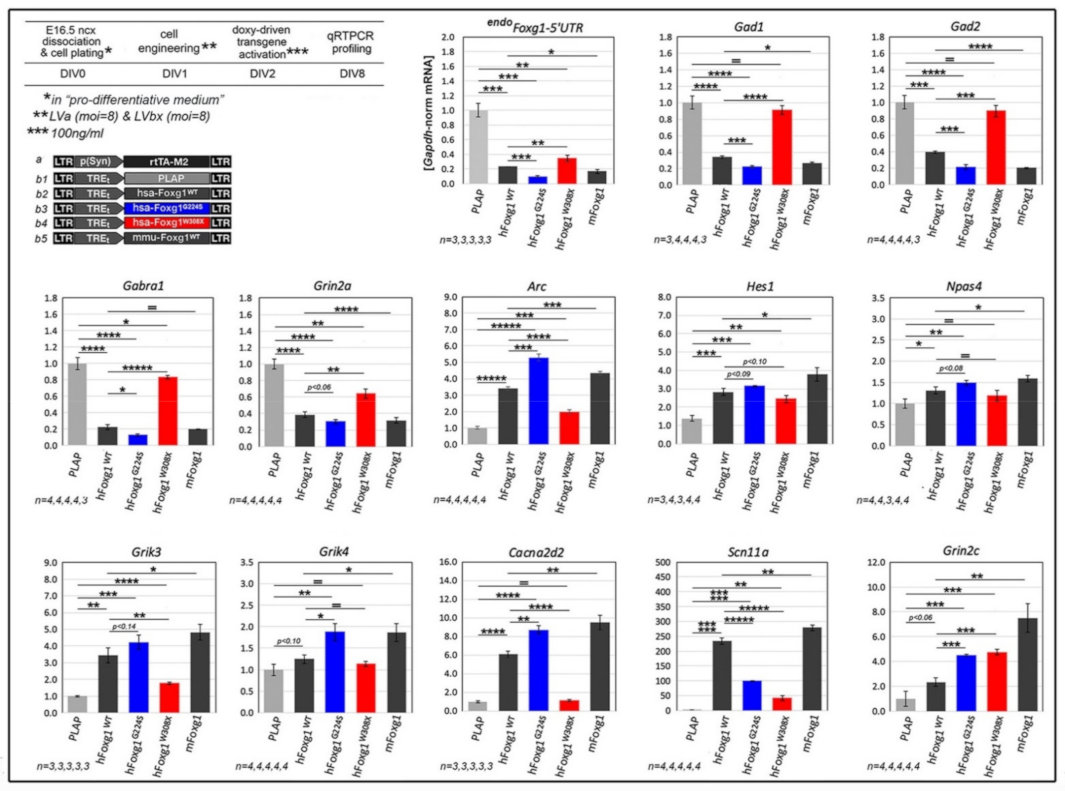
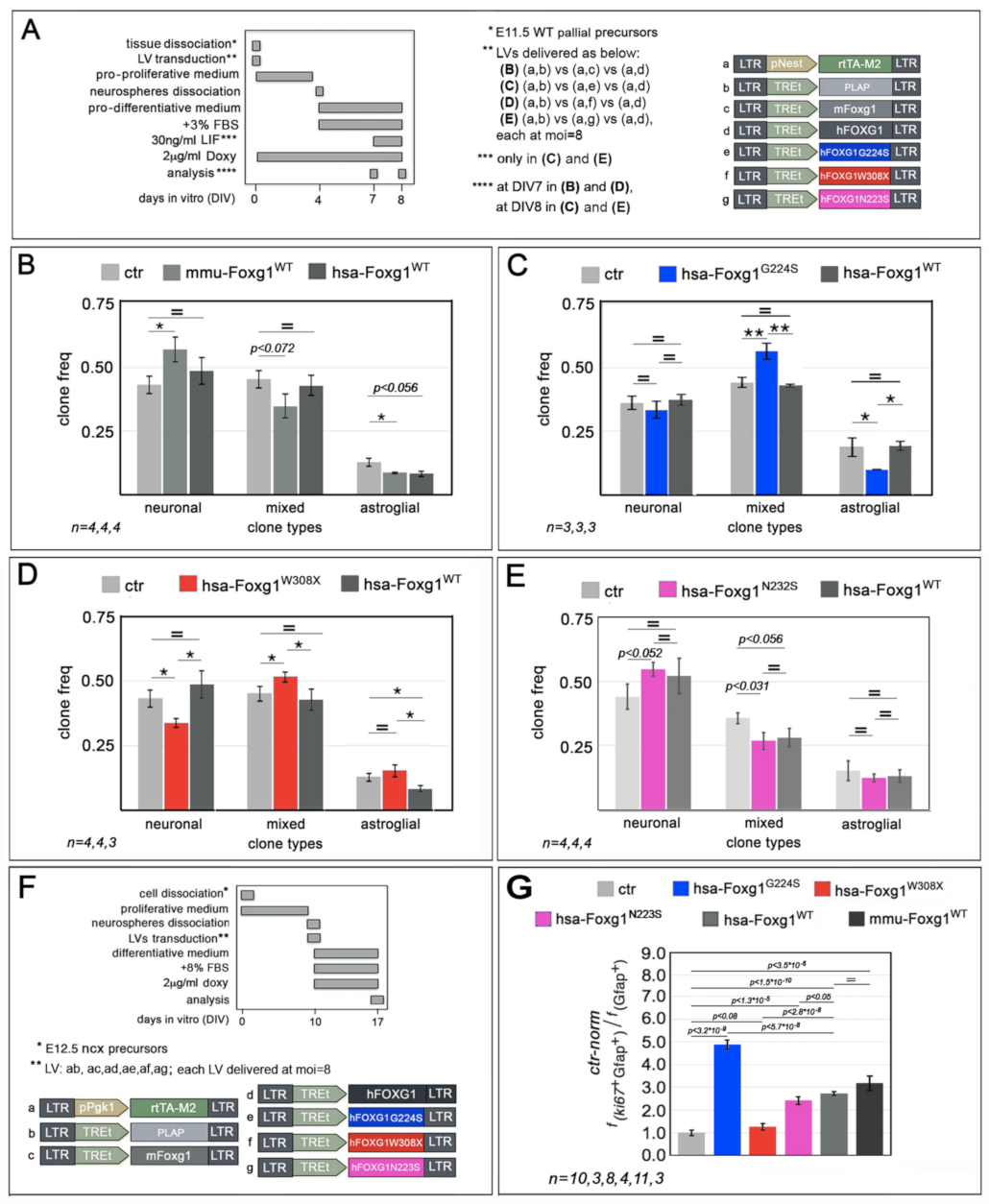
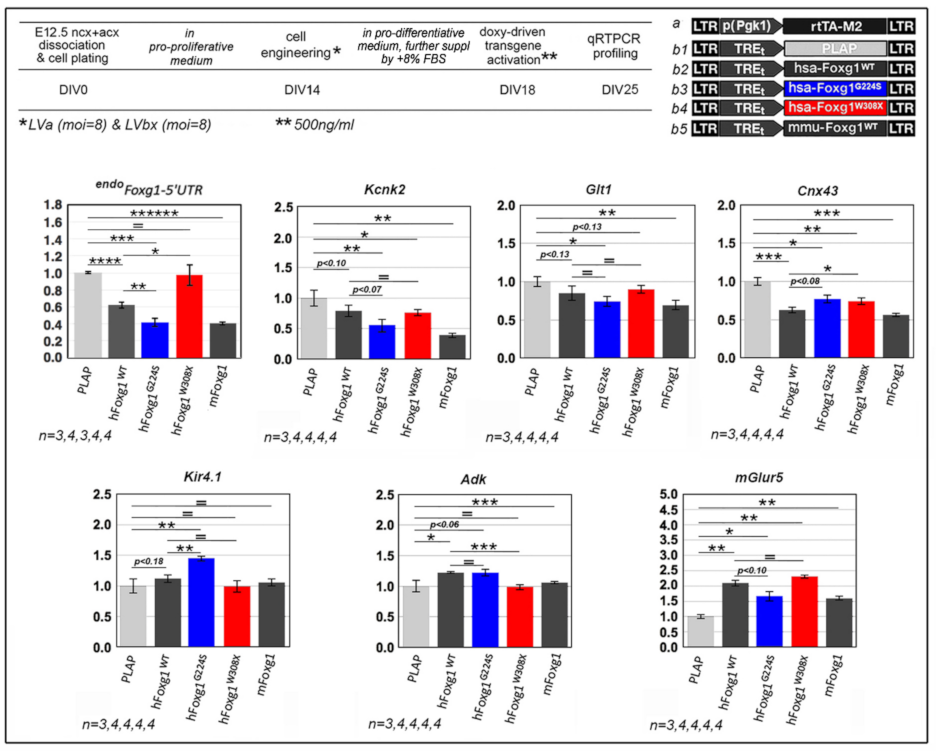
2.1. Functional Characterization of FOXG1G224S, FOXG1W308X and FOXG1N232S along the Neuronogenic Lineage
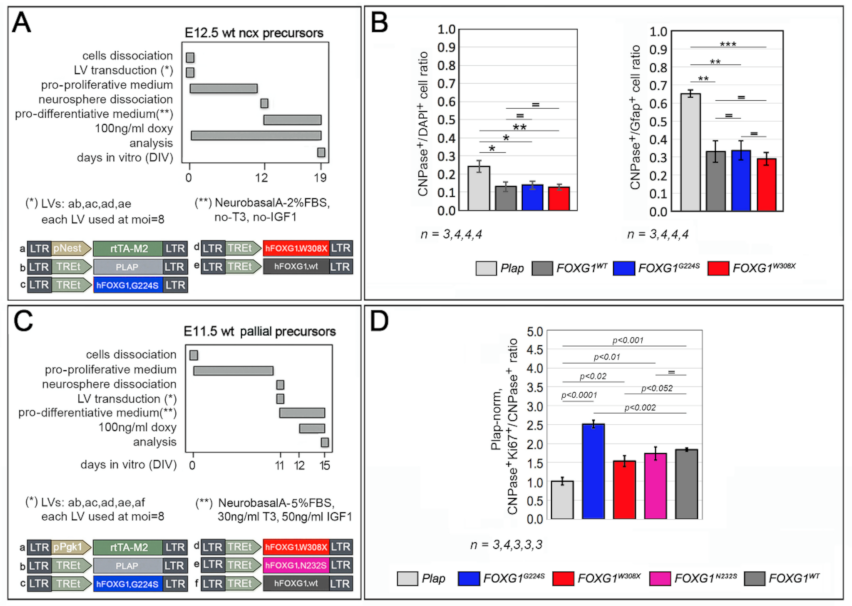
2.2. Impact of FOXG1G224S, FOXG1W308X and FOXG1N232S Alleles on Astrogenesis Progression
2.3. Functional Characterization of FOXG1G224S, FOXG1W308X and FOXG1N232S along the Oligodendrogenic Lineage
3. Discussion
4. Conclusions
- (a)
- Primary murine neural cultures can be succesfully employed for fast, functional multidimensional profiling of novel, human neuropathogenic FOXG1 alleles, propedeutically to timely delivery of appropriate precision therapies;
- (b)
- Among the three mutant FOXG1 alleles subject of the present pilot study, FOXG1G224S (encoding for a protein with altered DBD) and FOXG1W308X (encoding for a protein missing GBD and JBD) generally performed as a GOF and a LOF allele, respectively, while FOXG1N232S (encoding for another protein with altered DBD) acted as a mild LOF allele or phenocopied FOXG1WT;
- (c)
- Although de novo neuropathogenic functions associated to such mutations cannot be ruled out, the concordant functional dissimilarities displayed by each mutant allele, when ranked against its healthy counterpart in a variety of neurodevelopmental and physiological contexts, suggest that the neuropathological traits peculiar to patients heterozygous for FOXG1G224S, FOXG1W308X and FOXG1N232S mainly stem from abnormal quantitative tuning of FOXG1-controlled processes.
5. Materials and Methods
5.1. Mice and Embryo Dissection
5.2. Lentiviral Vectors Packaging and Titration
5.3. Primary Cortical Precursors Cultures
5.3.1. Depending on the Assay, Cultures Were Set as Follows
5.3.2. Main Media Composition Was as Follows
5.4. Dendrite Morphometry
5.5. Ca2+ Imaging Evaluation of Neuronal Activity
5.6. Extracellular Electrophysiological Recordings by MEAs
5.7. Quantitative RT-PCR
5.8. Immunofluorescent Assay
5.9. Image Acquisition
5.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanashima, C.; Fernandes, M.; Hebert, J.M.; Fishell, G. The Role of Foxg1 and Dorsal Midline Signaling in the Generation of Cajal-Retzius Subtypes. J. Neurosci. 2007, 27, 11103–11111. [Google Scholar] [CrossRef]
- Manuel, M.; Martynoga, B.; Yu, T.; West, J.D.; Mason, J.; Price, D. The transcription factor Foxg1 regulates the competence of telencephalic cells to adopt subpallial fates in mice. Development 2010, 137, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Muzio, L.; Mallamaci, A. Foxg1 Confines Cajal-Retzius Neuronogenesis and Hippocampal Morphogenesis to the Dorsomedial Pallium. J. Neurosci. 2005, 25, 4435–4441. [Google Scholar] [CrossRef]
- Martynoga, B.; Morrison, H.; Price, D.; Mason, J. Foxg1 is required for specification of ventral telencephalon and region-specific regulation of dorsal telencephalic precursor proliferation and apoptosis. Dev. Biol. 2005, 283, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, M.; Pivetta, C.; Granzotto, M.; Filippis, C.; Mallamaci, A. Emx2 and Foxg1 Inhibit Gliogenesis and Promote Neuronogenesis. Stem Cells 2010, 28, 1206–1218. [Google Scholar] [CrossRef]
- Falcone, C.; Santo, M.; Liuzzi, G.; Cannizzaro, N.; Grudina, C.; Valencic, E.; Peruzzotti-Jametti, L.; Pluchino, S.; Mallamaci, A. Foxg1 Antagonizes Neocortical Stem Cell Progression to Astrogenesis. Cereb. Cortex 2019, 29, 4903–4918. [Google Scholar] [CrossRef] [PubMed]
- Hanashima, C.; Li, S.C.; Shen, L.; Lai, E.; Fishell, G. Foxg1 Suppresses Early Cortical Cell Fate. Science 2004, 303, 56–59. [Google Scholar] [CrossRef]
- Miyoshi, G.; Fishell, G. Dynamic FoxG1 Expression Coordinates the Integration of Multipolar Pyramidal Neuron Precursors into the Cortical Plate. Neuron 2012, 74, 1045–1058. [Google Scholar] [CrossRef]
- Toma, K.; Kumamoto, T.; Hanashima, C. The Timing of Upper-Layer Neurogenesis Is Conferred by Sequential Derepression and Negative Feedback from Deep-Layer Neurons. J. Neurosci. 2014, 34, 13259–13276. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.-S.; Miyoshi, G.; Hanashima, C. Sensory cortex wiring requires preselection of short- and long-range projection neurons through an Egr-Foxg1-COUP-TFI network. Nat. Commun. 2019, 10, 3581. [Google Scholar] [CrossRef]
- Chiola, S.; Do, M.D.; Centrone, L.; Mallamaci, A. Foxg1 Overexpression in Neocortical Pyramids Stimulates Dendrite Elongation Via Hes1 and pCreb1 Upregulation. Cereb. Cortex 2018, 29, 1006–1019. [Google Scholar] [CrossRef]
- Yu, B.; Liu, J.; Su, M.; Wang, C.; Chen, H.; Zhao, C. Disruption of Foxg1 impairs neural plasticity leading to social and cognitive behavioral defects. Mol. Brain 2019, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhang, B.; Li, M.; Mo, F.; Mi, T.; Wu, Y.; Teng, Z.; Zhou, Q.; Li, W.; Hu, B. Precisely controlling endogenous protein dosage in hPSCs and derivatives to model FOXG1 syndrome. Nat. Commun. 2019, 10, 928. [Google Scholar] [CrossRef] [PubMed]
- Tigani, W.; Rossi, M.P.; Artimagnella, O.; Santo, M.; Rauti, R.; Sorbo, T.; Severino, F.P.U.; Provenzano, G.; Allegra, M.; Caleo, M.; et al. Foxg1 Upregulation Enhances Neocortical Activity. Cereb. Cortex 2020, 30, 5147–5165. [Google Scholar] [CrossRef] [PubMed]
- Fimiani, C.; Goina, E.; Su, Q.; Gao, G.; Mallamaci, A. RNA activation of haploinsufficient Foxg1 gene in murine neocortex. Sci. Rep. 2016, 6, 39311. [Google Scholar] [CrossRef]
- Shen, L.; Nam, H.-S.; Song, P.; Moore, H.; Anderson, S.A. FoxG1 haploinsufficiency results in impaired neurogenesis in the postnatal hippocampus and contextual memory deficits. Hippocampus 2006, 16, 875–890. [Google Scholar] [CrossRef]
- Miyoshi, G.; Ueta, Y.; Natsubori, A.; Hiraga, K.; Osaki, H.; Yagasaki, Y.; Kishi, Y.; Yanagawa, Y.; Fishell, G.; Machold, R.P.; et al. FoxG1 regulates the formation of cortical GABAergic circuit during an early postnatal critical period resulting in autism spectrum disorder-like phenotypes. Nat. Commun. 2021, 12, 3773. [Google Scholar] [CrossRef]
- Vegas, N.; Cavallin, M.; Maillard, C.; Boddaert, N.; Toulouse, J.; Schaefer, E.; Lerman-Sagie, T.; Lev, D.; Magalie, B.; Moutton, S.; et al. Delineating FOXG1 syndrome. Neurol. Genet. 2018, 4, e281. [Google Scholar] [CrossRef]
- SFARI Database_FOXG1. Available online: https://gene.sfari.org/database/human-gene/Foxg1#variants-tab (accessed on 15 December 2021).
- ClinVar Database_FOXG1. Available online: https://www.ncbi.nlm.nih.gov/clinvar/?term=Foxg1%5Bgene%5D (accessed on 15 December 2021).
- Shoichet, S.A.; Kunde, S.-A.; Viertel, P.; Schell-Apacik, C.; Von Voss, H.; Tommerup, N.; Ropers, H.-H.; Kalscheuer, V.M. Haploinsufficiency of novel FOXG1B variants in a patient with severe mental retardation, brain malformations and microcephaly. Qual. Life Res. 2005, 117, 536–544. [Google Scholar] [CrossRef]
- Eagleson, K.; McFadyen-Ketchum, L.S.; Ahrens, E.; Mills, P.; Does, M.; Nickols, J.; Levitt, P. Disruption of Foxg1 expression by knock-in of Cre recombinase: Effects on the development of the mouse telencephalon. Neuroscience 2007, 148, 385–399. [Google Scholar] [CrossRef]
- Siegenthaler, J.A.; Tremper-Wells, B.A.; Miller, M.W. Foxg1 Haploinsufficiency Reduces the Population of Cortical Intermediate Progenitor Cells: Effect of Increased p21 Expression. Cereb. Cortex 2007, 18, 1865–1875. [Google Scholar] [CrossRef]
- Bulstrode, H.; Johnstone, E.; Marques-Torrejon, M.A.; Ferguson, K.M.; Bressan, R.B.; Blin, C.; Grant, V.; Gogolok, S.; Gangoso, E.; Gagrica, S.; et al. Elevated FOXG1 and SOX2 in glioblastoma enforces neural stem cell identity through transcriptional control of cell cycle and epigenetic regulators. Genes Dev. 2017, 31, 757–773. [Google Scholar] [CrossRef] [PubMed]
- ClinVar_FOXG1(c.670G>A). Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/167092 (accessed on 15 December 2021).
- Trump, N.; McTague, A.; Brittain, H.; Papandreou, A.; Meyer, E.; Ngoh, A.; Palmer, R.; Morrogh, D.; Boustred, C.; Hurst, J.A.; et al. Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis. J. Med. Genet. 2016, 53, 310–317. [Google Scholar] [CrossRef]
- Papandreou, A.; Schneider, R.B.; Augustine, E.F.; Ng, J.; Mankad, K.; Meyer, E.; McTague, A.; Ngoh, A.; Hemingway, C.; Robinson, R.; et al. Delineation of the movement disorders associated withFOXG1mutations: Table 1. Neurology 2016, 86, 1794–1800. [Google Scholar] [CrossRef]
- Philippe, C.; Amsallem, D.; Francannet, C.; Lambert, L.; Saunier, A.; Verneau, F.; Jonveaux, P. Phenotypic variability in Rett syndrome associated with FOXG1 mutations in females. J. Med. Genet. 2009, 47, 59–65. [Google Scholar] [CrossRef] [PubMed][Green Version]
- ClinVar_FOXG1(c.924G>A). Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/13871 (accessed on 15 December 2021).
- Chen, T.-W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Artimagnella, O.; Mallamaci, A. RNASeq Profiling of Foxg1-GOF Neocortical Neurons. Available online: https://zenodo.org/record/2849657 (accessed on 25 May 2019).
- Dong, F.; Liu, D.; Jiang, F.; Liu, Y.; Wu, X.; Qu, X.; Liu, J.; Chen, Y.; Fan, H.; Yao, R. Conditional Deletion of Foxg1 Alleviates Demyelination and Facilitates Remyelination via the Wnt Signaling Pathway in Cuprizone-Induced Demyelinated Mice. Neurosci. Bull. 2021, 37, 15–30. [Google Scholar] [CrossRef]
- Chiola, S.; Santo, M.; Mallamaci, A. Intraventricular Transplantation of Engineered Neuronal Precursors for In Vivo Neuroarchitecture Studies. J. Vis. Exp. 2019, 59242, e59242. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, M.; Darbon, P.; Arsiero, M.; Lüscher, H.-R.; Streit, J. Single-Neuron Discharge Properties and Network Activity in Dissociated Cultures of Neocortex. J. Neurophysiol. 2004, 92, 977–996. [Google Scholar] [CrossRef][Green Version]
- Sin, S.T.K.; Jiang, P.; Deng, J.; Ji, L.; Cheng, S.H.; Dutta, A.; Leung, T.Y.; Chan, K.C.A.; Chiu, R.W.K.; Lo, Y.M.D. Identification and characterization of extrachromosomal circular DNA in maternal plasma. Proc. Natl. Acad. Sci. USA 2020, 117, 1658–1665. [Google Scholar] [CrossRef]
- Yu, S.C.Y.; Jiang, P.; Peng, W.; Cheng, S.H.; Cheung, Y.T.T.; Tse, O.Y.O.; Shang, H.; Poon, L.C.; Leung, T.Y.; Chan, K.C.A.; et al. Single-molecule sequencing reveals a large population of long cell-free DNA molecules in maternal plasma. Proc. Natl. Acad. Sci. USA 2021, 118, e2114937118. [Google Scholar] [CrossRef] [PubMed]
- Tucker, K.L.; Meyer, M.; Barde, Y. Neurotrophins are required for nerve growth during development. Nat. Neurosci. 2001, 4, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Spigoni, G.; Gedressi, C.; Mallamaci, A. Regulation of Emx2 Expression by Antisense Transcripts in Murine Cortico-Cerebral Precursors. PLoS ONE 2010, 5, e8658. [Google Scholar] [CrossRef] [PubMed]
- Raciti, M.; Granzotto, M.; Duc, M.D.; Fimiani, C.; Cellot, G.; Cherubini, E.; Mallamaci, A. Reprogramming fibroblasts to neural-precursor-like cells by structured overexpression of pallial patterning genes. Mol. Cell. Neurosci. 2013, 57, 42–53. [Google Scholar] [CrossRef]
- Falcone, C.; Filippis, C.; Granzotto, M.; Mallamaci, A. Emx2expression levels in NSCs modulate astrogenesis rates by regulatingEgfRandFgf9. Glia 2015, 63, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Longair, M.; Baker, D.A.; Armstrong, D. Simple Neurite Tracer: Open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics 2011, 27, 2453–2454. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.-Y.; Chao, C.-Y.; Huang, H.-L.; Chiu, T.-W.; Charoenkwan, P.; Hwang, E. NeurphologyJ: An automatic neuronal morphology quantification method and its application in pharmacological discovery. BMC Bioinform. 2011, 12, 230. [Google Scholar] [CrossRef]
- Edelstein, A.D.; Amodaj, N.; Hoover, K.H.; Vale, R.D.; Stuurman, N. Computer Control of Microscopes Using µManager. Curr. Protoc. Mol. Biol. 2010, 92, 14–20. [Google Scholar] [CrossRef]
- Emahmud, M.; Epulizzi, R.; Evasilaki, E.; Egiugliano, M. QSpike tools: A generic framework for parallel batch preprocessing of extracellular neuronal signals recorded by substrate microelectrode arrays. Front. Aging Neurosci. 2014, 8, 26. [Google Scholar] [CrossRef]
- Quiroga, R.Q.; Nadasdy, Z.; Ben-Shaul, Y. Unsupervised Spike Detection and Sorting with Wavelets and Superparamagnetic Clustering. Neural Comput. 2004, 16, 1661–1687. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Process/Neurodevelopmental Parameter | FOXG1WT | FOXG1G224S | FOXG1W3028X | FOXG1N232S |
|---|---|---|---|---|
| (vs-ctr) | (vs-FOXG1WT) | (vs-FOXG1WT) | (vs-FOXG1WT) | |
| NSC commitment to neuronogenesis | ↑ | GOF | LOF | = |
| neuron birth rate | ↓ | GOF | LOF | LOF |
| dendritic elongation and arborization | ↑ | GOF | LOF | na |
| spontaneous neuronal activity | ↑ | GOF | LOF | na |
| modulation of select neuronal genes (1) | ↓ | GOF | LOF | na |
| modulation of select neuronal genes (2) | ↑ | GOF | LOF | na |
| NSC commitment to astrogliogenesis | ↓ | GOF | LOF | = |
| astroblast proliferation rate | ↑ | GOF | LOF | LOF |
| modulation of select astroglial genes (1) | ↓ | GOF | LOF | na |
| modulation of select astroglial genes (2) | ↑ | V | V | na |
| NSC progression to oligodendrocytes | ↓ | = | = | na |
| oligodendroblast proliferation rate | ↑ | GOF | LOF | = |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frisari, S.; Santo, M.; Hosseini, A.; Manzati, M.; Giugliano, M.; Mallamaci, A. Multidimensional Functional Profiling of Human Neuropathogenic FOXG1 Alleles in Primary Cultures of Murine Pallial Precursors. Int. J. Mol. Sci. 2022, 23, 1343. https://doi.org/10.3390/ijms23031343
Frisari S, Santo M, Hosseini A, Manzati M, Giugliano M, Mallamaci A. Multidimensional Functional Profiling of Human Neuropathogenic FOXG1 Alleles in Primary Cultures of Murine Pallial Precursors. International Journal of Molecular Sciences. 2022; 23(3):1343. https://doi.org/10.3390/ijms23031343
Chicago/Turabian StyleFrisari, Simone, Manuela Santo, Ali Hosseini, Matteo Manzati, Michele Giugliano, and Antonello Mallamaci. 2022. "Multidimensional Functional Profiling of Human Neuropathogenic FOXG1 Alleles in Primary Cultures of Murine Pallial Precursors" International Journal of Molecular Sciences 23, no. 3: 1343. https://doi.org/10.3390/ijms23031343
APA StyleFrisari, S., Santo, M., Hosseini, A., Manzati, M., Giugliano, M., & Mallamaci, A. (2022). Multidimensional Functional Profiling of Human Neuropathogenic FOXG1 Alleles in Primary Cultures of Murine Pallial Precursors. International Journal of Molecular Sciences, 23(3), 1343. https://doi.org/10.3390/ijms23031343