

Calcium Signalling in Heart and Vessels: Role of Calmodulin and Downstream Calmodulin-Dependent Protein Kinases

 , , , and
, , , and 
Abstract
1. Introduction
The Role of Calcium in the Cardiovascular System
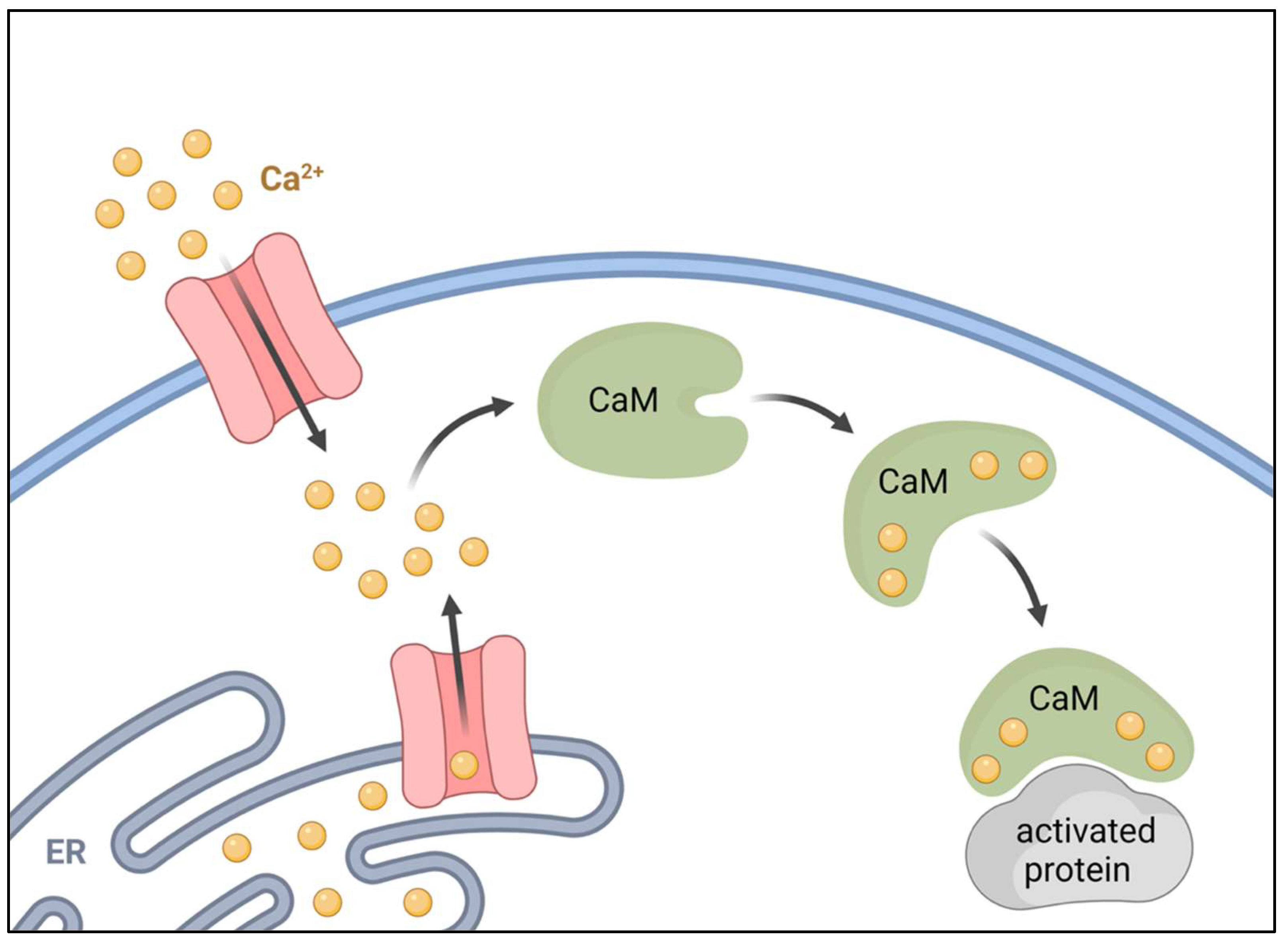
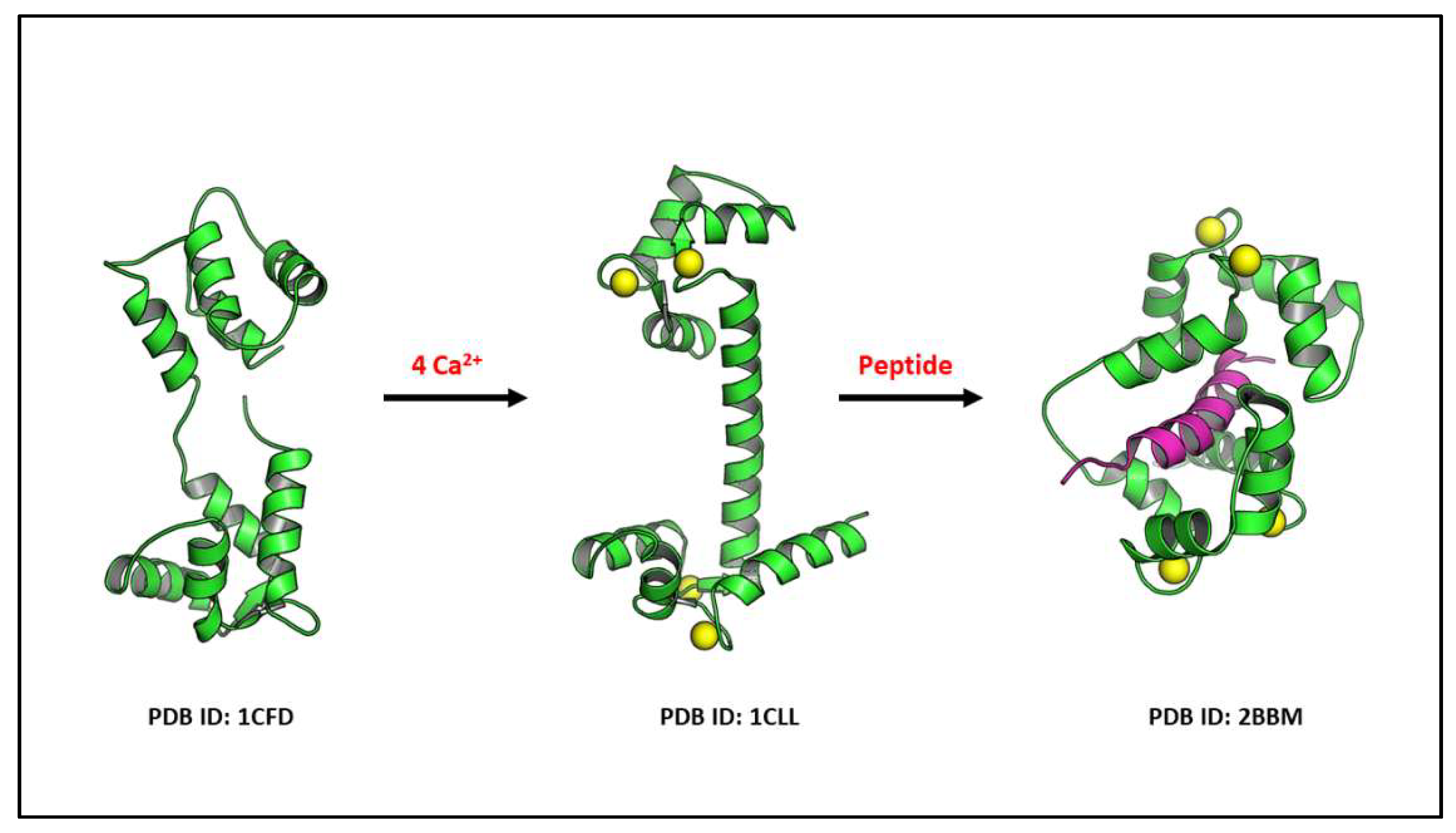
2. Calmodulin
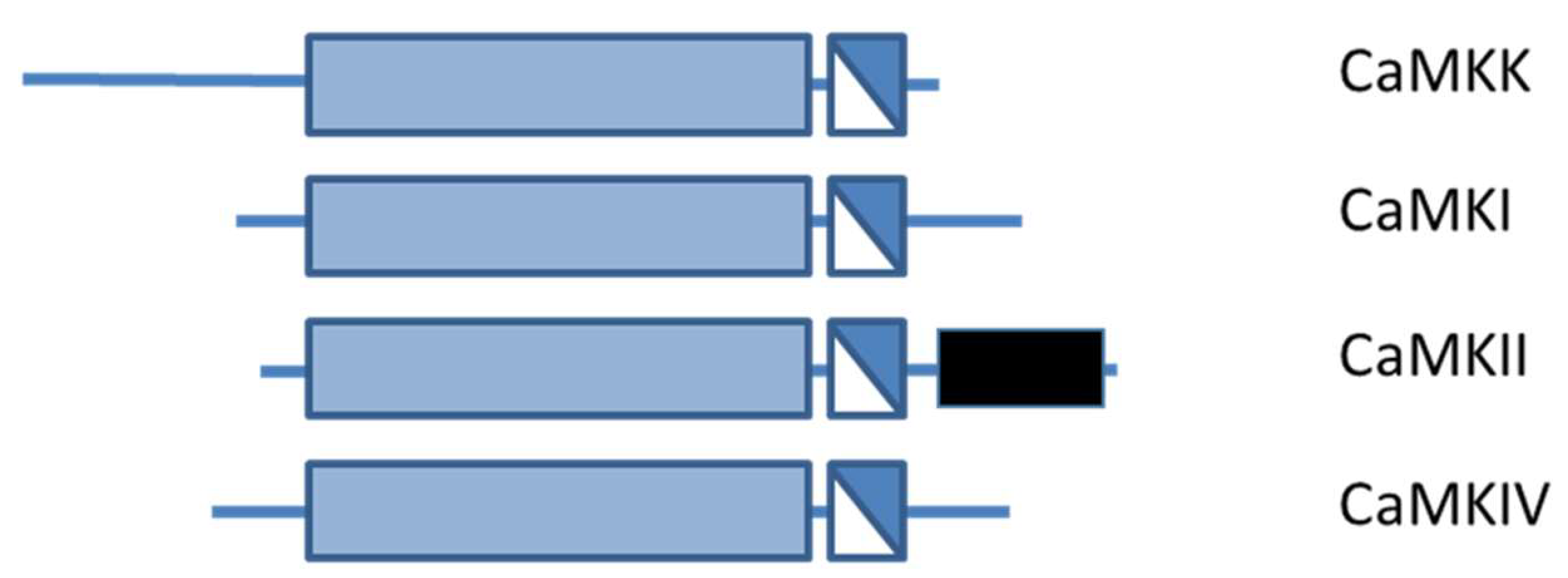
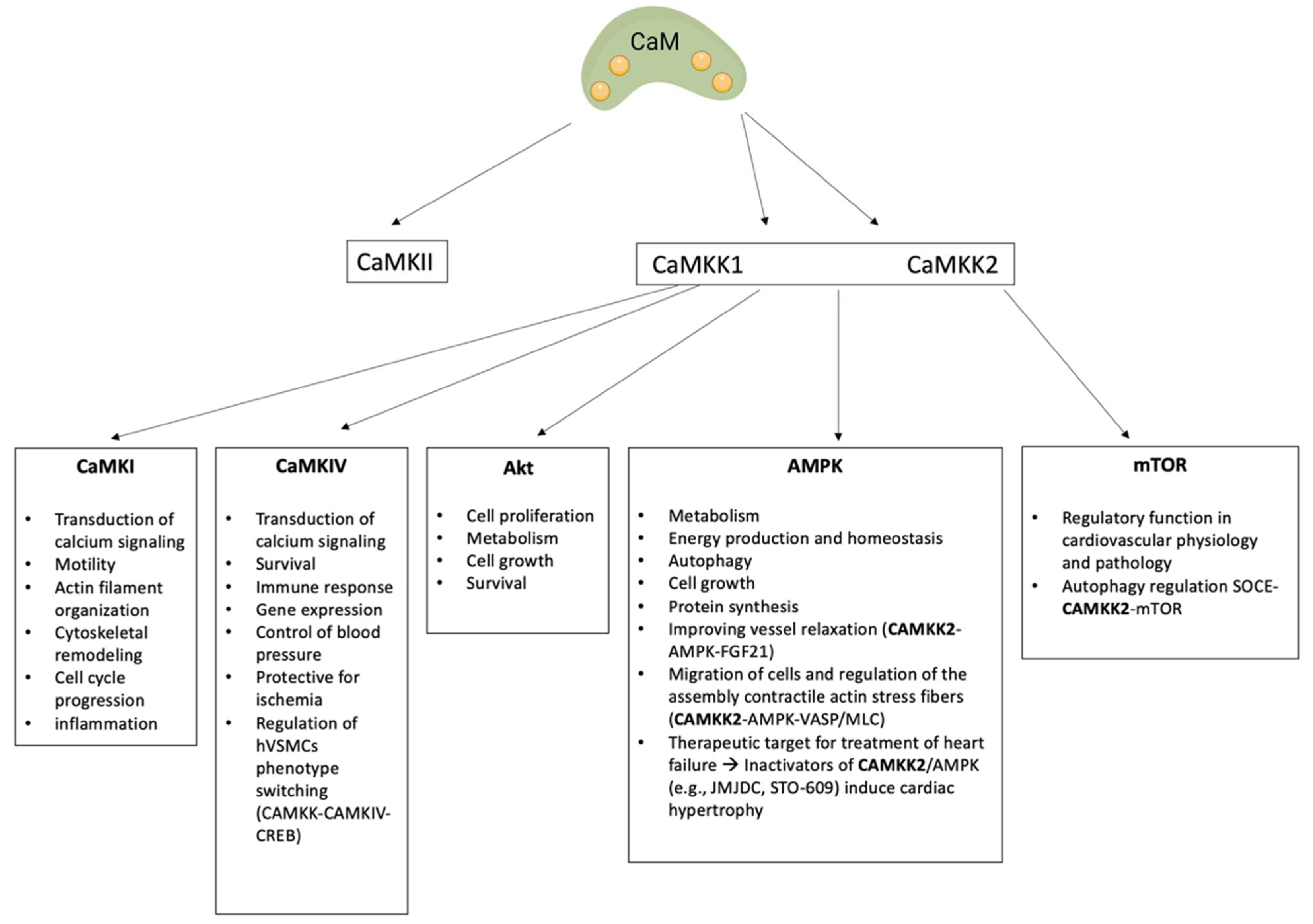
3. Calcium Calmodulin Kinases
3.1. CAMKII
3.2. Ca2+-CaM-Dependent Kinase Cascade
3.2.1. CaMKK Family
CAMKK1
CAMKK2
3.2.2. CaMKI and CaMKIV
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Poulter, N. Coronary Heart Disease Is a Multifactorial Disease. Am. J. Hypertens. 1999, 12, 92S–95S. [Google Scholar] [CrossRef]
- Dhingra, R.; Vasan, R.S. Biomarkers in Cardiovascular Disease: Statistical Assessment and Section on Key Novel Heart Failure Biomarkers. Trends Cardiovasc. Med. 2017, 27, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Soler-Botija, C.; Gálvez-Montón, C.; Bayés-Genís, A. Epigenetic Biomarkers in Cardiovascular Diseases. Front. Genet. 2019, 10, 950. [Google Scholar] [CrossRef] [PubMed]
- Ortaliza, J.; Orgera, K.; Amin, K.; Cox, C. COVID-19 Continues to Be a Leading Cause of Death in the U.S. in June 2021. Available online: https://www.healthsystemtracker.org (accessed on 27 August 2021).
- Libby, P. The Changing Landscape of Atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Tokumitsu, H.; Soderling, T.R. Requirements for Calcium and Calmodulin in the Calmodulin Kinase Activation Cascade. J. Biol. Chem. 1996, 271, 5617–5622. [Google Scholar] [CrossRef] [PubMed]
- Junho, C.V.C.; Caio-Silva, W.; Trentin-Sonoda, M.; Carneiro-Ramos, M.S. An Overview of the Role of Calcium/Calmodulin-Dependent Protein Kinase in Cardiorenal Syndrome. Front. Physiol. 2020, 11, 735. [Google Scholar] [CrossRef]
- Yu, X.; Murao, K.; Sayo, Y.; Imachi, H.; Cao, W.M.; Ohtsuka, S.; Niimi, M.; Tokumitsu, H.; Inuzuka, H.; Wong, N.C.W.; et al. The Role of Calcium/Calmodulin-Dependent Protein Kinase Cascade in Glucose Upregulation of Insulin Gene Expression. Diabetes 2004, 53, 1475–1481. [Google Scholar] [CrossRef][Green Version]
- Soderling, T.R. The Ca2+–Calmodulin-Dependent Protein Kinase Cascade. Trends Biochem. Sci. 1999, 24, 232–236. [Google Scholar] [CrossRef]
- Kitsos, C.M.; Sankar, U.; Illario, M.; Colomer-Font, J.M.; Duncan, A.W.; Ribar, T.J.; Reya, T.; Means, A.R. Calmodulin-Dependent Protein Kinase IV Regulates Hematopoietic Stem Cell Maintenance. J. Biol. Chem. 2005, 280, 33101–33108. [Google Scholar] [CrossRef]
- Racioppi, L.; Lento, W.; Huang, W.; Arvai, S.; Doan, P.L.; Harris, J.R.; Marcon, F.; Nakaya, H.I.; Liu, Y.; Chao, N. Calcium/Calmodulin-Dependent Kinase Kinase 2 Regulates Hematopoietic Stem and Progenitor Cell Regeneration. Cell Death Dis. 2017, 8, e3076. [Google Scholar] [CrossRef]
- Colomer, J.; Means, A.R. Physiological Roles of the Ca2+/CaM-Dependent Protein Kinase Cascade in Health and Disease. Calcium Signal. Dis. 2007, 45, 169–214. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of Smooth Muscle Cells in Vascular Calcification: Implications in Atherosclerosis and Arterial Stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Jaminon, A.; Reesink, K.; Kroon, A.; Schurgers, L. The Role of Vascular Smooth Muscle Cells in Arterial Remodeling: Focus on Calcification-Related Processes. Int. J. Mol. Sci. 2019, 20, 5694. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D. Molecular Mechanisms of Arterial Calcification. Artery Res. 2009, 3, 1423–1430. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, I.K.; Jeon, J.H. Vascular Calcification—New Insights into Its Mechanism. Int. J. Mol. Sci. 2020, 21, 2685. [Google Scholar] [CrossRef]
- Krohn, J.B.; Hutcheson, J.D.; Martínez-Martínez, E.; Aikawa, E. Extracellular Vesicles in Cardiovascular Calcification: Expanding Current Paradigms. J. Physiol. 2016, 594, 2895–2903. [Google Scholar] [CrossRef]
- Christensen, J.L.; Tan, S.; Chung, H.E.; Ghosalkar, D.S.; Qureshi, R.; Chu, A.; Yu, W.; Berus, J.; Shah, N.R.; Wu, W.C.; et al. Aortic Valve Calcification Predicts All-Cause Mortality Independent of Coronary Calcification and Severe Stenosis. Atherosclerosis 2020, 307, 16–20. [Google Scholar] [CrossRef]
- Veulemans, V.; Piayda, K.; Maier, O.; Bosbach, G.; Polzin, A.; Hellhammer, K.; Afzal, S.; Klein, K.; Dannenberg, L.; Zako, S.; et al. Aortic Valve Calcification Is Subject to Aortic Stenosis Severity and the Underlying Flow Pattern. Heart Vessel. 2021, 36, 242–251. [Google Scholar] [CrossRef]
- Di Lullo, L.; Tripepi, G.; Ronco, C.; D’Arrigo, G.; Barbera, V.; Russo, D.; di Iorio, B.R.; Uguccioni, M.; Paoletti, E.; Ravera, M.; et al. Cardiac Valve Calcification and Use of Anticoagulants: Preliminary Observation of a Potentially Modifiable Risk Factor. Int. J. Cardiol. 2019, 278, 243–249. [Google Scholar] [CrossRef]
- Terrar, D.A. Calcium Signaling in the Heart. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2020; Volume 1131. [Google Scholar] [CrossRef]
- Fearnley, C.J.; Llewelyn Roderick, H.; Bootman, M.D. Calcium Signaling in Cardiac Myocytes. Cold Spring Harb. Perspect Biol. 2011, 3, a004242. [Google Scholar] [CrossRef]
- Reid, I.R.; Birstow, S.M.; Bolland, M. Calcium and Cardiovascular Disease. Endocrinol. Metab. 2017, 32, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Schreckenberg, R.; Schlüter, K.-D. Calcium sensing receptor expression and signalling in cardiovascular physiology and disease. Vasc. Pharmacol. 2018, 107, 35–42. [Google Scholar] [CrossRef]
- Sundararaman, S.S.; van der Vorst, E.P.C. Calcium-Sensing Receptor (CaSR), Its Impact on Inflammation and the Consequences on Cardiovascular Health. Int. J. Mol. Sci. 2021, 22, 2478. [Google Scholar] [CrossRef] [PubMed]
- Alfadda, T.I.; Saleh, A.M.A.; Houillier, P.; Geibel, J.P. Calcium-sensing receptor 20 years later. Am. J. Physiol. Cell Physiol. 2014, 307, C221–C231. [Google Scholar] [CrossRef] [PubMed]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef]
- Zhang, C.; Miller, C.L.; Gorkhali, R.; Zou, J.; Huang, K.; Brown, E.M.; Yang, J.J. Molecular Basis of the Extracellular Ligands Mediated Signaling by the Calcium Sensing Receptor. Front. Physiol. 2016, 7, 441. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef]
- Cooper, D.; Dimri, M. Biochemistry, Calcium Channels; StatPearls: Treasure Island, FL, USA, 2022; PMID: 32965869. [Google Scholar]
- Muszbek, L.; Bereczky, Z.; Bagoly, Z.; Komáromi, I.; Katona, É. Factor XIII: A Coagulation Factor with Multiple Plasmatic and Cellular Functions. Physiol. Rev. 2011, 91, 931–972. [Google Scholar] [CrossRef]
- Singh, S.; Dodt, J.; Volkers, P.; Hethershaw, E.; Philippou, H.; Ivaskevicius, V.; Imhof, D.; Oldenburg, J.; Biswas, A. Structure functional insights into calcium binding during the activation of coagulation factor XIII A. Sci. Rep. 2019, 9, 11324. [Google Scholar] [CrossRef]
- Matchkov, V.V.; Kudryavtseva, O.; Aalkjaer, C. Intracellular Ca2+ Signalling and Phenotype of Vascular Smooth Muscle Cells. Basic Clin. Pharmacol. Toxicol. 2012, 110, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Petsophonsakul, P.; Furmanik, M.; Forsythe, R.; Dweck, M.; Schurink, G.W.; Natour, E.; Reutelingsperger, C.; Jacobs, M.; Mees, B.; Schurgers, L. Role of Vascular Smooth Muscle Cell Phenotypic Switching and Calcification in Aortic Aneurysm Formation Involvement of Vitamin K-Dependent Processes. Arter. Thromb. Vasc. Biol. 2019, 39, 1351–1368. [Google Scholar] [CrossRef] [PubMed]
- Furmanik, M.; Chatrou, M.; van Gorp, R.; Akbulut, A.; Willems, B.; Schmidt, H.; van Eys, G.; Bochaton-Piallat, M.-L.; Proudfoot, D.; Biessen, E.; et al. Reactive Oxygen-Forming Nox5 Links Vascular Smooth Muscle Cell Phenotypic Switching and Extracellular Vesicle-Mediated Vascular Calcification. Circ. Res. 2020, 127, 911–927. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zou, B.; Hou, Y.; Yan, W.; Chen, T.; Qu, S. Extracellular vesicles in vascular calcification. Clin. Chim. Acta 2019, 499, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, A.B.; Søndergaard, M.T.; Overgaard, M.T. Calmodulin in a Heartbeat. FEBS J. 2013, 280, 5511–5532. [Google Scholar] [CrossRef]
- Beghi, S.; Cavaliere, F.; Buschini, A. Gene polymorphisms in calcium-calmodulin pathway: Focus on cardiovascular disease. Mutat. Res. Mutat. Res. Rev. 2020, 786, 108325. [Google Scholar] [CrossRef]
- Babu, Y.S.; Sack, J.S.; Greenhough, T.J.; Bugg, C.E.; Means, A.R.; Cook, W.J. Three-dimensional structure of calmodulin. Nature 1985, 315, 37–40. [Google Scholar] [CrossRef]
- Finn, B.; Forsén, S. The evolving model of calmodulin structure, function and activation. Structure 1995, 3, 7–11. [Google Scholar] [CrossRef]
- Ye, Q.; Wei, Y.; Fischer, R.; Borner, C.; Berchtold, M.W. Expression of calmodulin and calmodulin binding proteins in rat fibroblasts stably transfected with protein kinase C and oncogenes. Biochim. Biophys. Acta Mol. Cell Res. 1997, 1359, 89–96. [Google Scholar] [CrossRef]
- Halling, D.B.; Liebeskind, B.J.; Hall, A.W.; Aldrich, R.W. Conserved properties of individual Ca 2+ -binding sites in calmodulin. Proc. Natl. Acad. Sci. USA 2016, 113, E1216–E1225. [Google Scholar] [CrossRef]
- Friedberg, F.; Rhoads, A.R.; Friedberg, A.R.R.F. Evolutionary Aspects of Calmodulin. IUBMB Life 2001, 51, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Brini, M. Plasma membrane Ca2+-ATPase: From a housekeeping function to a versatile signaling role. Pflugers Arch. 2009, 457, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, C.; Zhao, Q.; Li, D. Spectrin: Structure, function and disease. Sci. China Life Sci. 2013, 56, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Hartl, M.; Schneider, R. A Unique Family of Neuronal Signaling Proteins Implicated in Oncogenesis and Tumor Suppression. Front. Oncol. 2019, 9, 289. [Google Scholar] [CrossRef] [PubMed]
- Brozovich, F.; Nicholson, C.; Degen, C.; Gao, Y.Z.; Aggarwal, M.; Morgan, K. Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol. Rev. 2016, 68, 476–532. [Google Scholar] [CrossRef]
- Means, A.R.; VanBerkum, M.F.; Bagchi, I.; Lu, K.P.; Rasmussen, C.D. Regulatory functions of calmodulin. Pharmacol. Ther. 1991, 50, 255–270. [Google Scholar] [CrossRef]
- Abriel, H. Cardiac sodium channel Nav1.5 and interacting proteins: Physiology and pathophysiology. J. Mol. Cell. Cardiol. 2010, 48, 2–11. [Google Scholar] [CrossRef]
- Kang, P.W.; Westerlund, A.M.; Shi, J.; White, K.M.F.; Dou, A.K.; Cui, A.H.; Silva, J.R.; Delemotte, L.; Cui, J. Calmodulin acts as a state-dependent switch to control a cardiac potassium channel opening. Sci. Adv. 2020, 6, eabd6798. [Google Scholar] [CrossRef]
- Muir, W.W. Cardiovascular Physiology. In Veterinary Anesthesia and Analgesia: The Fifth Edition of Lumb and Jones; Wiley: Hoboken, NJ, USA, 2017; pp. 415–472. [Google Scholar] [CrossRef]
- Yang, D.; Song, L.-S.; Zhu, W.-Z.; Chakir, K.; Wang, W.; Wu, C.; Wang, Y.; Xiao, R.-P.; Chen, S.R.W.; Cheng, H. Calmodulin Regulation of Excitation-Contraction Coupling in Cardiac Myocytes. Circ. Res. 2003, 92, 659–667. [Google Scholar] [CrossRef]
- Pitt, G.S. Calmodulin and CaMKII as molecular switches for cardiac ion channels: Fig. 1. Cardiovasc. Res. 2007, 73, 641–647. [Google Scholar] [CrossRef]
- Ogawa, H.; Kurebayashi, N.; Yamazawa, T.; Murayama, T. Regulatory mechanisms of ryanodine receptor/Ca2+ release channel revealed by recent advancements in structural studies. J. Muscle Res. Cell Motil. 2021, 42, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation–contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhu, W.; Wang, S.; Yang, D.; Crow, M.T.; Xiao, R.-P.; Cheng, H. Sustained B1 -Adrenergic Stimulation Modulates Cardiac Contractility by Ca 2+ /Calmodulin Kinase Signaling Pathway. Circ. Res. 2004, 95, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Kotera, J. Overview of PDEs and Their Regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Crotti, L.; Johnson, C.N.; Graf, E.; de Ferrari, G.M.; Cuneo, B.F.; Ovadia, M.; Papagiannis, J.; Feldkamp, M.D.; Rathi, S.G.; Kunic, J.D.; et al. Calmodulin Mutations Associated with Recurrent Cardiac Arrest in Infants. Circulation 2013, 127, 1009–1017. [Google Scholar] [CrossRef]
- Baba, M.L.; Goodman, M.; Berger-Cohn, J.; Demaille, J.G.; Matsuda, G. The early adaptive evolution of calmodulin. Mol. Biol. Evol. 1984, 1, 442–455. [Google Scholar] [CrossRef][Green Version]
- Kotta, M.-C.; Sala, L.; Ghidoni, A.; Badone, B.; Ronchi, C.; Parati, G.; Zaza, A.; Crotti, L. Calmodulinopathy: A Novel, Life-Threatening Clinical Entity Affecting the Young. Front. Cardiovasc. Med. 2018, 5, 175. [Google Scholar] [CrossRef]
- Makita, N.; Yagihara, N.; Crotti, L.; Johnson, C.N.; Beckmann, B.-M.; Roh, M.S.; Shigemizu, D.; Lichtner, P.; Ishikawa, T.; Aiba, T.; et al. Novel Calmodulin Mutations Associated with Congenital Arrhythmia Susceptibility. Circ. Cardiovasc. Genet. 2014, 7, 466–474. [Google Scholar] [CrossRef]
- Reed, G.J.; Boczek, N.J.; Etheridge, S.P.; Ackerman, M.J. CALM3 mutation associated with long QT syndrome. Heart Rhythm. 2015, 12, 419–422. [Google Scholar] [CrossRef]
- Pipilas, D.C.; Johnson, C.N.; Webster, G.; Schlaepfer, J.; Fellmann, F.; Sekarski, N.; Wren, L.M.; Ogorodnik, K.V.; Chazin, D.M.; Chazin, W.J.; et al. Novel calmodulin mutations associated with congenital long QT syndrome affect calcium current in human cardiomyocytes. Heart Rhythm. 2016, 13, 2012–2019. [Google Scholar] [CrossRef]
- Chaix, M.-A.; Koopmann, T.T.; Goyette, P.; Alikashani, A.; Latour, F.; Fatah, M.; Hamilton, R.M.; Rioux, J.D. Novel CALM3 mutations in pediatric long QT syndrome patients support a CALM3 -specific calmodulinopathy. Heart Rhythm Case Rep. 2016, 2, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Nyegaard, M.; Overgaard, M.T.; Søndergaard, M.T.; Vranas, M.; Behr, E.R.; Hildebrandt, L.L.; Lund, J.; Hedley, P.L.; Camm, A.J.; Wettrell, G.; et al. Mutations in Calmodulin Cause Ventricular Tachycardia and Sudden Cardiac Death. Am. J. Hum. Genet. 2012, 91, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Marsman, R.F.; Barc, J.; Beekman, L.; Alders, M.; Dooijes, D.; van den Wijngaard, A.; Ratbi, I.; Sefiani, A.; Bhuiyan, Z.A.; Wilde, A.A.; et al. A Mutation in CALM1 Encoding Calmodulin in Familial Idiopathic Ventricular Fibrillation in Childhood and Adolescence. J. Am. Coll. Cardiol. 2014, 63, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Tester, D.J.; Will, M.L.; Ackerman, M.J. Whole-Exome Molecular Autopsy After Exertion-Related Sudden Unexplained Death in the Young. Circ. Cardiovasc. Genet. 2016, 9, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Marsman, R.F.; Barc, J.G.; Beekman, L.; Bhuijan, Z.A.; Wilde, A.A.M.; Bezzina, C.R. A mutation in CALM1 encoding calmodulin causes sudden cardiac death in childhood and adolescence. Eur. Heart J. 2013, 34, P2298. [Google Scholar] [CrossRef]
- Koledova, V.V.; Khalil, R.A. Ca2+, Calmodulin, and Cyclins in Vascular Smooth Muscle Cell Cycle. Circ. Res. 2006, 98, 1240–1243. [Google Scholar] [CrossRef]
- Hughes, A. Calcium Channels in Vascular Smooth Muscle Cells. J. Vasc. Res. 1995, 32, 353–370. [Google Scholar] [CrossRef]
- Clunn, G.F.; Sever, P.S.; Hughes, A.D. Calcium channel regulation in vascular smooth muscle cells: Synergistic effects of statins and calcium channel blockers. Int. J. Cardiol. 2010, 139, 2–6. [Google Scholar] [CrossRef]
- Zhu, Y.; Qu, J.; He, L.; Zhang, F.; Zhou, Z.; Yang, S.; Zhou, Y. Calcium in Vascular Smooth Muscle Cell Elasticity and Adhesion: Novel Insights into the Mechanism of Action. Front. Physiol. 2019, 10, 852. [Google Scholar] [CrossRef]
- Swulius, M.T.; Waxham, M.N. Ca2+/Calmodulin-dependent Protein Kinases. Cell. Mol. Life Sci. 2008, 65, 2637–2657. [Google Scholar] [CrossRef]
- Brzozowski, J.S.; Skelding, K.A. The Multi-Functional Calcium/Calmodulin Stimulated Protein Kinase (CaMK) Family: Emerging Targets for Anti-Cancer Therapeutic Intervention. Pharmaceuticals 2019, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Westphal, R.S.; Anderson, K.A.; Means, A.R.; Wadzinski, B.E. A Signaling Complex of Ca2+-Calmodulin-Dependent Protein Kinase IV and Protein Phosphatase 2A. Science 1998, 280, 1258–1261. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.; Hanson, P.I.; Stryer, L.; Schulman, H. Calmodulin Trapping by Calcium-Calmodulin-Dependent Protein Kinase. Science 1992, 256, 1199–1202. [Google Scholar] [CrossRef] [PubMed]
- Beckendorf, J.; van den Hoogenhof, M.M.G.; Backs, J. Physiological and unappreciated roles of CaMKII in the heart. Basic Res. Cardiol. 2018, 113, 29. [Google Scholar] [CrossRef]
- Joseph, J.D.; Means, A.R. Identification and Characterization of Two Ca2+/CaM-dependent Protein Kinases Required for Normal Nuclear Division in Aspergillus nidulans. J. Biol. Chem. 2000, 275, 38230–38238. [Google Scholar] [CrossRef]
- Skelding, K.A.; Rostas, J.A.P.; Verrills, N.M. Controlling the cell cycle: The role of calcium/calmodulin-stimulated protein kinases I and II. Cell Cycle 2011, 10, 631–639. [Google Scholar] [CrossRef]
- Wayman, G.A.; Kaech, S.; Grant, W.F.; Davare, M.; Impey, S.; Tokumitsu, H.; Nozaki, N.; Banker, G.; Soderling, T.R. Regulation of Axonal Extension and Growth Cone Motility by Calmodulin-Dependent Protein Kinase I. J. Neurosci. 2004, 24, 3786–3794. [Google Scholar] [CrossRef]
- Condon, J.C.; Pezzi, V.; Drummond, B.M.; Yin, S.; Rainey, W.E. Calmodulin-Dependent Kinase I Regulates Adrenal Cell Expression of Aldosterone Synthase. Endocrinology 2002, 143, 3651–3657. [Google Scholar] [CrossRef]
- Schmitt, J.M.; Guire, E.S.; Saneyoshi, T.; Soderling, T.R. Calmodulin-Dependent Kinase Kinase/Calmodulin Kinase I Activity Gates Extracellular-Regulated Kinase-Dependent Long-Term Potentiation. J. Neurosci. 2005, 25, 1281–1290. [Google Scholar] [CrossRef]
- Ang, E.S.M.; Zhang, P.; Steer, J.H.; Tan, J.W.-Y.; Yip, K.; Zheng, M.H.; Joyce, D.A.; Xu, J. Calcium/calmodulin-dependent kinase activity is required for efficient induction of osteoclast differentiation and bone resorption by receptor activator of nuclear factor kappa B ligand (RANKL). J. Cell. Physiol. 2007, 212, 787–795. [Google Scholar] [CrossRef]
- Soderling, T.R.; Derkach, V.A. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 2000, 23, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Von Hertzen, L.S.J.; Giese, K.P. Alpha-isoform of Ca2+/calmodulin-dependent kinase II autophosphorylation is required for memory consolidation-specific transcription. NeuroReport 2005, 16, 1411–1414. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-W. Regulation of Synaptic Transmission by Presynaptic CaMKII and BK Channels. Mol. Neurobiol. 2008, 38, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Fink, C.C.; Bayer, K.-U.; Myers, J.W.; Ferrell, J.E.; Schulman, H.; Meyer, T. Selective Regulation of Neurite Extension and Synapse Formation by the β but not the α Isoform of CaMKII. Neuron 2003, 39, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Rostas, J.A.P.; Spratt, N.J.; Hoffman, A.; Murtha, L.A.; Pepperall, D.; McLeod, D.D.; Dickson, P.W.; Skelding, K.A. The Role of Ca2+-Calmodulin Stimulated Protein Kinase II in Ischaemic Stroke—A Potential Target for Neuroprotective Therapies. Neurochem. Int. 2017, 107, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Rostas, J.A.P.; Hoffman, A.; Murtha, L.A.; Pepperall, D.; McLeod, D.D.; Dickson, P.W.; Spratt, N.J.; Skelding, K.A. Ischaemia- and excitotoxicity-induced CaMKII-Mediated neuronal cell death: The relative roles of CaMKII autophosphorylation at T286 and T253. Neurochem. Int. 2017, 104, 6–10. [Google Scholar] [CrossRef]
- Liu, W.; Xu, C.; Ran, D.; Wang, Y.; Zhao, H.; Gu, J.; Liu, X.; Bian, J.; Yuan, Y.; Liu, Z. CaMKⅡ mediates cadmium induced apoptosis in rat primary osteoblasts through MAPK activation and endoplasmic reticulum stress. Toxicology 2018, 406–407, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.K.; Kim, M.-K.; Bae, Y.-S.; Jo, I.; Lee, S.-J.; Chung, C.-P.; Park, Y.-J.; Min, D.S. A novel collagen-binding peptide promotes osteogenic differentiation via Ca2+/calmodulin-dependent protein kinase II/ERK/AP-1 signaling pathway in human bone marrow-derived mesenchymal stem cells. Cell. Signal. 2008, 20, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Takemura, M.; Mishima, T.; Wang, Y.; Kasahara, J.; Fukunaga, K.; Ohashi, K.; Mizuno, K. Ca2+/Calmodulin-dependent Protein Kinase IV-mediated LIM Kinase Activation Is Critical for Calcium Signal-induced Neurite Outgrowth. J. Biol. Chem. 2009, 284, 28554–28562. [Google Scholar] [CrossRef]
- Mattiazzi, A.; Kranias, E.G. The role of CaMKII regulation of phospholamban activity in heart disease. Front. Pharmacol. 2014, 5, 5. [Google Scholar] [CrossRef]
- Luczak, E.D.; Anderson, M.E. CaMKII oxidative activation and the pathogenesis of cardiac disease. J. Mol. Cell. Cardiol. 2014, 73, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.E.; Brown, J.H.; Bers, D.M. CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2011, 51, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.S.; Bers, D.M. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation–contraction coupling in the heart. Cardiovasc. Res. 2007, 73, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Lars, S.M. Role of CaMKII for signaling and regulation in the heart. Front. Biosci. 2009, 14, 486–496. [Google Scholar] [CrossRef]
- Chen, H.H.; Zhang, C.; Lyu, R. The Role of RyR2 Phosphorylation in Heart Failure. Fudan Univ. J. Med. Sci. 2017, 44. [Google Scholar] [CrossRef]
- Bers, D.M.; Morotti, S. Ca2+ current facilitation is CaMKII-dependent and has arrhythmogenic consequences. Front. Pharmacol. 2014, 5, 144. [Google Scholar] [CrossRef]
- Tong, C.W.; Wu, X.; Liu, Y.; Rosas, P.C.; Sadayappan, S.; Hudmon, A.; Muthuchamy, M.; Powers, P.A.; Valdivia, H.H.; Moss, R.L. Phosphoregulation of Cardiac Inotropy via Myosin Binding Protein-C During Increased Pacing Frequency or B1-Adrenergic Stimulation. Circ. Heart Fail. 2015, 8, 595–604. [Google Scholar] [CrossRef]
- Eikemo, H.; Moltzau, L.R.; Hussain, R.I.; Nguyen, C.H.; Qvigstad, E.; Levy, F.O.; Skomedal, T.; Osnes, J.-B. CaMKII in addition to MLCK contributes to phosphorylation of regulatory light chain in cardiomyocytes. Biochem. Biophys. Res. Commun. 2016, 471, 219–225. [Google Scholar] [CrossRef]
- Perkin, J.; Slater, R.; Del Favero, G.; Lanzicher, T.; Hidalgo, C.; Anderson, B.; Smith, J.E.; Sbaizero, O.; Labeit, S.; Granzier, H. Phosphorylating Titin’s Cardiac N2B Element by ERK2 or CaMKIIδ Lowers the Single Molecule and Cardiac Muscle Force. Biophys. J. 2015, 109, 2592–2601. [Google Scholar] [CrossRef]
- Hamdani, N.; Krysiak, J.; Kreusser, M.M.; Neef, S.; dos Remedios, C.G.; Maier, L.S.; Krüger, M.; Backs, J.; Linke, W.A. Crucial Role for Ca 2+ /Calmodulin-Dependent Protein Kinase-II in Regulating Diastolic Stress of Normal and Failing Hearts via Titin Phosphorylation. Circ. Res. 2013, 112, 664–674. [Google Scholar] [CrossRef]
- Bussey, C.; Erickson, J. Physiology and pathology of cardiac CaMKII. Curr. Opin. Physiol. 2018, 1, 52–58. [Google Scholar] [CrossRef]
- Guilbert, A.; Lim, H.J.; Cheng, J.; Wang, Y. CaMKII-dependent myofilament Ca2+ desensitization contributes to the frequency-dependent acceleration of relaxation. Cell Calcium 2015, 58, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Ashpole, N.M.; Herren, A.W.; Ginsburg, K.S.; Brogan, J.D.; Johnson, D.E.; Cummins, T.R.; Bers, D.M.; Hudmon, A. Ca2+/Calmodulin-dependent Protein Kinase II (CaMKII) Regulates Cardiac Sodium Channel NaV1.5 Gating by Multiple Phosphorylation Sites. J. Biol. Chem. 2012, 287, 19856–19869. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.S. CaMKII regulation of voltage-gated sodium channels and cell excitability. Heart Rhythm. 2011, 8, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Mustroph, J.; Maier, L.S.; Wagner, S. CaMKII regulation of cardiac K channels. Front. Pharmacol. 2014, 5, 20. [Google Scholar] [CrossRef]
- Alday, A.; Ahyayauch, H.; Fernández-López, V.; Echeazarra, L.; Urrutia, J.; Casis, O.; Gallego, M. CaMKII Modulates the Cardiac Transient Outward K+ Current through its Association with Kv4 Channels in Non-Caveolar Membrane Rafts. Cell. Physiol. Biochem. 2020, 54, 27–39. [Google Scholar] [CrossRef]
- Bers, D.M.; Grandi, E. CaMKII Regulation of Cardiac Ion Channels. J. Cardiovasc. Pharmacol. 2009, 54, 180–187. [Google Scholar] [CrossRef]
- Myers, J.B.; Zaegel, V.; Coultrap, S.J.; Miller, A.P.; Bayer, K.U.; Reichow, S.L. The CaMKII holoenzyme structure in activation-competent conformations. Nat. Commun. 2017, 8, 15742. [Google Scholar] [CrossRef]
- Wu, Y.; Temple, J.; Zhang, R.; Dzhura, I.; Zhang, W.; Trimble, R.; Roden, D.M.; Passier, R.; Olson, E.N.; Colbran, R.J.; et al. Calmodulin Kinase II and Arrhythmias in a Mouse Model of Cardiac Hypertrophy. Circulation 2002, 106, 1288–1293. [Google Scholar] [CrossRef]
- Li, L.; Satoh, H.; Ginsburg, K.S.; Bers, D.M. The effect of Ca2+-calmodulin-dependent protein kinase II on cardiac excitation-contraction coupling in ferret ventricular myocytes. J. Physiol. 1997, 501, 17–31. [Google Scholar] [CrossRef]
- Bers, D.M. Calcium Cycling and Signaling in Cardiac Myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Dewald, O.; Frangogiannis, N.G. Inflammatory mechanisms in myocardial infarction. Curr. Drug Targets Inflamm. Allergy 2003, 2, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Finet, J.E.; Wolfram, J.A.; Anderson, M.E.; Ai, X.; Donahue, J.K. Calcium/calmodulin-dependent protein kinase II causes atrial structural remodeling associated with atrial fibrillation and heart failure. Heart Rhythm. 2019, 16, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Rajtik, T.; Carnicka, S.; Szobi, A.; Giricz, Z.; O-Uchi, J.; Hassova, V.; Svec, P.; Ferdinandy, P.; Ravingerova, T.; Adameova, A. Oxidative activation of CaMKIIδ in acute myocardial ischemia/reperfusion injury: A role of angiotensin AT1 receptor-NOX2 signaling axis. Eur. J. Pharmacol. 2016, 771, 114–122. [Google Scholar] [CrossRef]
- Cipolletta, E.; Rusciano, M.R.; Maione, A.S.; Santulli, G.; Sorriento, D.; Del Giudice, C.; Ciccarelli, M.; Franco, A.; Crola, C.; Campiglia, P.; et al. Targeting the CaMKII/ERK Interaction in the Heart Prevents Cardiac Hypertrophy. PLoS ONE 2015, 10, e0130477. [Google Scholar] [CrossRef]
- Rokita, A.G.; Anderson, M.E. New Therapeutic Targets in Cardiology: Arrhythmias and Ca2+/Calmodulin- Dependent Kinase II (CaMKII). Circulation 2012, 126, 2125–2139. [Google Scholar] [CrossRef]
- Ma, H.; Groth, R.D.; Cohen, S.M.; Emery, J.F.; Li, B.; Hoedt, E.; Zhang, G.; Neubert, T.A.; Tsien, R.W. ΓCaMKII Shuttles Ca2+/CaM to the Nucleus to Trigger CREB Phosphorylation and Gene Expression. Cell 2014, 159, 281–294. [Google Scholar] [CrossRef]
- Cipolletta, E.; Monaco, S.; Maione, A.S.; Vitiello, L.; Campiglia, P.; Pastore, L.; Franchini, C.; Novellino, E.; Limongelli, V.; Bayer, K.U.; et al. Calmodulin-Dependent Kinase II Mediates Vascular Smooth Muscle Cell Proliferation and Is Potentiated by Extracellular Signal Regulated Kinase. Endocrinology 2010, 151, 2747–2759. [Google Scholar] [CrossRef]
- Ma, C.; Gu, R.; Wang, X.; He, S.; Bai, J.; Zhang, L.; Zhang, J.; Li, Q.; Qu, L.; Xin, W.; et al. circRNA CDR1as Promotes Pulmonary Artery Smooth Muscle Cell Calcification by Upregulating CAMK2D and CNN3 via Sponging miR-7-5p. Mol. Ther. Nucleic Acids 2020, 22, 530–541. [Google Scholar] [CrossRef]
- Skelding, K.A.; Rostas, J.A.P. Regulation of Multifunctional Calcium/Calmodulin Stimulated Protein Kinases by Molecular Targeting. Adv. Exp. Med. Biol. 2020, 1131, 649–679. [Google Scholar] [CrossRef]
- Tokumitsu, H.; Wayman, G.A.; Muramatsu, M.; Soderling, T.R. Calcium/Calmodulin-Dependent Protein Kinase Kinase: Identification of Regulatory Domains. Biochemistry 1997, 36, 12823–12827. [Google Scholar] [CrossRef] [PubMed]
- Santiago, A.D.S.; Couñago, R.M.; Ramos, P.Z.; Godoi, P.H.C.; Massirer, K.B.; Gileadi, O.; Elkins, J.M. Structural Analysis of Inhibitor Binding to CAMKK1 Identifies Features Necessary for Design of Specific Inhibitors. Sci. Rep. 2018, 8, 14800. [Google Scholar] [CrossRef] [PubMed]
- Wayman, G.A.; Tokumitsu, H.; Soderling, T.R. Inhibitory Cross-talk by cAMP Kinase on the Calmodulin-dependent Protein Kinase Cascade. J. Biol. Chem. 1997, 272, 16073–16076. [Google Scholar] [CrossRef] [PubMed]
- Racioppi, L.; Means, A.R. Calcium/Calmodulin-dependent Protein Kinase Kinase 2: Roles in Signaling and Pathophysiology. J. Biol. Chem. 2012, 287, 31658–31665. [Google Scholar] [CrossRef] [PubMed]
- Sallé-Lefort, S.; Miard, S.; Nolin, M.-A.; Boivin, L.; Paré, M.-È.; Debigaré, R.; Picard, F. Hypoxia upregulates Malat1 expression through a CaMKK/AMPK/HIF-1α axis. Int. J. Oncol. 2016, 49, 1731–1736. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- O’Byrne, S.N.; Scott, J.W.; Pilotte, J.R.; da Santiago, A.S.; Langendorf, C.G.; Oakhill, J.S.; Eduful, B.J.; Couñago, R.M.; Wells, C.I.; Zuercher, W.J.; et al. In Depth Analysis of Kinase Cross Screening Data to Identify CAMKK2 Inhibitory Scaffolds. Molecules 2020, 25, 325. [Google Scholar] [CrossRef]
- Takemoto-Kimura, S.; Suzuki, K.; Horigane, S.-I.; Kamijo, S.; Inoue, M.; Sakamoto, M.; Fujii, H.; Bito, H. Calmodulin kinases: Essential regulators in health and disease. J. Neurochem. 2017, 141, 808–818. [Google Scholar] [CrossRef]
- Ferey, J.L.A.; Brault, J.J.; Smith, C.A.S.; Witczak, C.A. Constitutive activation of CaMKKα signaling is sufficient but not necessary for mTORC1 activation and growth in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E686–E694. [Google Scholar] [CrossRef]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New Insights into the Role of mTOR Signaling in the Cardiovascular System. Circ. Res. 2018, 122, 489–505. [Google Scholar] [CrossRef]
- Dong, F.; Patnaik, S.; Duan, Z.-H.; Kiedrowski, M.; Penn, M.S.; Mayorga, M.E. A Novel Role for CAMKK1 in the Regulation of the Mesenchymal Stem Cell Secretome. Stem Cells Transl. Med. 2017, 6, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Beghi, S.; Cavaliere, F.; Manfredini, M.; Ferrarese, S.; Corazzari, C.; Beghi, C.; Buschini, A. Polymorphism rs7214723 in CAMKK1: A new genetic variant associated with cardiovascular diseases. Biosci. Rep. 2021, 41, BSR20210326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-H.; Xu, Q.; Zhao, Z.; Wu, J.; Liu, W.-X.; Wang, H.; Jin, L.; Wang, J.-C. Polymorphism rs7214723 in CAMKK1 and lung cancer risk in Chinese population. Tumor Biol. 2013, 34, 3147–3152. [Google Scholar] [CrossRef]
- Chen, D.; Zhong, F.; Chen, Y. Association of calcium/calmodulin-dependent protein kinase kinase1 rs7214723 polymorphism with lung cancer risk in a Chinese population. Biosci. Rep. 2017, 37, BSR2017076. [Google Scholar] [CrossRef]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; Flicek, P.; et al. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Hurley, R.L.; Anderson, K.A.; Franzone, J.M.; Kemp, B.E.; Means, A.R.; Witters, L.A. The Ca2+/Calmodulin-dependent Protein Kinase Kinases Are AMP-activated Protein Kinase Kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Salt, I.; Hardie, D.G. AMP-Activated Protein Kinase: An Ubiquitous Signaling Pathway with Key Roles in the Cardiovascular System. Circ. Res. 2017, 120, 1825–1841. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, Y.; Zhao, H.; Wang, Q.; Chen, P. The Histone Demethylase JMJD1C Regulates CAMKK2-AMPK Signaling to Participate in Cardiac Hypertrophy. Front. Physiol. 2020, 11, 539. [Google Scholar] [CrossRef]
- Watanabe, S.; Horie, T.; Nagao, K.; Kuwabara, Y.; Baba, O.; Nishi, H.; Sowa, N.; Narazaki, M.; Matsuda, T.; Takemura, G.; et al. Cardiac-Specific Inhibition of Kinase Activity in Calcium/Calmodulin-Dependent Protein Kinase Kinase-β Leads to Accelerated Left Ventricular Remodeling and Heart Failure after Transverse Aortic Constriction in Mice. PLoS ONE 2014, 9, e108201. [Google Scholar] [CrossRef]
- Horie, T.; Ono, K.; Nagao, K.; Nishi, H.; Kinoshita, M.; Kawamura, T.; Wada, H.; Shimatsu, A.; Kita, T.; Hasegawa, K. Oxidative stress induces GLUT4 translocation by activation of PI3-K/Akt and dual AMPK kinase in cardiac myocytes. J. Cell. Physiol. 2008, 215, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Tojkander, S.; Ciuba, K.; Lappalainen, P. CaMKK2 Regulates Mechanosensitive Assembly of Contractile Actin Stress Fibers. Cell Rep. 2018, 24, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Shi, X.; Zhang, Y.; Wen, Z.; Cai, J.; Gao, W.; Xu, J.; Zheng, Y.; Ji, B.; Cui, Y.; et al. Myosin-18B Promotes Mechanosensitive CaMKK2-AMPK-VASP Regulation of Contractile Actin Stress Fibers. iScience 2020, 23, 100975. [Google Scholar] [CrossRef] [PubMed]
- Jiu, Y.; Kumari, R.; Fenix, A.M.; Schaible, N.; Liu, X.; Varjosalo, M.; Krishnan, R.; Burnette, D.T.; Lappalainen, P. Myosin-18B Promotes the Assembly of Myosin II Stacks for Maturation of Contractile Actomyosin Bundles. Curr. Biol. 2019, 29, 81–92.e5. [Google Scholar] [CrossRef]
- Ying, L.; Li, N.; He, Z.; Zeng, X.; Nan, Y.; Chen, J.; Miao, P.; Ying, Y.; Lin, W.; Zhao, X.; et al. Fibroblast growth factor 21 Ameliorates diabetes-induced endothelial dysfunction in mouse aorta via activation of the CaMKK2/AMPKα signaling pathway. Cell Death Dis. 2019, 10, 665. [Google Scholar] [CrossRef]
- Jardin, I.; Lopez, J.J.; Sanchez-Collado, J.; Gomez, L.J.; Salido, G.M.; Rosado, J.A. Store-Operated Calcium Entry and Its Implications in Cancer Stem Cells. Cells 2022, 11, 1332. [Google Scholar] [CrossRef]
- Yang, J.; Yu, J.; Li, D.; Yu, S.; Ke, J.; Wang, L.; Wang, Y.; Qiu, Y.; Gao, X.; Zhang, J.; et al. Store-operated calcium entry-activated autophagy protects EPC proliferation via the CAMKK2-MTOR pathway in ox-LDL exposure. Autophagy 2017, 13, 82–98. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Kim, H.K.; Ko, T.H.; Song, I.-S.; Jeong, Y.J.; Heo, H.J.; Jeong, S.H.; Kim, M.; Park, N.M.; Seo, D.Y.; Kha, P.T.; et al. BH4 activates CaMKK2 and rescues the cardiomyopathic phenotype in rodent models of diabetes. Life Sci. Alliance 2020, 3, e201900619. [Google Scholar] [CrossRef]
- Zhan, J.-K.; Wang, Y.-J.; Wang, Y.; Tang, Z.-Y.; Tan, P.; Huang, W.; Liu, Y.-S. Adiponectin attenuates the osteoblastic differentiation of vascular smooth muscle cells through the AMPK/mTOR pathway. Exp. Cell Res. 2014, 323, 352–358. [Google Scholar] [CrossRef]
- Lu, Y.; Yuan, T.; Min, X.; Yuan, Z.; Cai, Z. AMPK: Potential Therapeutic Target for Vascular Calcification. Front. Cardiovasc. Med. (IF 3.915) 2021, 8, 670222. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.; Bundy, K.; Roach, C.; Douglas, H.; Ventura, V.; Segars, M.; Schwartz, O.; Simpson, C. Mechanisms of the Osteogenic Switch of Smooth Muscle Cells in Vascular Calcification: WNT Signaling, BMPs, Mechanotransduction, and EndMT. Bioengineering 2020, 7, 88. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Li, F.-X.; Lin, X.; Zhong, J.-Y.; Wu, F.; Shan, S.-K.; Tan, C.-M.; Yuan, L.-Q.; Liao, X.-B. Adipose tissue-derived omentin-1 attenuates arterial calcification via AMPK/Akt signaling pathway. Aging 2019, 11, 8760–8776. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-H.; Chang, C.-W.; Lee, F.-T.; Kuo, C.-H.; Hsu, J.-H.; Liu, C.-P.; Wu, H.-L.; Yeh, J.-L. Targeting vascular smooth muscle cell dysfunction with xanthine derivative KMUP-3 inhibits abdominal aortic aneurysm in mice. Atherosclerosis 2020, 297, 16–24. [Google Scholar] [CrossRef]
- Haribabu, B.; Hook, S.S.; Selbert, M.A.; Goldstein, E.G.; Tomhave, E.D.; Edelman, A.M.; Snyderman, R.; Means, A.R. Human calcium-calmodulin dependent protein kinase I: cDNA cloning, domain structure and activation by phosphorylation at threonine-177 by calcium-calmodulin dependent protein kinase I kinase. EMBO J. 1995, 14, 3679–3686. [Google Scholar] [CrossRef]
- Zha, M.; Zhong, C.; Ou, Y.; Han, L.; Wang, J.; Ding, J. Crystal Structures of Human CaMKIα Reveal Insights into the Regulation Mechanism of CaMKI. PLoS ONE 2012, 7, e44828. [Google Scholar] [CrossRef]
- Bleier, J.; Toliver, A. Exploring the Role of CaMKIV in Homeostatic Plasticity. J. Neurosci. 2017, 37, 11520–11522. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Z.; Zhang, L.; Lü, G.; Zhou, C.; Wang, D.; Tang, Z.; Wang, L.; Qin, L.; Zhai, Z. CAMK4 gene variation is associated with hypertension in a Uygur population. Genet. Mol. Res. 2016, 15, gmr.15017207. [Google Scholar] [CrossRef]
- Levy, D.; Larson, M.G.; Benjamin, E.J.; Newton-Cheh, C.; Wang, T.J.; Hwang, S.-J.; Vasan, R.S.; Mitchell, G.F. Framingham Heart Study 100K Project: Genome-wide associations for blood pressure and arterial stiffness. BMC Med Genet. 2007, 8, S3–S11. [Google Scholar] [CrossRef]
- Santulli, G.; Cipolletta, E.; Sorriento, D.; Del Giudice, C.; Anastasio, A.; Monaco, S.; Maione, A.S.; Condorelli, G.; Puca, A.; Trimarco, B.; et al. CaMK4 Gene Deletion Induces Hypertension. J. Am. Heart Assoc. 2012, 1, e001081. [Google Scholar] [CrossRef]
- McCullough, L.D.; Tarabishy, S.; Liu, L.; Benashski, S.; Xu, Y.; Ribar, T.; Means, A.; Li, J. Inhibition of Calcium/Calmodulin-Dependent Protein Kinase Kinase β and Calcium/Calmodulin-Dependent Protein Kinase IV Is Detrimental in Cerebral Ischemia. Stroke 2013, 44, 2559–2566. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; McCullough, L.; Li, J. Genetic deletion of calcium/calmodulin-dependent protein kinase kinase β (CaMKK β) or CaMK IV exacerbates stroke outcomes in ovariectomized (OVXed) female mice. BMC Neurosci. 2014, 15, 118. [Google Scholar] [CrossRef] [PubMed]
- Wamhoff, B.R.; Bowles, D.; Owens, G.K. Excitation–Transcription Coupling in Arterial Smooth Muscle. Circ. Res. 2006, 98, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Najwer, I.; Lilly, B. Ca2+/calmodulin-dependent protein kinase IV activates cysteine-rich protein 1 through adjacent CRE and CArG elements. Am. J. Physiol. Cell Physiol. 2005, 289, C785–C793. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sun, P.; Enslen, H.; Myung, P.S.; Maurer, R.A. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994, 8, 2527–2539. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, L.-Y.; Singer, D.V.; Ginnan, R.; Singer, H.A. CaMKIIδ-dependent Inhibition of cAMP-response Element-binding Protein Activity in Vascular Smooth Muscle. J. Biol. Chem. 2013, 288, 33519–33529. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Nucleotide Change | Amino Acid Change | Associated Phenotype | Refs. |
|---|---|---|---|---|
| CALM1 | c.389A>G | p.D130G | LQTS | [59,61] |
| CALM1 | c.426C>G | p.F142L | LQTS | [59,61] |
| CALM1 | c.161A>T | p.N54I | CPVT | [61,66] |
| CALM1 | c.293A>G | p.N98S | LQTS, CPVT | [61,66] |
| CALM1 | c.268T>C | p.F90L | IVF, SUD | [67,69] |
| CALM1 | c.395A>T | p.D132V | LQTS | [64] |
| CALM2 | c.293A>G | p.N98S | LQTS, SUD | [61,62,68] |
| CALM2 | c.287A>T | p.D96V | LQTS | [59,61] |
| CALM2 | c.293A>T | p.N98I | LQTS | [61,62] |
| CALM2 | c.400G>C | p.D134H | LQTS | [38,39] |
| CALM2 | c.389A>G | p.D130G | LQTS | [61] |
| CALM2 | c.396T>G | p.D132E | LQTS, CPVT | [61,62] |
| CALM2 | c.394G>C | p.D132H | LQTS | [64] |
| CALM2 | c.407A>C | p.Q136P | LQTS, CPVT | [62] |
| CALM3 | c.389A>G | p.D130G | LQTS | [61,63] |
| CALM3 | c.308C>T | p.A103V | CPVT | [61] |
| CALM3 | c.286G>C | p.D96H | LQTS | [61,65] |
| CALM3 | c.426T>G | p.F142L | LQTS | [61,65] |
| Kinase | In Heart Pathology | In Vasculature Pathology |
|---|---|---|
| CaM | Polymorphisms linked to calmodulinopathy, arrhythmia [44,45,59,60,61,62,63,64,65,66,67,68,69]. | Unknown |
| CaMKII | Implicated in myocardial injury [116], atrial fibrillation [117], cardiac hypertrophy, ischaemia/reperfusion injury [118], heart failures, contributing to apoptosis, arrhythmias [113], defective ECC and ETC [53,114,115], pathological hypertrophy [96] and contractile dysfunction during heart failure [54,96,114,115]. | Regulation of VSMCs phenotype switching through the inhibition of CREB [34]. |
| CaMKK1 | Polymorphism linked to the higher risk to develop CVD [136]. | Regulation of VSMCs phenotype switching (CaMKK-CaMKIV-CREB) [34]. |
| CaMKK2 | Inactivation of CaMKK2 indirectly results in the development of metabolic dysfunction and cardiac hypertrophy [143]. | Regulation of VSMCs phenotype switching (CaMKK-CaMKIV-CREB) [34]. |
| CAMKI | Unknown | Unknown |
| CAMKIV | Polymorphisms linked to elevated diastolic blood pressure [162,163,164]. | Regulation of VSMCs phenotype switching (CaMKK-CaMKIV-CREB) [34]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beghi, S.; Furmanik, M.; Jaminon, A.; Veltrop, R.; Rapp, N.; Wichapong, K.; Bidar, E.; Buschini, A.; Schurgers, L.J. Calcium Signalling in Heart and Vessels: Role of Calmodulin and Downstream Calmodulin-Dependent Protein Kinases. Int. J. Mol. Sci. 2022, 23, 16139. https://doi.org/10.3390/ijms232416139
Beghi S, Furmanik M, Jaminon A, Veltrop R, Rapp N, Wichapong K, Bidar E, Buschini A, Schurgers LJ. Calcium Signalling in Heart and Vessels: Role of Calmodulin and Downstream Calmodulin-Dependent Protein Kinases. International Journal of Molecular Sciences. 2022; 23(24):16139. https://doi.org/10.3390/ijms232416139
Chicago/Turabian StyleBeghi, Sofia, Malgorzata Furmanik, Armand Jaminon, Rogier Veltrop, Nikolas Rapp, Kanin Wichapong, Elham Bidar, Annamaria Buschini, and Leon J. Schurgers. 2022. "Calcium Signalling in Heart and Vessels: Role of Calmodulin and Downstream Calmodulin-Dependent Protein Kinases" International Journal of Molecular Sciences 23, no. 24: 16139. https://doi.org/10.3390/ijms232416139
APA StyleBeghi, S., Furmanik, M., Jaminon, A., Veltrop, R., Rapp, N., Wichapong, K., Bidar, E., Buschini, A., & Schurgers, L. J. (2022). Calcium Signalling in Heart and Vessels: Role of Calmodulin and Downstream Calmodulin-Dependent Protein Kinases. International Journal of Molecular Sciences, 23(24), 16139. https://doi.org/10.3390/ijms232416139