
Treatment of Erythroid Precursor Cells from β-Thalassemia Patients with Cinchona Alkaloids: Induction of Fetal Hemoglobin Production

 , ,
, ,  ,
, 

Abstract
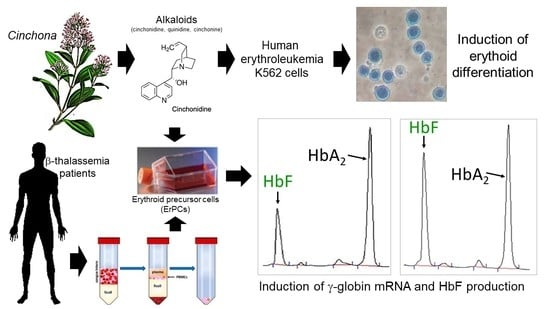
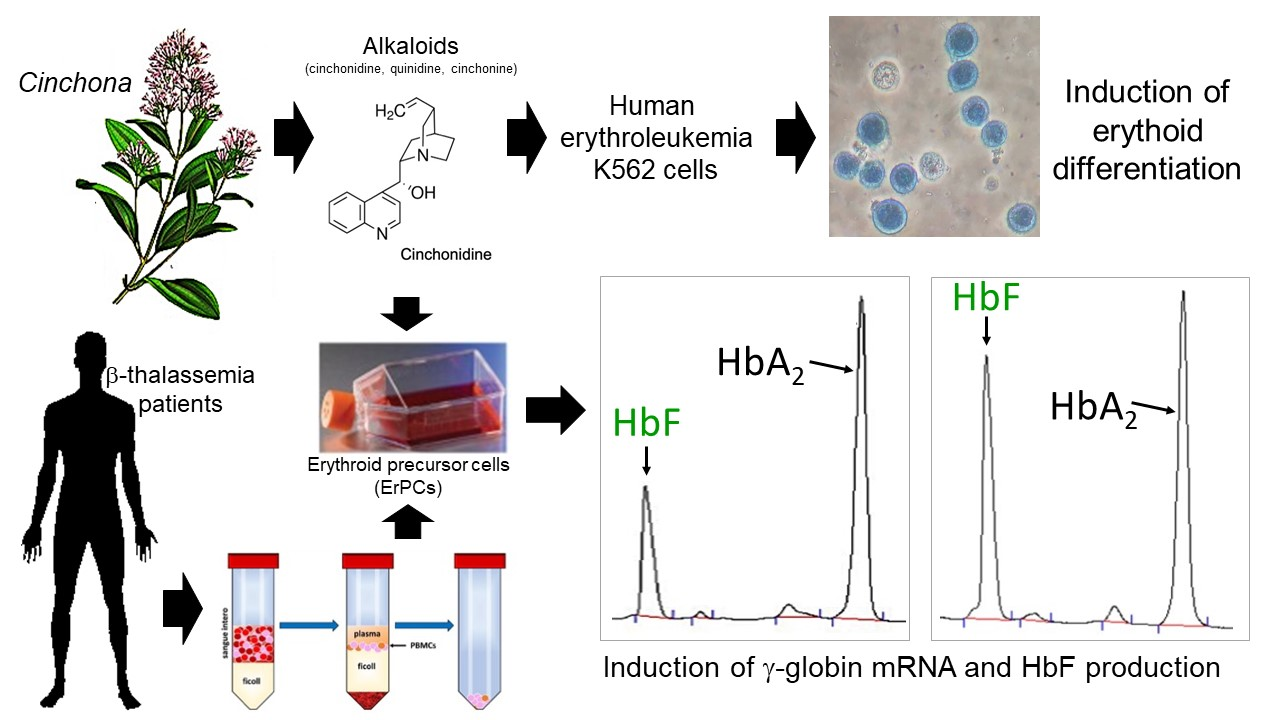
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
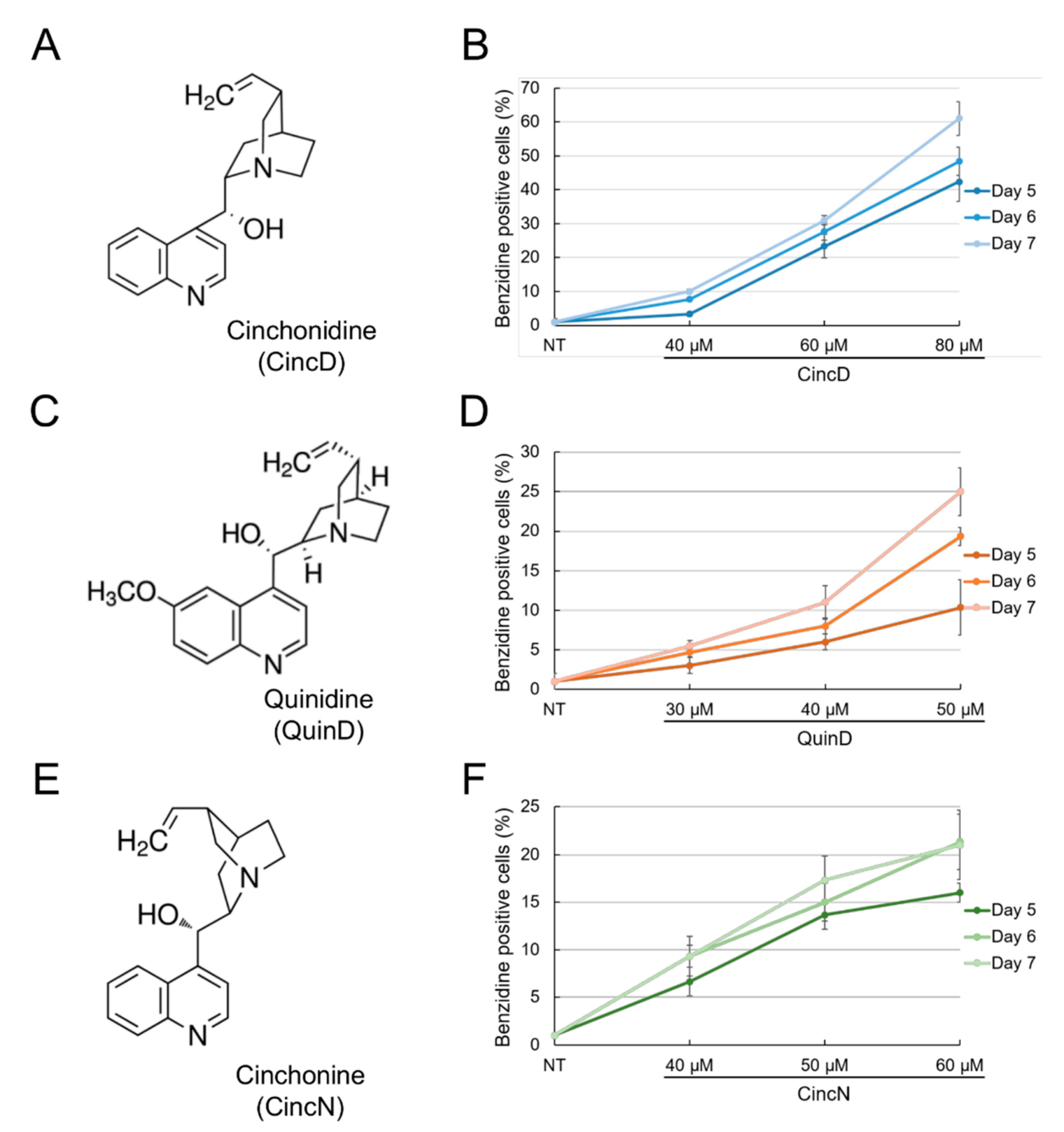
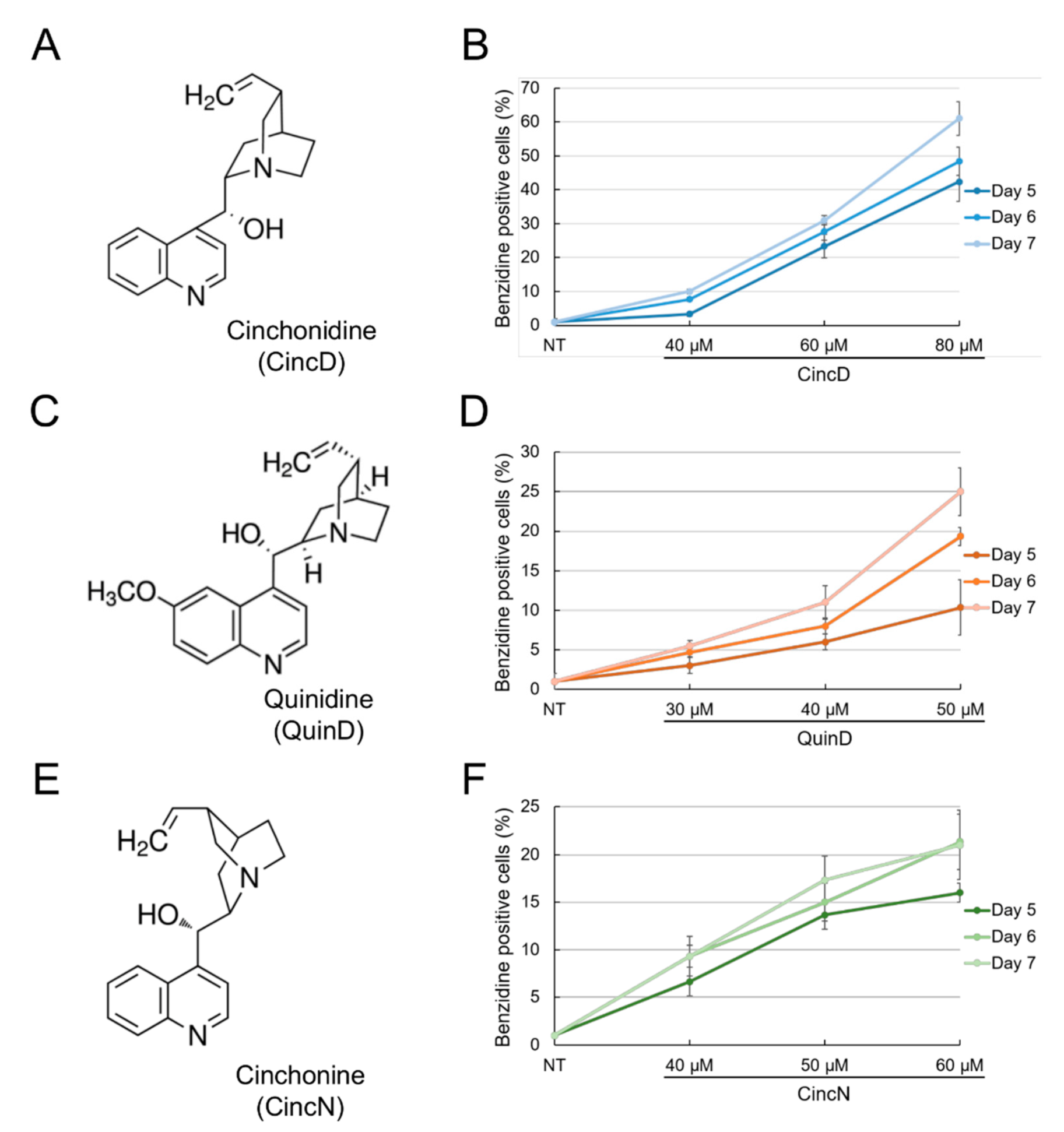
2.1. Cinchonidine, Quinidine and Cinchonine Induce Differentiation of K562 Erythroleukemia Cells
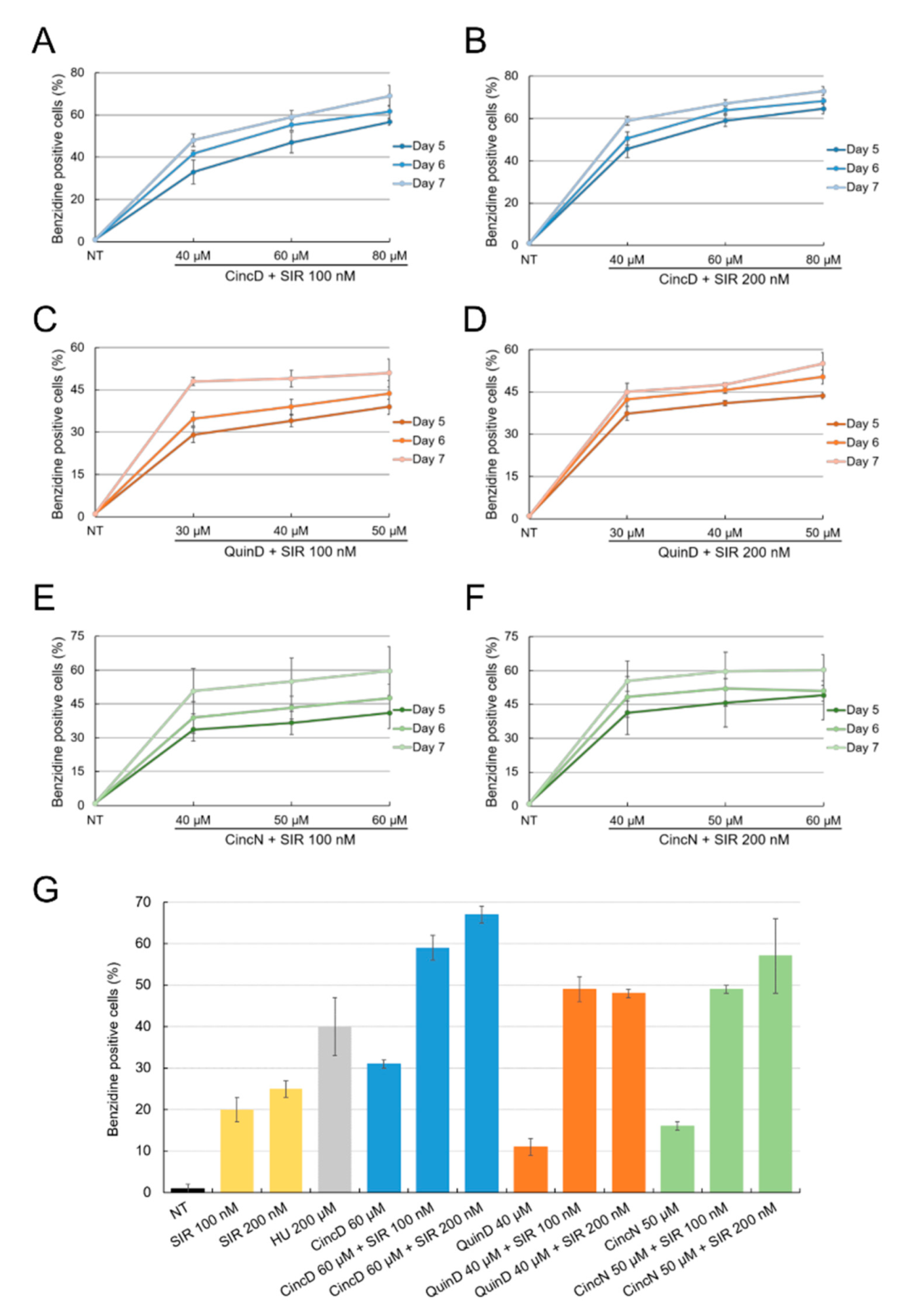
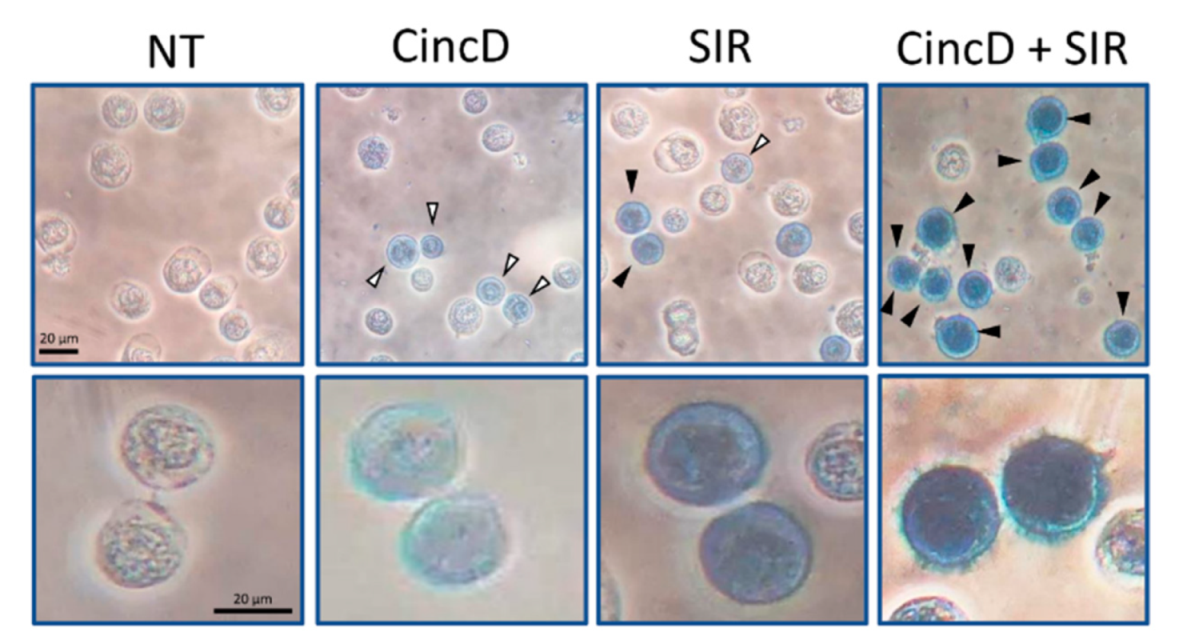
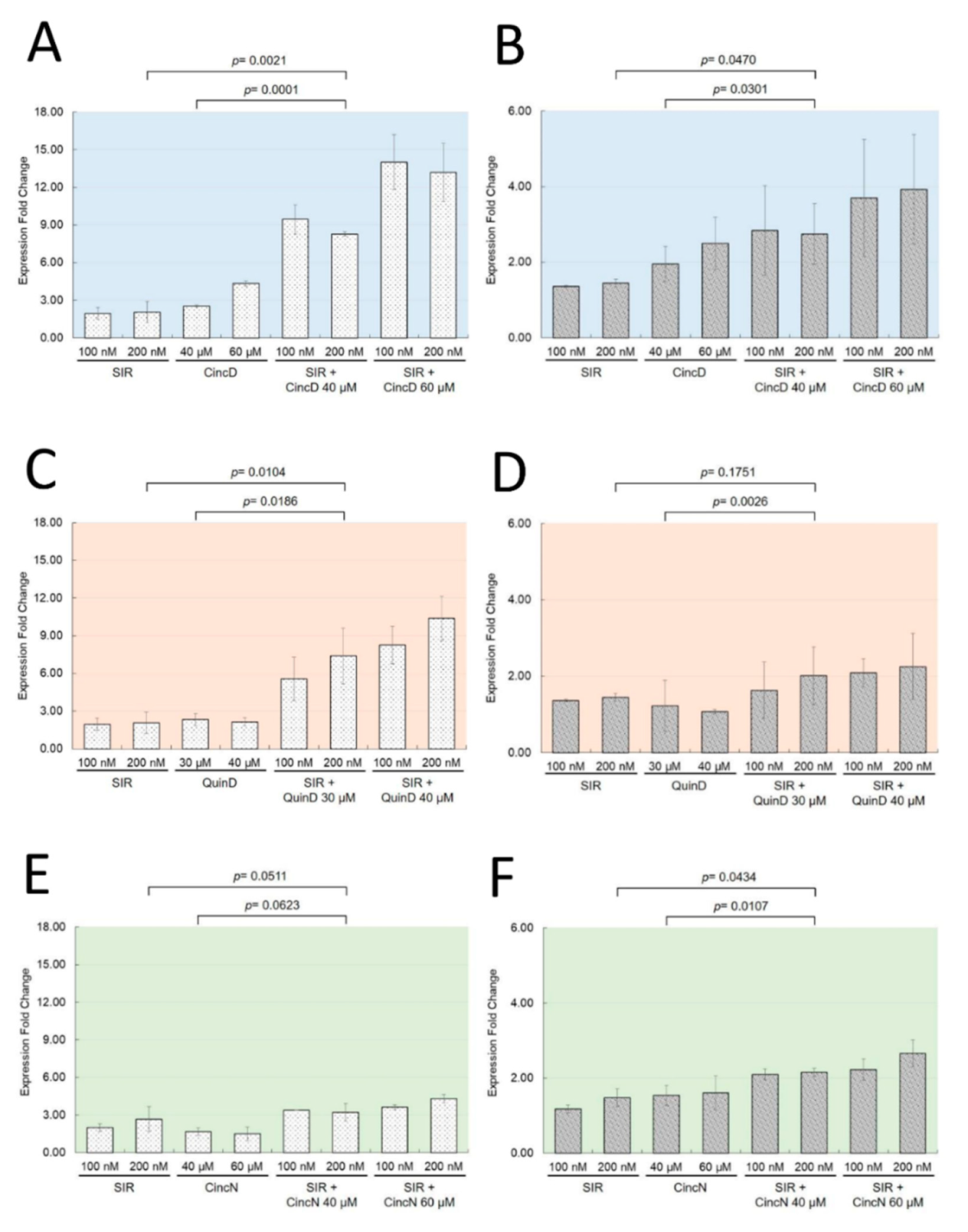
2.2. Cinchonidine, Quinidine and Cinchonine Potentiate Sirolimus-Induced Differentiation of K562 Erythroleukemia Cells
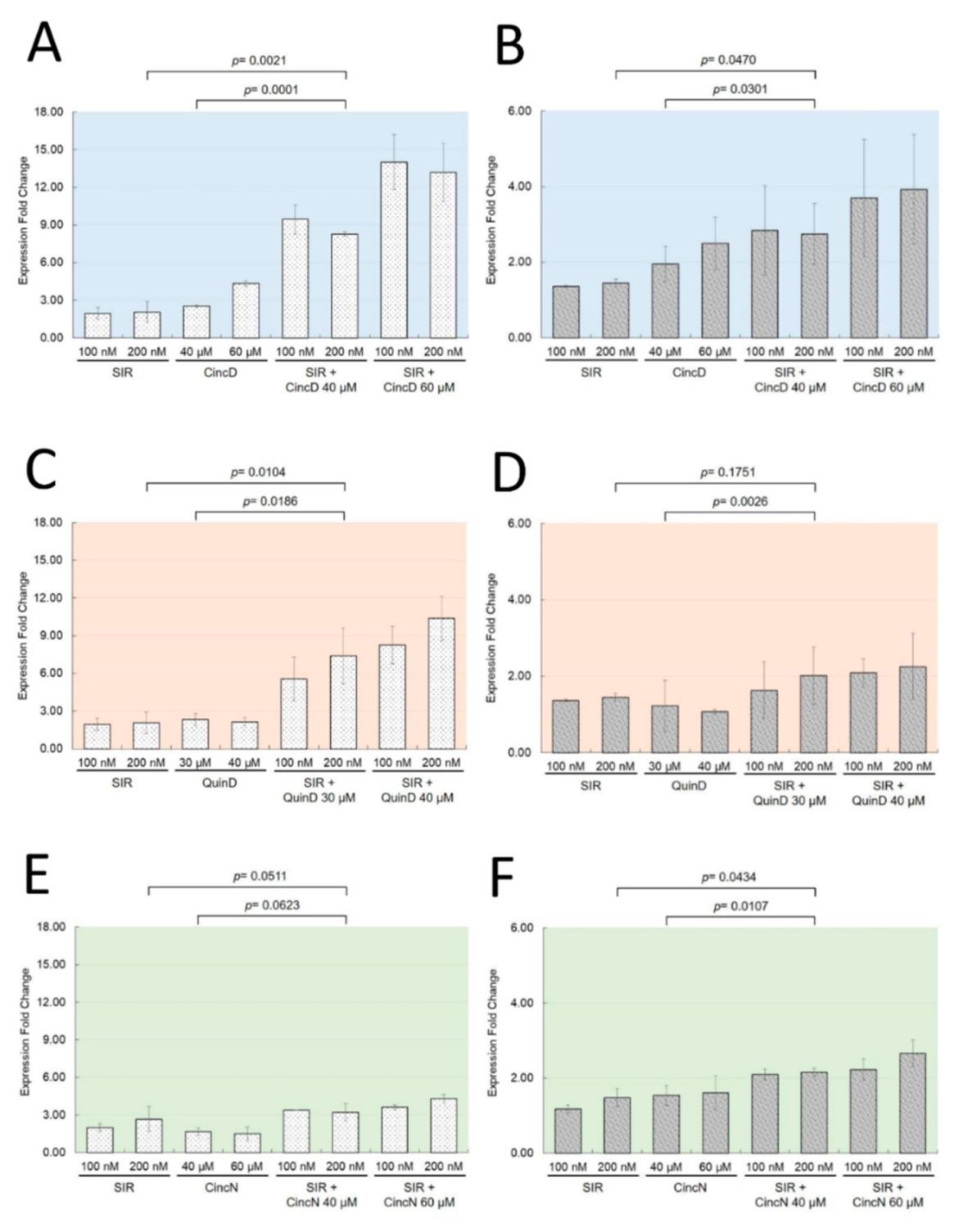
2.3. The Effects of Cinchonidine, Quinidine and Cinchonine on K562 Erythroid Differentiation Are Associated with a Modulation of Expression of α-Globin and γ-Globin Genes
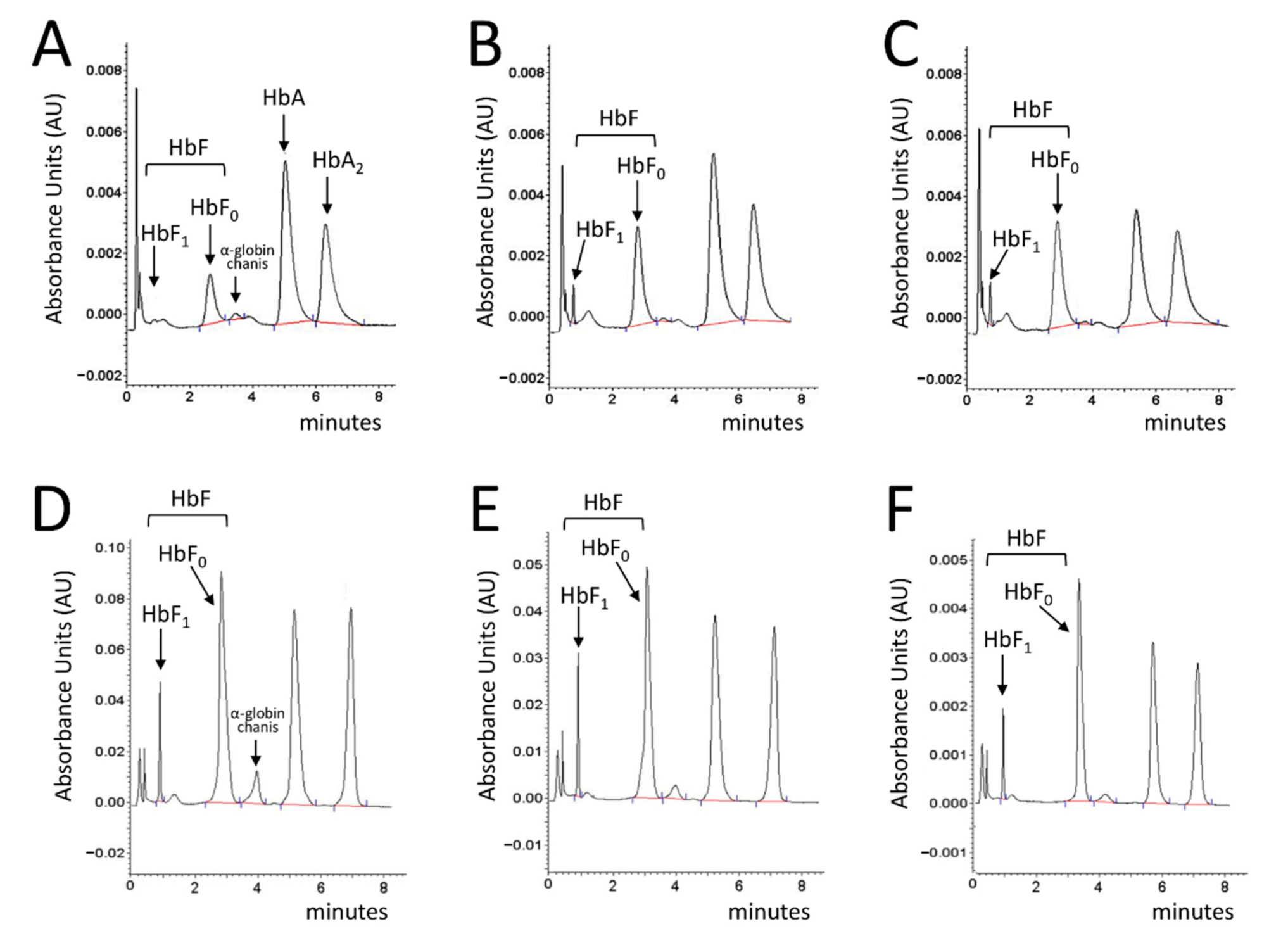
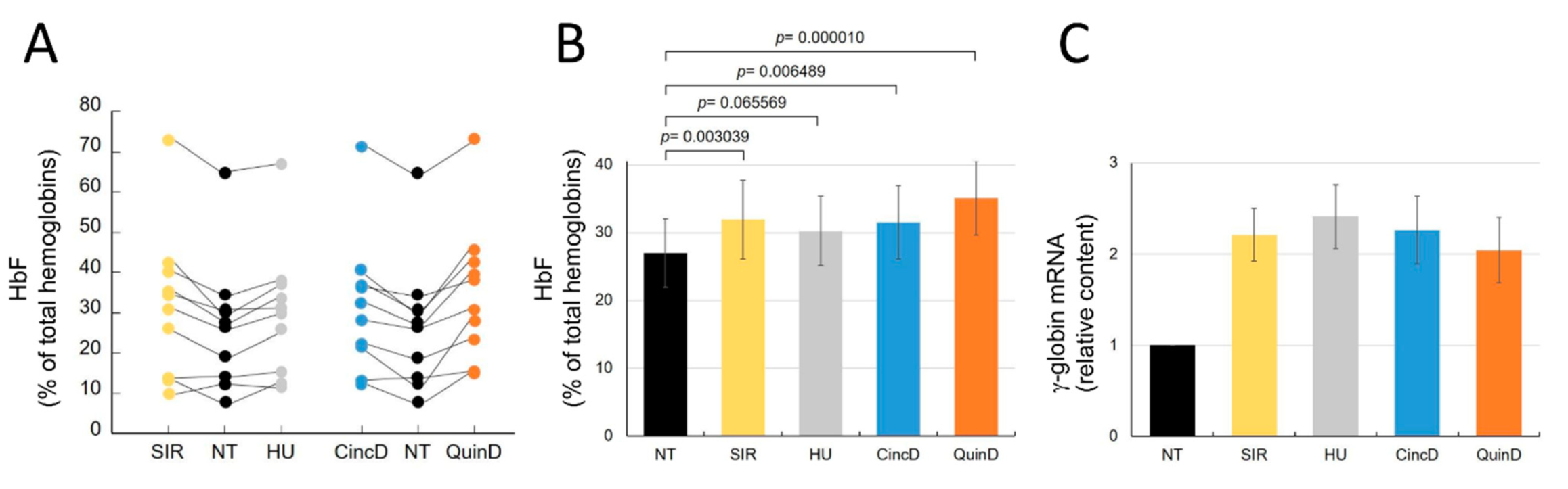
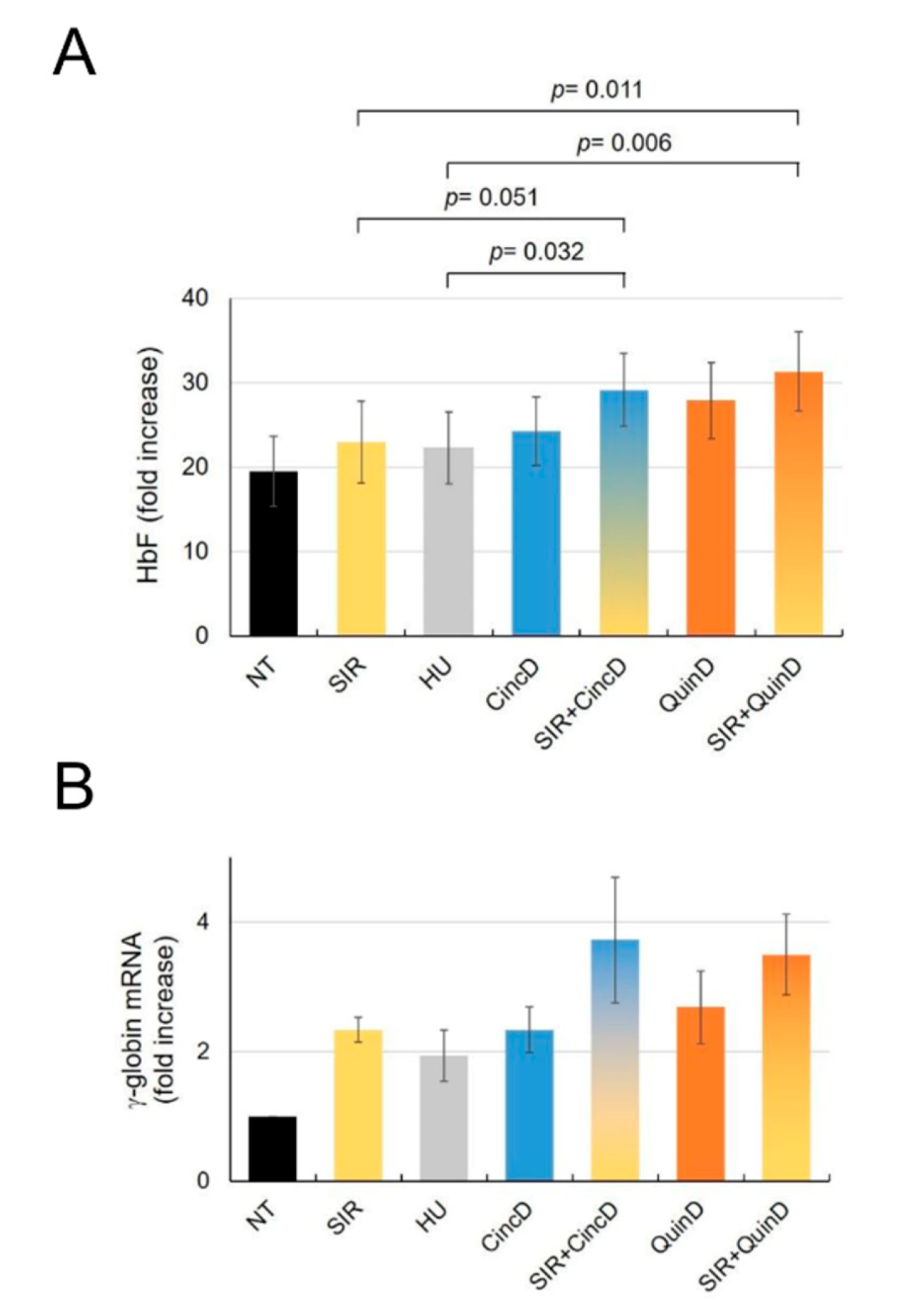
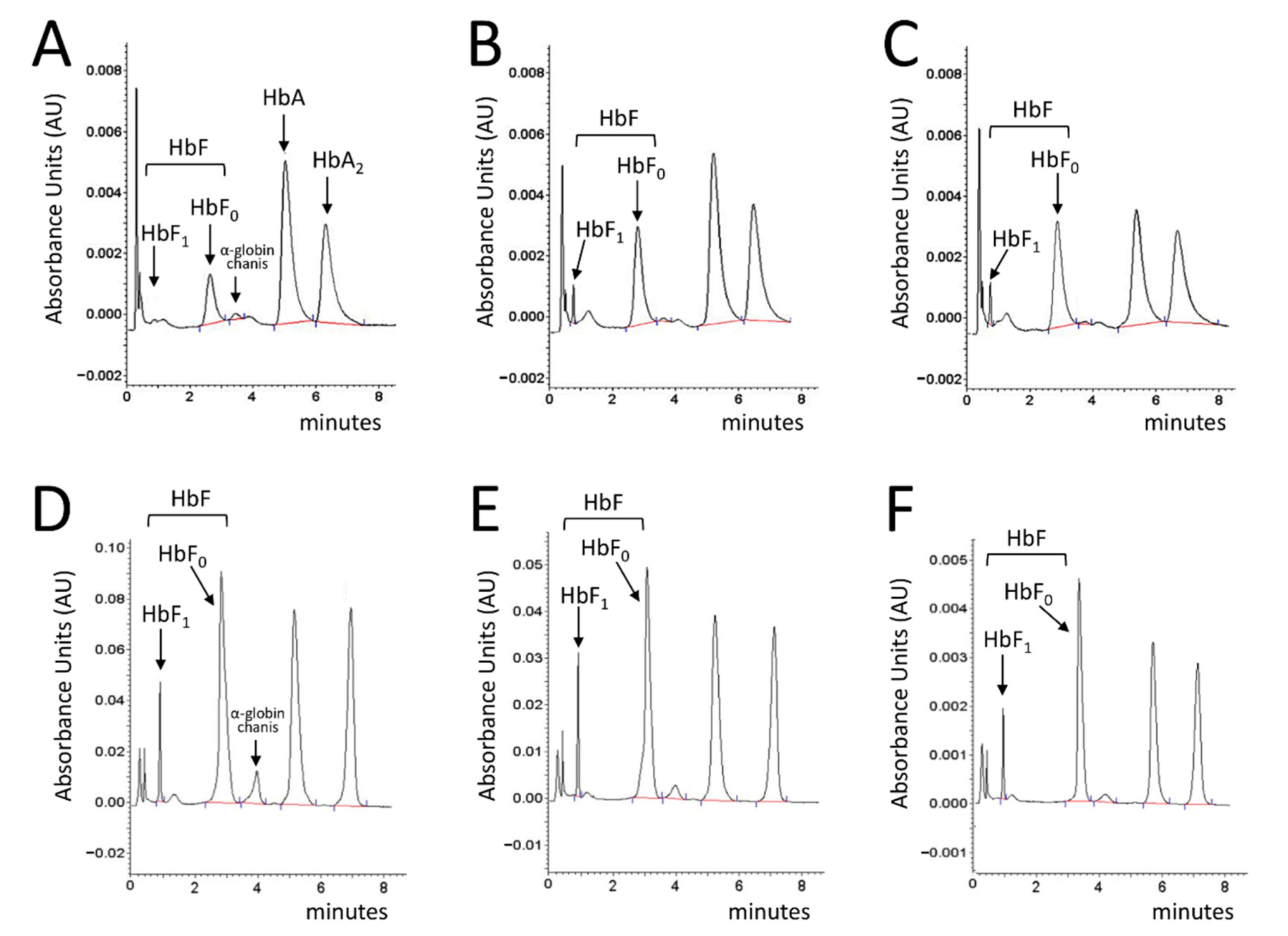
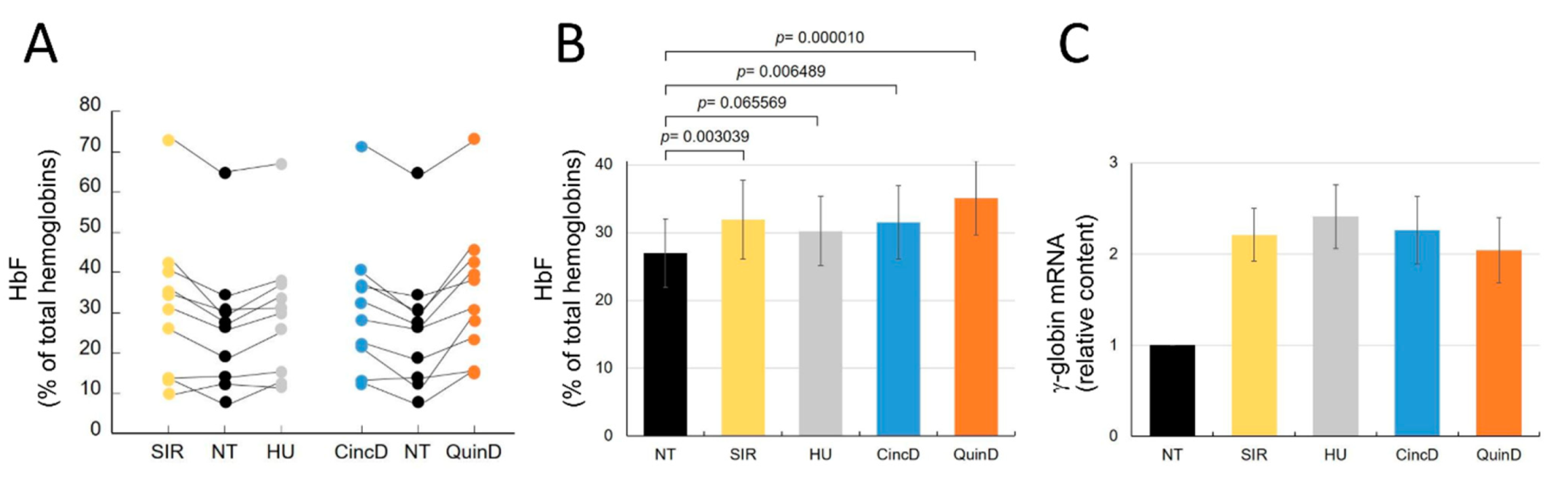
2.4. Cinchonidine and Quinidine Induce HbF and γ-Globin mRNA in Erythroid Precursor Cells (ErPCs) from β-Thalassemia Patients
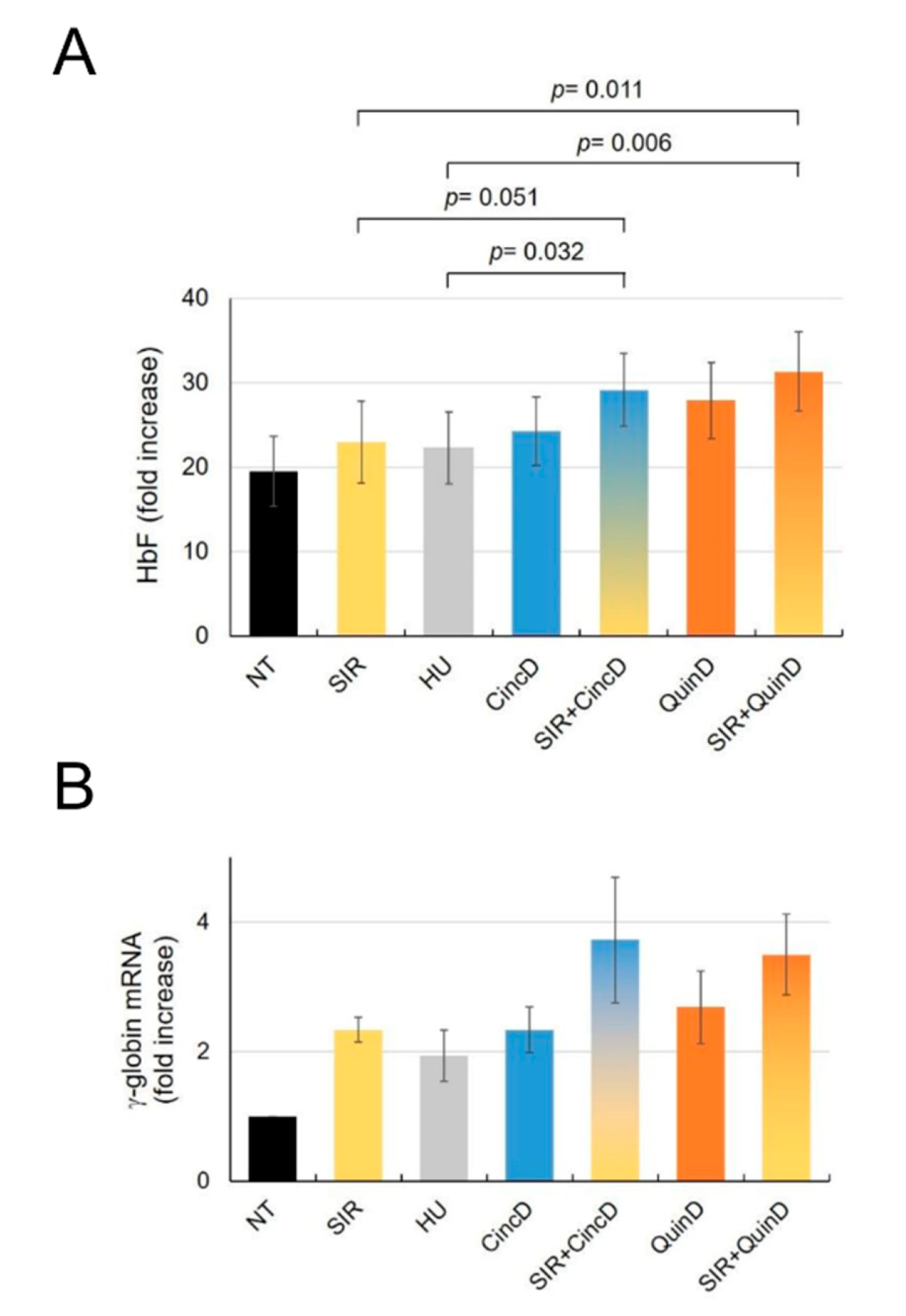
2.5. Cinchonidine and Quinidine Potentiate Sirolimus-Mediated Induction of HbF and γ-Globin mRNA in ErPCs from β-Thalassemia Patients
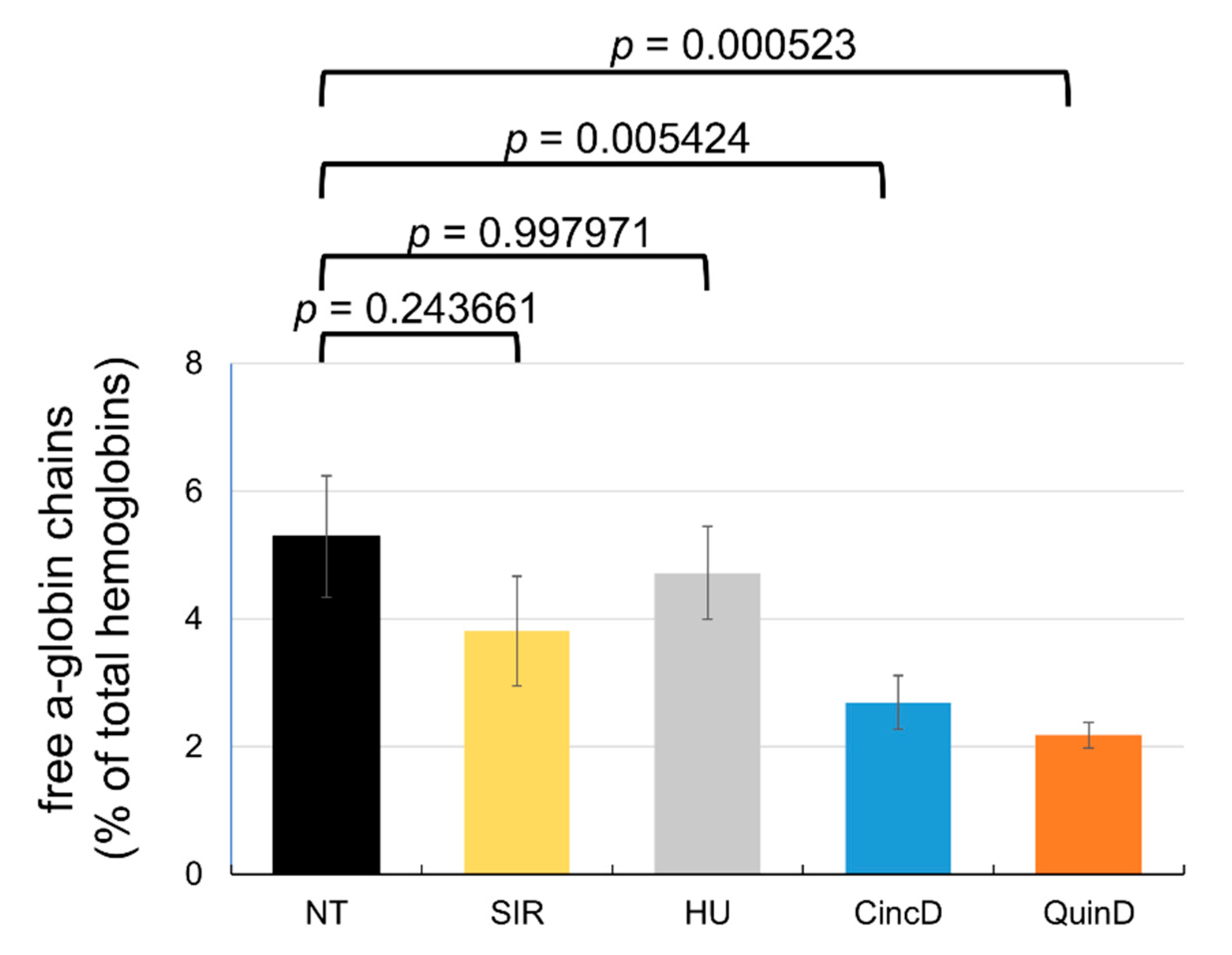
2.6. Treatment of ErPCs from β-Thalassemia Patients with Cinchonidine and Quinidine Is Associated with a Sharp Decrease in the Free α-Globin Chains
3. Discussion
4. Materials and Methods
4.1. Patients Recruitment
4.2. Chemical Reagents for Cell Culture Treatments
4.3. Human K562 Cell Cultures
4.4. In Vitro Culture of Erythroid Progenitors from β-Thalassemia Patients
4.5. Reverse Transcription and Quantitative Real-Time PCR (RT-qPCR)
4.6. HPLC Analysis of Hemoglobins
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Weatherall, D.J. Phenotype-genotype relationships in monogenic disease: Lessons from the thalassaemias. Nat. Rev. Genet. 2001, 2, 245–255. [Google Scholar] [CrossRef]
- Origa, R. β-Thalassemia. Genet. Med. 2017, 19, 609–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fucharoen, S.; Weatherall, D.J. Progress Toward the Control and Management of the Thalassemias. Hematol. Oncol. Clin. N. Am. 2016, 30, 359–371. [Google Scholar] [CrossRef]
- Modell, B.; Darlison, M.; Birgens, H.; Cario, H.; Faustino, P.; Giordano, P.C.; Gulbis, B.; Hopmeier, P.; Lena-Russo, D.; Romao, L.; et al. Epidemiology of haemoglobin disorders in Europe: An overview. Scand. J. Clin. Lab. Investig. 2007, 67, 39–69. [Google Scholar] [CrossRef] [PubMed]
- Galanello, R.; Origa, R. Β-thalassemia. Orphanet. J. Rare Dis. 2010, 5, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thein, S.L. The molecular basis of β-thalassemia. Cold Spring Harb. Perspect. Med. 2013, 3, a011700. [Google Scholar]
- Sripichai, O.; Fucharoen, S. Fetal hemoglobin regulation in β-thalassemia: Heterogeneity, modifiers and therapeutic approaches. Expert Rev. Hematol. 2016, 9, 1129–1137. [Google Scholar] [CrossRef]
- Forget, B.G. Molecular basis of hereditary persistence of fetal hemoglobin. Ann. N. Y. Acad. Sci. 1998, 850, 38–44. [Google Scholar] [CrossRef]
- Musallam, K.M.; Sankaran, V.G.; Cappellini, M.D.; Duca, L.; Nathan, D.G.; Taher, A.T. Fetal hemoglobin levels and morbidity in untransfused patients with β-thalassemia intermedia. Blood 2012, 119, 364–367. [Google Scholar] [CrossRef] [Green Version]
- Nuinoon, M.; Makarasara, W.; Mushiroda, T.; Setianingsih, I.; Wahidiyat, P.A.; Sripichai, O.; Kumasaka, N.; Takahashi, A.; Svasti, S.; Munkongdee, T.; et al. A genome-wide association identified the common genetic variants influence disease severity in β0-thalassemia/hemoglobin E. Hum. Genet. 2010, 127, 303–314. [Google Scholar] [CrossRef]
- Danjou, F.; Anni, F.; Perseu, L.; Satta, S.; Dessì, C.; Lai, M.E.; Fortina, P.; Devoto, M.; Galanello, R. Genetic modifiers of b-thalassemia and clinical severity as assessed by age at first transfusion. Haematologica 2012, 97, 989–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badens, C.; Joly, P.; Agouti, I.; Thuret, I.; Gonnet, K.; Fattoum, S.; Francina, A.; Simeoni, M.C.; Loundou, A.; Pissard, S. Variants in genetic modifiers of β-thalassemia can help to predict the major or intermedia type of the disease. Haematologica 2011, 96, 1712–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uda, M.; Galanello, R.; Sanna, S.; Lettre, G.; Sankaran, V.G.; Chen, W.; Usala, G.; Busonero, F.; Maschio, A.; Albai, G.; et al. Genome wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of β-thalassemia. Proc. Natl. Acad. Sci. USA 2008, 105, 1620–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breveglieri, G.; Bianchi, N.; Cosenza, L.C.; Gamberini, M.R.; Chiavilli, F.; Zuccato, C.; Montagner, G.; Borgatti, M.; Lampronti, I.; Finotti, A.; et al. An Agamma-globin G->A gene polymorphism associated with beta(0)39 thalassemia globin gene and high fetal hemoglobin production. BMC Med. Genet. 2017, 18, 93. [Google Scholar] [CrossRef] [Green Version]
- Antoniani, C.; Meneghini, V.; Lattanzi, A.; Felix, T.; Romano, O.; Magrin, E.; Weber, L.; Pavani, G.; El Hoss, S.; Kurita, R.; et al. Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood 2018, 131, 1960–1973. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zeng, J.; Roscoe, B.P.; Liu, P.; Yao, Q.; Lazzarotto, C.R.; Clement, K.; Cole, M.A.; Luk, K.; Baricordi, C.; et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019, 25, 776–783. [Google Scholar] [CrossRef]
- Métais, J.Y.; Doerfler, P.A.; Mayuranathan, T.; Bauer, D.E.; Fowler, S.C.; Hsieh, M.M.; Katta, V.; Keriwala, S.; Lazzarotto, C.R.; Luk, K.; et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019, 3, 3379–3392. [Google Scholar] [CrossRef] [Green Version]
- Mingoia, M.; Caria, C.A.; Ye, L.; Asunis, I.; Marongiu, M.F.; Manunza, L.; Sollaino, M.C.; Wang, J.; Cabriolu, A.; Kurita, R.; et al. Induction of therapeutic levels of HbF in genome-edited primary β(0) 39-thalassaemia haematopoietic stem and progenitor cells. Br. J. Haematol. 2021, 192, 395–404. [Google Scholar] [CrossRef]
- Frangou, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- NCT03655678 (A Safety and Efficacy Study Evaluating CTX001 in Subjects with Transfusion-Dependent β-Thalassemia). Available online: https://clinicaltrials.gov/ct2/show/NCT03655678 (accessed on 28 November 2021).
- Sherkow, J.S. CRISPR, Patents, and the Public Health. Yale J. Biol. Med. 2017, 90, 667–672. [Google Scholar]
- Rigter, T.; Klein, D.; Weinreich, S.S.; Cornel, M.C. Moving somatic gene editing to the clinic: Routes to market access and reimbursement in Europe. Eur. J. Hum. Genet. 2021, 29, 1477–1484. [Google Scholar] [CrossRef]
- Cornel, M.C.; Howard, H.C.; Lim, D.; Bonham, V.L.; Wartiovaara, K. Moving towards a cure in genetics: What is needed to bring somatic gene therapy to the clinic? Eur. J. Hum. Genet. 2019, 27, 484–487. [Google Scholar] [CrossRef] [Green Version]
- Finotti, A.; Gambari, R. Recent trends for novel options in experimental biological therapy of β-thalassemia. Expert Opin. Biol. Ther. 2014, 14, 1443–1454. [Google Scholar] [CrossRef]
- Mukherjee, M.; Rahaman, M.; Ray, S.K.; Shukla, P.C.; Dolai, T.K.; Chakravorty, N. Revisiting fetal hemoglobin inducers in beta-hemoglobinopathies: A review of natural products, conventional and combinatorial therapies. Mol. Biol. Rep. 2021. Online ahead of print.
- Lampronti, I.; Bianchi, N.; Zuccato, C.; Dall’Acqua, F.; Vedaldi, D.; Viola, G.; Potenza, R.; Chiavilli, F.; Breveglieri, G.; Borgatti, M.; et al. Increase in gamma-globin mRNA content in human erythroid cells treated with angelicin analogs. Int. J. Hematol. 2009, 90, 318–327. [Google Scholar] [CrossRef]
- Finotti, A.; Bianchi, N.; Fabbri, E.; Borgatti, M.; Breveglieri, G.; Gasparello, J.; Gambari, R. Erythroid induction of K562 cells treated with mithramycin is associated with inhibition of raptor gene transcription and mammalian target of rapamycin complex 1 (mTORC1) functions. Pharmacol. Res. 2015, 91, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iftikhar, F.; Ali, H.; Musharraf, S.G. Cinchona alkaloids as natural fetal hemoglobin inducing agents in human erythroleukemia cells. RSC Adv. 2019, 9, 17551. [Google Scholar] [CrossRef] [Green Version]
- Warhurst, D.C. Cinchona alkaloids and malaria. Lancet 1981, 2, 1346. [Google Scholar]
- Uzor, P.F. Alkaloids from Plants with Antimalarial Activity: A Review of Recent Studies. Evid. Based Complement. Alternat. Med. 2020, 2020, 8749083. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, C.; Barnes, C.J.; Cornett, C.; Holmfred, E.; Hansen, S.H.; Persson, C.; Antonelli, A.; Rønsted, N. Phylogeny Predicts the Quantity of Antimalarial Alkaloids within the Iconic Yellow Cinchona Bark (Rubiaceae: Cinchona calisaya). Front. Plant. Sci. 2017, 8, 391. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, A.; Larson, J.; Ghannam, M.; Saeed, M.; Cunnane, R.; Ghanbari, H.; Latchamsetty, R.; Crawford, T.; Jongnarangsin, K.; Pelosi, F.; et al. Efficacy and tolerability of quinidine as salvage therapy for monomorphic ventricular tachycardia in patients with structural heart disease. J. Cardiovasc. Electrophysiol. 2021. Online ahead of print.
- Vitali Serdoz, L.; Rittger, H.; Furlanello, F.; Bastian, D. Quinidine-A legacy within the modern era of antiarrhythmic therapy. Pharmacol. Res. 2019, 144, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Li, D.L.; Cox, Z.L.; Richardson, T.D.; Kanagasundram, A.N.; Saavedra, P.J.; Shen, S.T.; Montgomery, J.A.; Murray, K.T.; Roden, D.M.; Stevenson, W.G. Quinidine in the Management of Recurrent Ventricular Arrhythmias: A Reappraisal. JACC Clin. Electrophysiol. 2021, 7, 1254–1263. [Google Scholar] [CrossRef]
- Sehgal, S.N. Sirolimus: Its discovery, biological properties, and mechanism of action. Transplant. Proc. 2003, 35, 7S–14S. [Google Scholar] [CrossRef]
- Mischiati, C.; Sereni, A.; Lampronti, I.; Bianchi, N.; Borgatti, M.; Prus, E.; Fibach, E.; Gambari, R. Rapamycin-mediated induction of gamma-globin mRNA accumulation in human erythroid cells. Br. J. Haematol. 2004, 126, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Fibach, E.; Bianchi, N.; Borgatti, M.; Zuccato, C.; Finotti, A.; Lampronti, I.; Prus, E.; Mischiati, C.; Gambari, R. Effects of rapamycin on accumulation of alpha-, β- and gamma-globin mRNAs in erythroid precursor cells from β-thalassaemia patients. Eur. J. Haematol. 2006, 77, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Bianchi, N.; Borgatti, M.; Lampronti, I.; Massei, F.; Favre, C.; Gambari, R. Everolimus is a potent inducer of erythroid differentiation and gamma-globin gene expression in human erythroid cells. Acta Haematol. 2007, 117, 168–176. [Google Scholar] [CrossRef]
- Pecoraro, A.; Troia, A.; Calzolari, R.; Scazzone, C.; Rigano, P.; Martorana, A.; Sacco, M.; Maggio, A.; Di Marzo, R. Efficacy of Rapamycin as Inducer of Hb F in Primary Erythroid Cultures from Sickle Cell Disease and β-Thalassemia Patients. Hemoglobin 2015, 39, 225–229. [Google Scholar] [CrossRef]
- Khaibullina, A.; Almeida, L.E.; Wang, L.; Kamimura, S.; Wong, E.C.; Nouraie, M.; Maric, I.; Albani, S.; Finkel, J.; Quezado, Z.M. Rapamycin increases fetal hemoglobin and ameliorates the nociception phenotype in sickle cell mice. Blood Cells Mol. Dis. 2015, 55, 363–372. [Google Scholar] [CrossRef]
- Wang, J.; Tran, J.; Wang, H.; Guo, C.; Harro, D.; Campbell, A.D.; Eitzman, D.T. mTOR Inhibition improves anaemia and reduces organ damage in a murine model of sickle cell disease. Br. J. Haematol. 2016, 174, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Lechauve, C.; Keith, J.; Khandros, E.; Fowler, S.; Mayberry, K.; Freiwan, A.; Thom, C.S.; Delbini, P.; Romero, E.B.; Zhang, J.; et al. The autophagy-activating kinase ULK1 mediates clearance of free α-globin in β-thalassemia. Sci. Transl. Med. 2019, 11, eaav4881. [Google Scholar] [CrossRef]
- Gaudre, N.; Cougoul, P.; Bartolucci, P.; Dörr, G.; Bura-Riviere, A.; Kamar, N.; Del Bello, A. Improved Fetal Hemoglobin With mTOR Inhibitor-Based Immunosuppression in a Kidney Transplant Recipient with Sickle Cell Disease. Am. J. Transplant. 2017, 17, 2212–2214. [Google Scholar] [CrossRef] [Green Version]
- Al-Khatti, A.A.; Alkhunaizi, A.M. Additive effect of sirolimus and hydroxycarbamide on fetal haemoglobin level in kidney transplant patients with sickle cell disease. Br. J. Haematol. 2019, 185, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Gamberini, M.R.; Prosdocimi, M.; Gambari, R. Sirolimus for Treatment of β-Thalassemia: From Pre-Clinical Studies to the Design of Clinical Trials. Health Educ. Public Health 2021, 4, 425–435. [Google Scholar]
- Kahan, B.D. Sirolimus: A new agent for clinical renal transplantation. Transplant. Proc. 1997, 29, 48–50. [Google Scholar] [CrossRef]
- Vasquez, E.M. Sirolimus: A new agent for prevention of renal allograft rejection. Am. J. Health Syst. Pharm. 2000, 57, 437–448. [Google Scholar] [CrossRef]
- Schaffer, S.A.; Ross, H.J. Everolimus: Efficacy and safety in cardiac transplantation. Expert Opin. Drug Saf. 2010, 9, 843–854. [Google Scholar] [CrossRef]
- Tang, C.Y.; Shen, A.; Wei, X.F.; Li, Q.D.; Liu, R.; Deng, H.J.; Wu, Y.Z.; Wu, Z.J. Everolimus in de novo liver transplant recipients: A systematic review. Hepatobiliary Pancreat. Dis. Int. 2015, 14, 461–469. [Google Scholar] [CrossRef]
- Ji, L.; Xie, W.; Zhang, Z. Efficacy and safety of sirolimus in patients with systemic lupus erythematosus: A systematic review and meta-analysis. Semin. Arthritis Rheum. 2020, 50, 1073–1080. [Google Scholar] [CrossRef]
- Wang, Q.; Luo, M.; Xiang, B.; Chen, S.; Ji, Y. The efficacy and safety of pharmacological treatments for lymphangioleiomyomatosis. Respir. Res. 2020, 21, 55. [Google Scholar] [CrossRef] [Green Version]
- Sasongko, T.H.; Ismail, N.F.; Zabidi-Hussin, Z. Rapamycin and rapalogs for tuberous sclerosis complex. Cochrane Database Syst. Rev. 2016, 7, CD011272. [Google Scholar] [CrossRef] [PubMed]
- Graillon, T.; Sanson, M.; Campello, C.; Idbaih, A.; Peyre, M.; Peyrière, H.; Basset, N.; Autran, D.; Roche, C.; Kalamarides, M.; et al. Everolimus and Octreotide for Patients with Recurrent Meningioma: Results from the Phase II CEVOREM Trial. Clin. Cancer Res. 2020, 26, 552–557. [Google Scholar] [CrossRef]
- Gallo, M.; Malandrino, P.; Fanciulli, G.; Rota, F.; Faggiano, A.; Colao, A.; NIKE Group. Everolimus as first line therapy for pancreatic neuroendocrine tumours: Current knowledge and future perspectives. J. Cancer Res. Clin. Oncol. 2017, 143, 1209–1224. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma: Final results and analysis of prognostic factors. Cancer 2010, 116, 4256–4265. [Google Scholar] [CrossRef] [PubMed]
- Algiraigri, A.H.; Wright, N.A.M.; Paolucci, E.O.; Kassam, A. Hydroxyurea for lifelong transfusion-dependent β-thalassemia: A meta-analysis. Pediatr. Hematol. Oncol. 2017, 34, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, N.; Finotti, A.; Ferracin, M.; Lampronti, I.; Zuccato, C.; Breveglieri, G.; Brognara, E.; Fabbri, E.; Borgatti, M.; Negrini, M.; et al. Increase of microRNA-210, decrease of raptor gene expression and alteration of mammalian target of rapamycin regulated proteins following mithramycin treatment of human erythroid cells. PLoS ONE 2015, 10, e0121567. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, N.; Ongaro, F.; Chiarabelli, C.; Gualandi, L.; Mischiati, C.; Bergamini, P.; Gambari, R. Induction of erythroid differentiation of human K562 cells by cisplatin analogs. Biochem. Pharmacol. 2000, 60, 31–40. [Google Scholar] [CrossRef]
- Cosenza, L.C.; Breda, L.; Breveglieri, G.; Zuccato, C.; Finotti, A.; Lampronti, I.; Borgatti, M.; Chiavilli, F.; Gamberini, M.R.; Satta, S.; et al. A validated cellular biobank for β-thalassemia. J. Transl. Med. 2016, 14, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambari, R.; Fibach, E. Medicinal chemistry of fetal hemoglobin inducers for treatment of beta-thalassemia. Curr. Med. Chem. 2007, 14, 199–212. [Google Scholar] [CrossRef]
- Bianchi, N.; Chiarabelli, C.; Zuccato, C.; Lampronti, I.; Borgatti, M.; Amari, G.; Delcanale, M.; Chiavilli, F.; Prus, E.; Fibach, E.; et al. Erythroid differentiation ability of butyric acid analogues: Identification of basal chemical structures of new inducers of foetal haemoglobin. Eur. J. Pharmacol. 2015, 752, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, Z.; Ebrahimzadeh, M.A. Hemoglobin F (HbF) inducers; History, Structure and Efficacies. Mini Rev. Med. Chem. 2021. Online ahead of print.
- Ansari, S.H.; Lassi, Z.S.; Khowaja, S.M.; Adil, S.O.; Shamsi, T.S. Hydroxyurea (hydroxycarbamide) for transfusion-dependent beta-thalassaemia. Cochrane Database Syst. Rev. 2019, 3, CD012064. [Google Scholar]
- McLaughlin, L.A.; Paine, M.J.; Kemp, C.A.; Maréchal, J.D.; Flanagan, J.U.; Ward, C.J.; Sutcliffe, M.J.; Roberts, G.C.; Wolf, C.R. Why is quinidine an inhibitor of cytochrome P450 2D6? The role of key active-site residues in quinidine binding. J. Biol. Chem. 2005, 280, 38617–38624. [Google Scholar] [CrossRef] [Green Version]
- Fromm, M.F.; Kim, R.B.; Stein, C.M.; Wilkinson, G.R.; Roden, D.M. Inhibition of P-glycoprotein-mediated drug transport: A unifying mechanism to explain the interaction between digoxin and quinidine [see comments]. Circulation 1999, 99, 552–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballou, L.M.; Lin, R.Z. Rapamycin and mTOR kinase inhibitors. J. Chem. Biol. 2008, 1, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuccato, C.; Cosenza, L.C.; Zurlo, M.; Lampronti, I.; Borgatti, M.; Scapoli, C.; Gambari, R.; Finotti, A. Treatment of Erythroid Precursor Cells from β-Thalassemia Patients with Cinchona Alkaloids: Induction of Fetal Hemoglobin Production. Int. J. Mol. Sci. 2021, 22, 13433. https://doi.org/10.3390/ijms222413433
Zuccato C, Cosenza LC, Zurlo M, Lampronti I, Borgatti M, Scapoli C, Gambari R, Finotti A. Treatment of Erythroid Precursor Cells from β-Thalassemia Patients with Cinchona Alkaloids: Induction of Fetal Hemoglobin Production. International Journal of Molecular Sciences. 2021; 22(24):13433. https://doi.org/10.3390/ijms222413433
Chicago/Turabian StyleZuccato, Cristina, Lucia Carmela Cosenza, Matteo Zurlo, Ilaria Lampronti, Monica Borgatti, Chiara Scapoli, Roberto Gambari, and Alessia Finotti. 2021. "Treatment of Erythroid Precursor Cells from β-Thalassemia Patients with Cinchona Alkaloids: Induction of Fetal Hemoglobin Production" International Journal of Molecular Sciences 22, no. 24: 13433. https://doi.org/10.3390/ijms222413433
APA StyleZuccato, C., Cosenza, L. C., Zurlo, M., Lampronti, I., Borgatti, M., Scapoli, C., Gambari, R., & Finotti, A. (2021). Treatment of Erythroid Precursor Cells from β-Thalassemia Patients with Cinchona Alkaloids: Induction of Fetal Hemoglobin Production. International Journal of Molecular Sciences, 22(24), 13433. https://doi.org/10.3390/ijms222413433