
Evaluation of the Impact of Esterases and Lipases from the Circulatory System against Substrates of Different Lipophilicity



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. General Considerations Related with Assay Development
2.2. Esterase and Lipase Activity Overview and Selection of Representatives
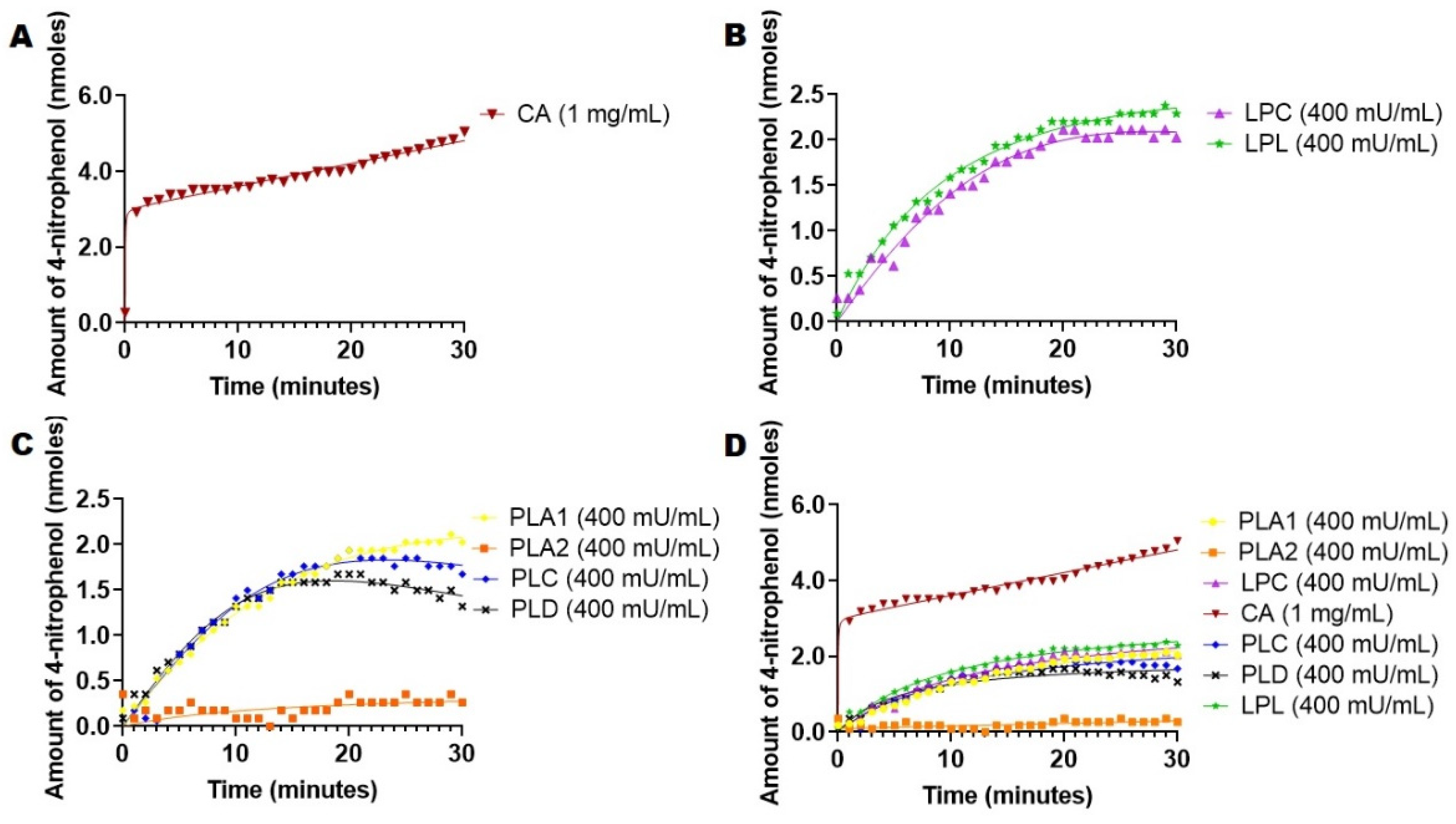
2.3. Hydrolysis of 4-Nitrophenyl Acetate Mediated by Esterases and Lipases
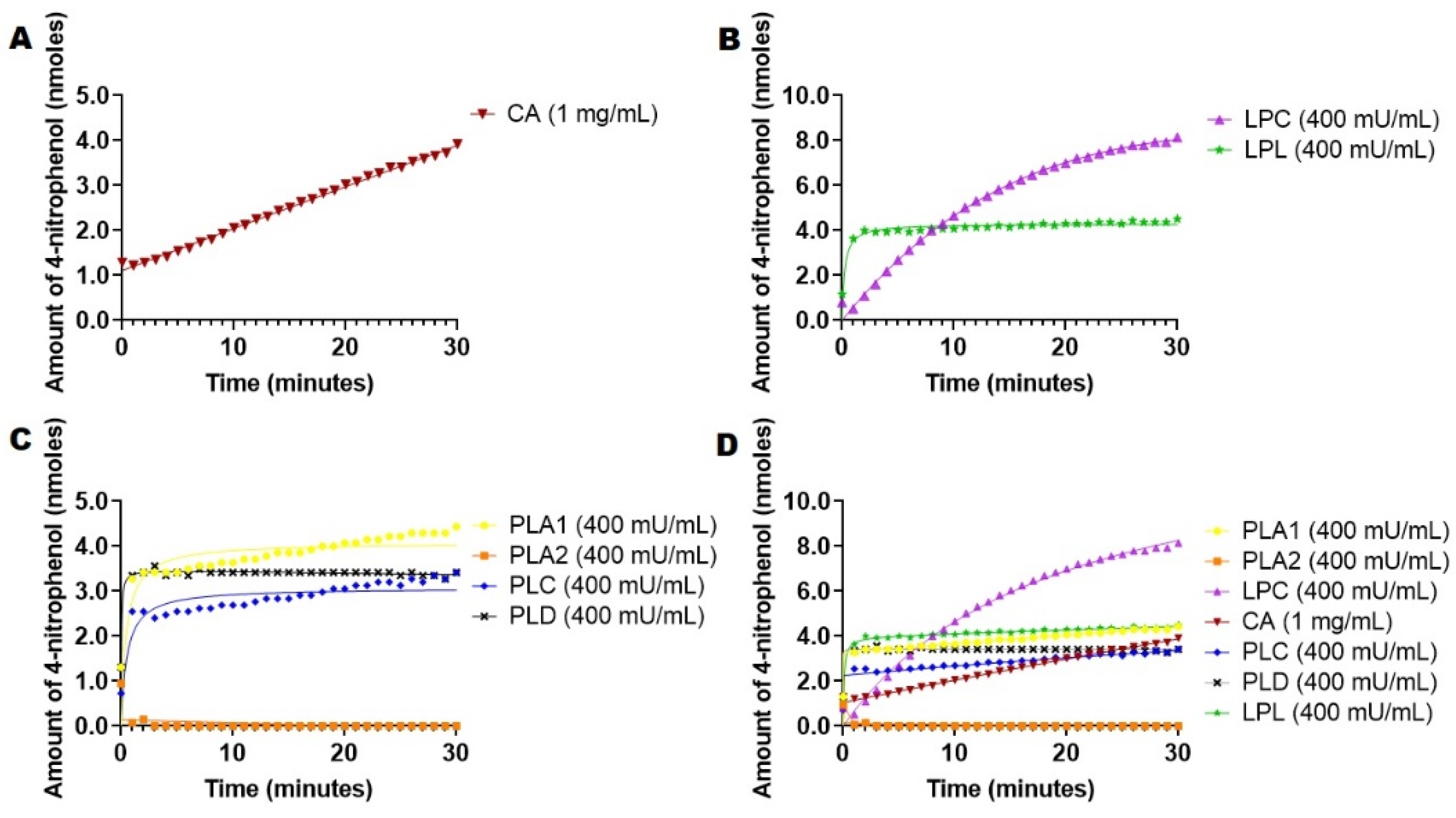
2.4. Hydrolysis of 4-Nitrophenyl Palmitate Mediated by Esterases and Lipases
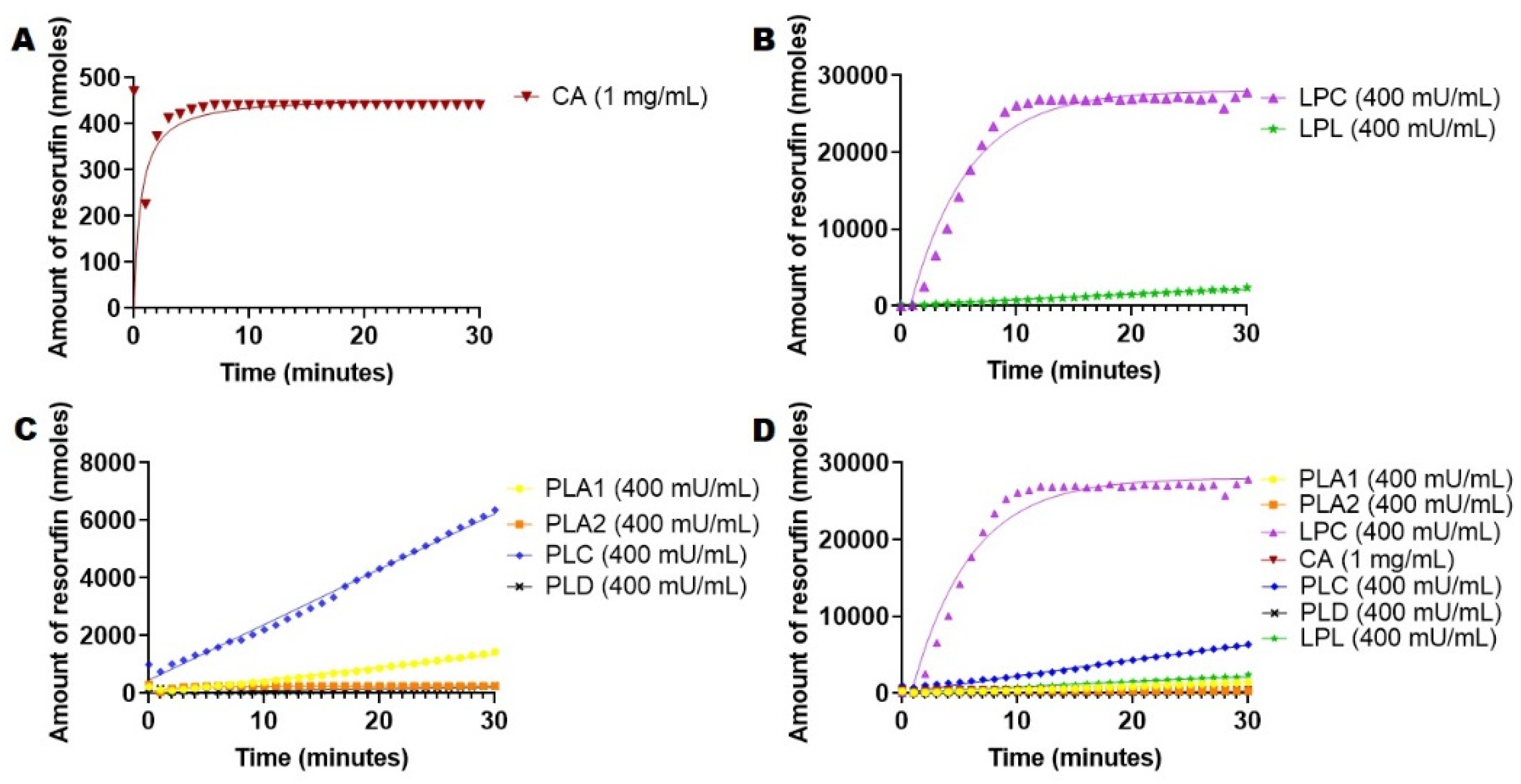
2.5. Hydrolysis of 1,2-Di-O-Lauryl-rac-glycero-3-glutaric acid 6′-methylresorufin ester (DGGR) Mediated by Esterases and Lipases
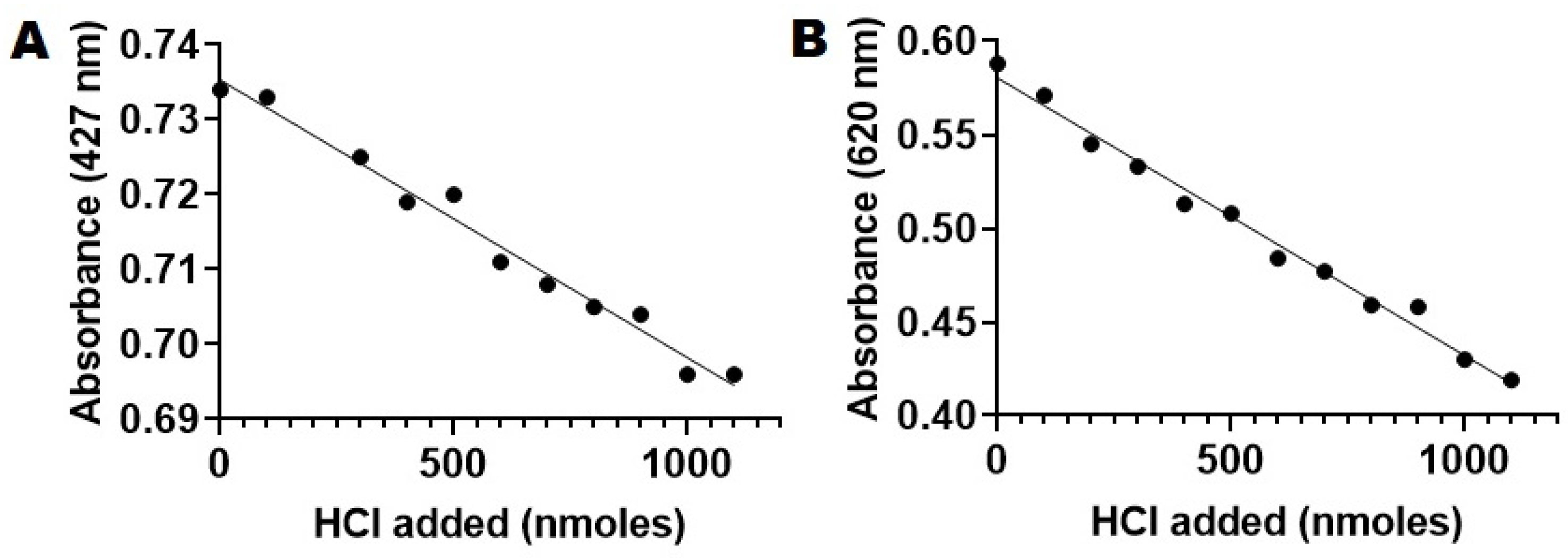
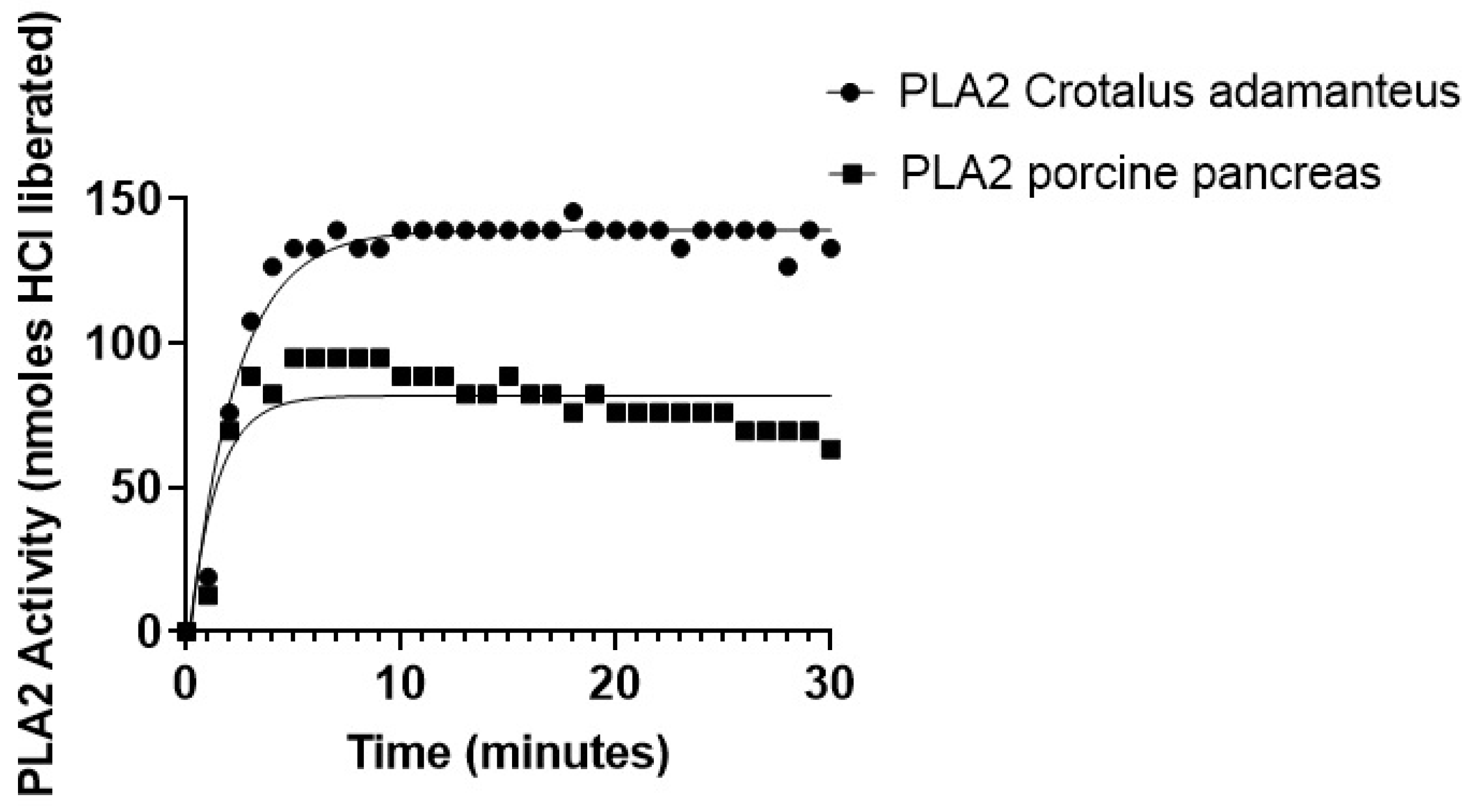
2.6. PLA2-Specific Hydrolysis of Phospholipids
3. Materials and Methods
3.1. Materials
3.2. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fukami, T.; Yokoi, T. The emerging role of human esterases. Drug Metab. Pharmacokinet. 2012, 27, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Fojan, P.; Jonson, P.H.; Petersen, M.T.N.; Petersen, S.B. What distinguishes an esterase from a lipase: A novel structural approach. Biochimie 2000, 82, 1033–1041. [Google Scholar] [CrossRef]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Unraveling the mysteries of serum albumin—More than just a serum protein. Front. Physiol. 2014, 5, 299. [Google Scholar] [CrossRef] [PubMed]
- Goncharov, N.V.; Belinskaia, D.A.; Shmurak, V.I.; Terpilowski, M.A.; Jenkins, R.O.; Avdonin, P.V. Serum Albumin Binding and Esterase Activity: Mechanistic Interactions with Organophosphates. Molecules 2017, 22, 1201. [Google Scholar] [CrossRef] [PubMed]
- Zamanova, S.; Shabana, A.M.; Mondal, U.K.; Ilies, M.A. Carbonic anhydrases as disease markers. Expert Opin. Ther. Pat. 2019, 29, 509–533. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Lemon, N.; Canepa, E.; Ilies, M.A.; Fossati, S. Carbonic Anhydrases as Potential Targets Against Neurovascular Unit Dysfunction in Alzheimer’s Disease and Stroke. Front. Aging Neurosci. 2021, 13, 785. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, M.H.; Péterfy, M. Mechanisms of lipase maturation. Clin. Lipidol. 2010, 5, 71–85. [Google Scholar] [CrossRef]
- Xiang, J.; Liu, X.; Yuan, G.; Zhang, R.; Zhou, Q.; Xie, T.; Shen, Y. Nanomedicine from amphiphilized prodrugs: Concept and clinical translation. Adv. Drug Deliv. Rev. 2021, 179, 114027. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Thompson, D.H. Stimuli-responsive liposomes for drug delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1450. [Google Scholar] [CrossRef] [PubMed]
- Satyal, U.; Sharma, V.D.; Shif, J.A.; Ilies, M.A. Interface-Engineered Amphiphilic Block Copolymers with Tuned Enzymatic Resistance for Controlled Delivery of Chemotherapeutic Drugs. In Control of Amphiphile Self-Assembling at the Molecular Level: Supra-Molecular Assemblies with Tuned Physicochemical Properties for Delivery Applications; Ilies, M.A., Ed.; American Chemical Society: Washington, DC, USA, 2017; Volume 1271, pp. 211–229. [Google Scholar]
- Acker, M.G.; Auld, D.S. Considerations for the design and reporting of enzyme assays in high-throughput screening applications. Perspect. Sci. 2014, 1, 56–73. [Google Scholar] [CrossRef]
- Innocenti, A.; Scozzafava, A.; Parkkila, S.; Puccetti, L.; De Simone, G.; Supuran, C.T. Investigations of the esterase, phosphatase, and sulfatase activities of the cytosolic mammalian carbonic anhydrase isoforms I, II, and XIII with 4-nitrophenyl esters as substrates. Bioorg. Med. Chem. Lett. 2008, 18, 2267–2271. [Google Scholar] [CrossRef] [PubMed]
- Baird, T.T., Jr.; Waheed, A.; Okuyama, T.; Sly, W.S.; Fierke, C.A. Catalysis and inhibition of human carbonic anhydrase IV. Biochemistry 1997, 36, 2669–2678. [Google Scholar] [CrossRef] [PubMed]
- Mansfeld, J.; Brandt, W.; Haftendorn, R.; Schöps, R.; Ulbrich-Hofmann, R. Discrimination between the regioisomeric 1,2- and 1,3-diacylglycerophosphocholines by phospholipases. Chem. Phys. Lipids 2011, 164, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Orasanu, G.; Nehra, V.; Asatryan, L.; Rader, D.J.; Ziouzenkova, O.; Plutzky, J. High-density lipoprotein hydrolysis by endothelial lipase activates PPARalpha: A candidate mechanism for high-density lipoprotein-mediated repression of leukocyte adhesion. Circ. Res. 2006, 98, 490–498. [Google Scholar] [CrossRef]
- Chang, C.L. Lipoprotein lipase: New roles for an “old” enzyme. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 111–115. [Google Scholar] [CrossRef] [PubMed]
- De Caro, J.D.; Rouimi, P.; Rovery, M. Hydrolysis of p-nitrophenyl acetate by the peptide chain fragment (336–449) of porcine pancreatic lipase. Eur. J. Biochem. 1986, 158, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, L.; Wang, Q.; Wu, W.; Zhang, H.; Fang, Y.; Dong, S. Bio-inspired nanozyme: A hydratase mimic in a zeolitic imidazolate framework. Nanoscale 2019, 11, 5960–5966. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, B.; Tessari, M.; Boelens, R.; Dijkman, R.; Kaptein, R.; de Haas, G.H.; Verheij, H.M. Solution structure of porcine pancreatic phospholipase A2 complexed with micelles and a competitive inhibitor. J. Biomol. NMR 1995, 5, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Kramer, R.M.; Hession, C.; Johansen, B.; Hayes, G.; McGray, P.; Chow, E.P.; Tizard, R.; Pepinsky, R.B. Structure and properties of a human non-pancreatic phospholipase A2. J. Biol. Chem. 1989, 264, 5768–5775. [Google Scholar] [CrossRef]
- Thorslund, A.; Lindskog, S. Studies of the Esterase Activity and the Anion Inhibition of Bovine Zinc and Cobalt Carbonic Anhydrases. Eur. J. Biochem. 1967, 3, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Gutfreund, H.; Sturtevant, J.M. The mechanism of the reaction of chymotrypsin with p-nitrophenyl acetate. Biochem. J. 1956, 63, 656–661. [Google Scholar] [CrossRef]
- Nelson, R.K.; Frohman, M.A. Physiological and pathophysiological roles for phospholipase D. J. Lipid Res. 2015, 56, 2229–2237. [Google Scholar] [CrossRef] [PubMed]
- Burdette, R.A.; Quinn, D.M. Interfacial reaction dynamics and acyl-enzyme mechanism for lipoprotein lipase-catalyzed hydrolysis of lipid p-nitrophenyl esters. J. Biol. Chem. 1986, 261, 12016–12021. [Google Scholar] [CrossRef]
- Dennis, E.A.; Cao, J.; Hsu, Y.H.; Magrioti, V.; Kokotos, G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef] [PubMed]
- Gururaj, P.; Ramalingam, S.; Nandhini Devi, G.; Gautam, P. Process optimization for production and purification of a thermostable, organic solvent tolerant lipase from Acinetobacter sp. AU07. Braz. J. Microbiol. 2016, 47, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, H.J.; Park, H.W.; Youn, S.H.; Choi, D.Y.; Shin, C.S. Development of inhibitors against lipase and alpha-glucosidase from derivatives of monascus pigment. FEMS Microbiol. Lett. 2007, 276, 93–98. [Google Scholar] [CrossRef]
- Thumser, A.E.; Voysey, J.E.; Wilton, D.C. The binding of lysophospholipids to rat liver fatty acid-binding protein and albumin. Biochem. J. 1994, 301 Pt 3, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Ganasen, P.; Khan, M.R.; Kalam, M.A.; Mahmud, M.S. Effect of visible light on catalytic hydrolysis of p-nitrophenyl palmitate by the Pseudomonas cepacia lipase immobilized on sol-gel support. Bioprocess Biosyst. Eng. 2014, 37, 2353–2359. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Koga, Y.; Nakano, H.; Yamane, T. Modifying the chain-length selectivity of the lipase from Burkholderia cepacia KWI-56 through in vitro combinatorial mutagenesis in the substrate-binding site. Protein Eng. Des. Sel. 2002, 15, 147–152. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bornscheuer, U.; Reif, O.-W.; Lausch, R.; Freitag, R.; Scheper, T.; Kolisis, F.N.; Menge, U. Lipase of Pseudomonas cepacia for biotechnological purposes: Purification, crystallization and characterization. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1994, 1201, 55–60. [Google Scholar] [CrossRef]
- Reis, P.; Holmberg, K.; Watzke, H.; Leser, M.E.; Miller, R. Lipases at interfaces: A review. Adv. Colloid Interface Sci. 2009, 147–148, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Sarda, L.; Desnuelle, P. Actions of pancreatic lipase on esters in emulsions. Biochim. Biophys. Acta 1958, 30, 513–521. [Google Scholar] [CrossRef]
- Jaeger, K.E.; Ransac, S.; Dijkstra, B.W.; Colson, C.; van Heuvel, M.; Misset, O. Bacterial lipases. FEMS Microbiol. Rev. 1994, 15, 29–63. [Google Scholar] [CrossRef]
- Shirai, K.; Jackson, R.L. Lipoprotein lipase-catalyzed hydrolysis of p-nitrophenyl butyrate. Interfacial activation by phospholipid vesicles. J. Biol. Chem. 1982, 257, 1253–1258. [Google Scholar] [CrossRef]
- Arima, N.; Inoue, A.; Makide, K.; Nonaka, T.; Aoki, J. Surface loops of extracellular phospholipase A(1) determine both substrate specificity and preference for lysophospholipids. J. Lipid Res. 2012, 53, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Martin, D. Phospholipases A1 from Armillaria ostoyae Provide Insight into the Substrate Recognition of α/β-Hydrolase Fold Enzymes. J. Am. Oil Chem. Soc. 2012, 89, 1435–1448. [Google Scholar]
- Lehner, R.; Verger, R. Purification and characterization of a porcine liver microsomal triacylglycerol hydrolase. Biochemistry 1997, 36, 1861–1868. [Google Scholar] [CrossRef]
- Okawa, Y.; Yamaguchi, T. Studies on phospholipases from Streptomyces. II. Purification and properties of Streptomyces hachijoensis phospholipase D. J. Biochem. 1975, 78, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.; Lee, J.; Koh, E.; Choi, M. Enzymatic Hydrolysis of p-Nitrophenyl Phsphoryl Derivatives by Phospholipase D. Bull. Korean Chem. Soc. 1994, 15, 1001–1003. [Google Scholar]
- Kim, J.; Yoon, J.; Hayward, R.C. Dynamic display of biomolecular patterns through an elastic creasing instability of stimuli-responsive hydrogels. Nat. Mater. 2010, 9, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Panteghini, M.; Bonora, R.; Pagani, F. Measurement of pancreatic lipase activity in serum by a kinetic colorimetric assay using a new chromogenic substrate. Ann. Clin. Biochem. 2001, 38 Pt 4, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Garrett, C.K.; Broadwell, L.J.; Hayne, C.K.; Neher, S.B. Modulation of the Activity of Mycobacterium tuberculosis LipY by Its PE Domain. PLoS ONE 2015, 10, e0135447. [Google Scholar] [CrossRef] [PubMed]
- Lafferty, M.J.; Bradford, K.C.; Erie, D.A.; Neher, S.B. Angiopoietin-like protein 4 inhibition of lipoprotein lipase: Evidence for reversible complex formation. J. Biol. Chem. 2013, 288, 28524–28534. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.K.; Maliwal, B.P.; DeHaas, G.H.; Slotboom, A.J. Anchoring of phospholipase A2: The effect of anions and deuterated water, and the role of N-terminus region. Biochim. Biophys. Acta 1986, 860, 448–461. [Google Scholar] [CrossRef]
- Khan, A.K.; Ho, J.C.S.; Roy, S.; Liedberg, B.; Nallani, M. Facile Mixing of Phospholipids Promotes Self-Assembly of Low-Molecular-Weight Biodegradable Block Co-Polymers into Functional Vesicular Architectures. Polymers 2020, 12, 979. [Google Scholar] [CrossRef]
- Lindström, M.B.; Persson, J.; Thurn, L.; Borgström, B. Effect of pancreatic phospholipase A2 and gastric lipase on the action of pancreatic carboxyl ester lipase against lipid substrates in vitro. Biochim. Biophys. Acta 1991, 1084, 194–197. [Google Scholar] [CrossRef]
- Koops, B.C.; Slotboom, A.J.; Verheij, H.M. Comparative studies on selective modification of ε-amino groups in Upases and phospholipase A2. In Progress in Biotechnology; Ballesteros, A., Plou, F.J., Iborra, J.L., Halling, P.J., Eds.; Elsevier: Amsterdam, The Netherlands, 1998; Volume 15, pp. 127–134. [Google Scholar]
- Salhi, A.; Amara, S.; Mansuelle, P.; Puppo, R.; Lebrun, R.; Gontero, B.; Aloulou, A.; Carrière, F. Characterization of all the lipolytic activities in pancreatin and comparison with porcine and human pancreatic juices. Biochimie 2020, 169, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Venuti, E.; Shishmarev, D.; Kuchel, P.W.; Dutt, S.; Blumenthal, C.S.; Gaskin, K.J. Bile salt stimulated lipase: Inhibition by phospholipids and relief by phospholipase A(2). J. Cyst. Fibros. 2017, 16, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.A.; Hanahan, D.J. Phospholipase A. I. Isolation and characterization of two enzymes from Crotalus adamanteus venom. Biochemistry 1969, 8, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Price, J.A., 3rd. A colorimetric assay for measuring phospholipase A2 degradation of phosphatidylcholine at physiological pH. J. Biochem. Biophys. Methods 2007, 70, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Wang, M.H.; Zhao, K.Y.; Wang, C.C. Assay for enzyme activity by following the absorbance change of pH-indicators. J. Biochem. Biophys. Methods 1998, 36, 119–130. [Google Scholar] [CrossRef]
- El Nahhal, I.M.; Zourab, S.M.; Kodeh, F.S.; Qudaih, A.I. Thin film optical BTB pH sensors using sol–gel method in presence of surfactants. Int. Nano Lett. 2012, 2, 16. [Google Scholar] [CrossRef]
- Cunningham, T.J.; Yao, L.; Lucena, A. Product inhibition of secreted phospholipase A2 may explain lysophosphatidylcholines' unexpected therapeutic properties. J. Inflamm. 2008, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.R.; Liu, D.J. Direct Measurement of Lipase Inhibition by Orlistat Using a Dissolution Linked In Vitro Assay. Clin. Pharmacol. Biopharm. 2012, 1, 1000103. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Rathi, P.; Gupta, R. Simplified para-nitrophenyl palmitate assay for lipases and esterases. Anal. Biochem. 2002, 311, 98–99. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lam, L.; Ilies, M.A. Evaluation of the Impact of Esterases and Lipases from the Circulatory System against Substrates of Different Lipophilicity. Int. J. Mol. Sci. 2022, 23, 1262. https://doi.org/10.3390/ijms23031262
Lam L, Ilies MA. Evaluation of the Impact of Esterases and Lipases from the Circulatory System against Substrates of Different Lipophilicity. International Journal of Molecular Sciences. 2022; 23(3):1262. https://doi.org/10.3390/ijms23031262
Chicago/Turabian StyleLam, Leslie, and Marc A. Ilies. 2022. "Evaluation of the Impact of Esterases and Lipases from the Circulatory System against Substrates of Different Lipophilicity" International Journal of Molecular Sciences 23, no. 3: 1262. https://doi.org/10.3390/ijms23031262
APA StyleLam, L., & Ilies, M. A. (2022). Evaluation of the Impact of Esterases and Lipases from the Circulatory System against Substrates of Different Lipophilicity. International Journal of Molecular Sciences, 23(3), 1262. https://doi.org/10.3390/ijms23031262