

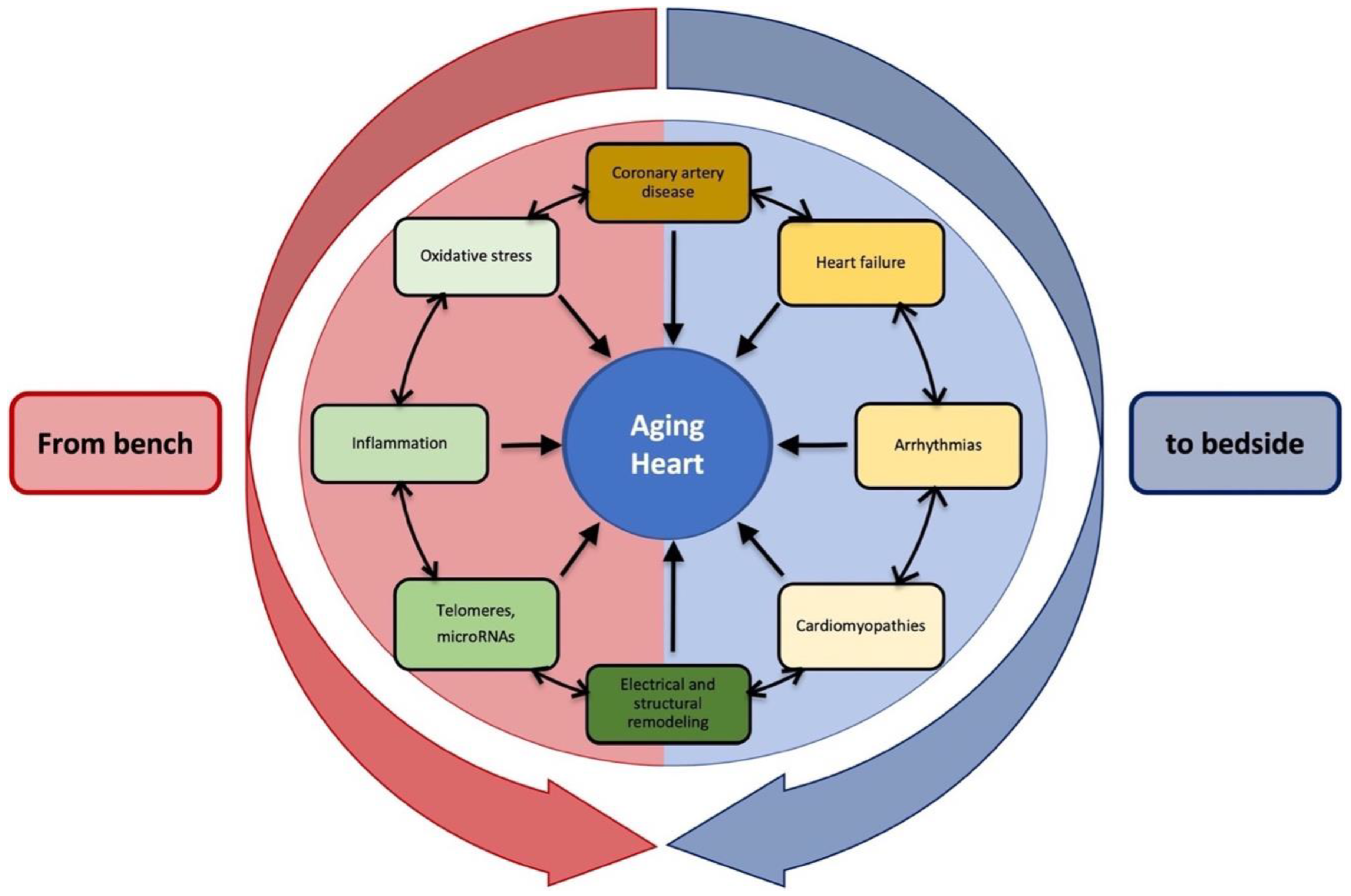
The Aging Heart: A Molecular and Clinical Challenge

 ,
,  ,
, 

Abstract
1. Introduction
2. Definition of the Aging Heart
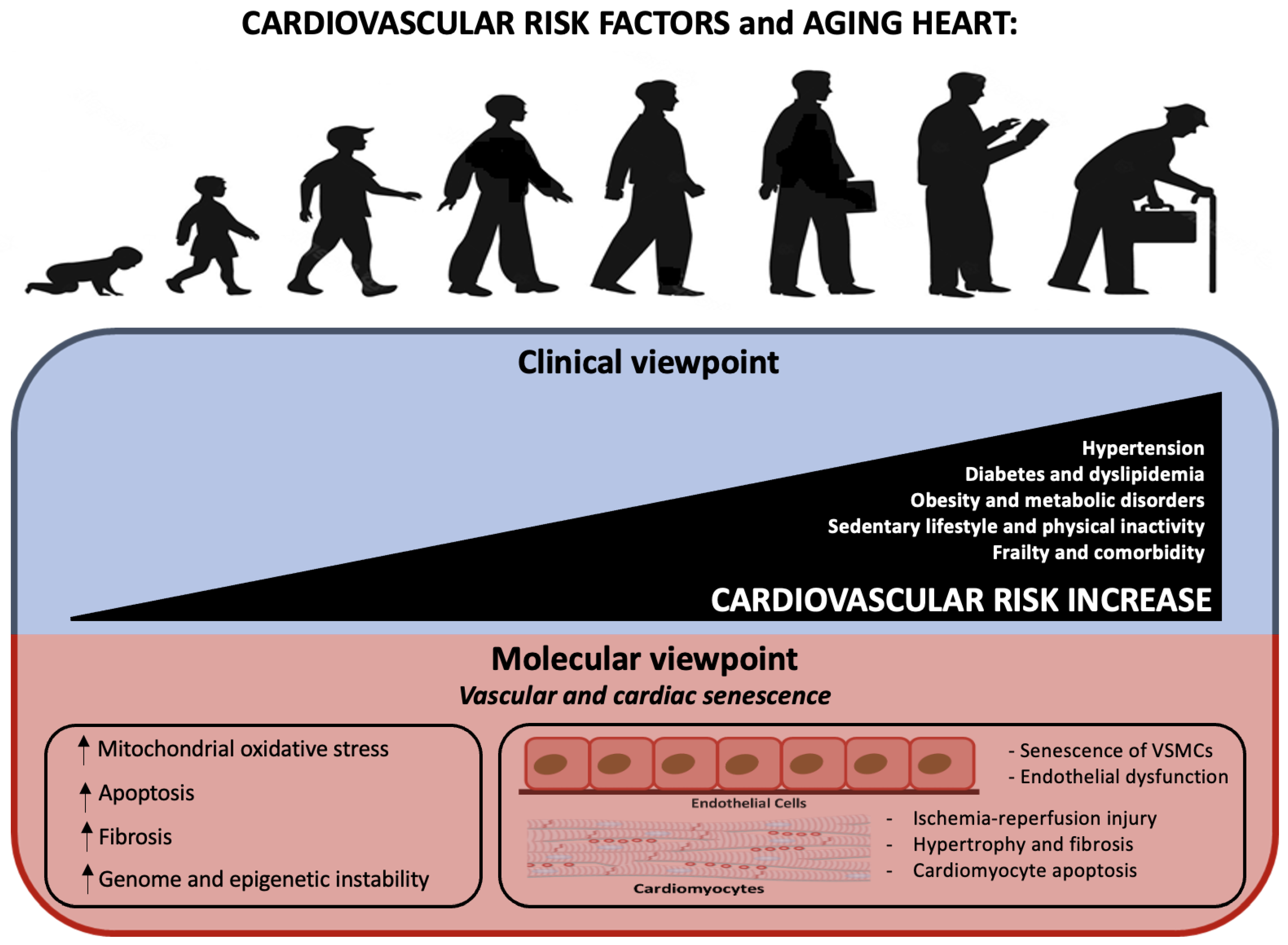
3. Cardiovascular Risk Factors
- Age as an independent risk factor
- MicroRNAs
- P66shc
- mTOR pathway: AMPK
- NAD-dependent proteins: SIRT1
- NF-κB
4. Ischemic Cardiomyopathy
- Inflammatory markers and cardiovascular risk
- T cells and cardiovascular risk
- Telomere shortening and cardiovascular risk

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Mechanisms and Intracellular Modifications Underlying Ischemic Cardiomyopathy | Studies |
|---|---|
| - Increase of cytokines (IL-6; TNFα), microRNA, plasminogen activator inhibitor-1 (PAI-1) | [38,41,42] |
| - Impairment of autophagy | [44] |
| - The release of IFN-γ, perforin and granzyme by senescent T cells | [45,46,47] |
| - Reduction of NAD+ levels | [48] |
| - Telomere shortening and dysfunction | [49,50] |
5. Heart Failure
6. Arrhythmias
| Molecular Mechanisms and Intracellular Modifications Underlying Arrhythmias | Studies |
|---|---|
| - Accumulation of amyloid, lipid, and lipofuscin, which leads to bradyarrhythmia | [69,70,74,82,83] |
| - Replacement of pacemaker cells with collagen and elastin fibers | [84] |
| - Delay of action potential duration and repolarization | [74,104,105] |
| - Impairment of Ca2+ homeostasis | [78,118,119,120,121,122,123] |
7. Cardiomyopathies
8. Clinical Management of CVD in Older People
9. Molecular Therapies
- mTOR pathway inhibitors
- SIRT1 stimulation
- Telomere-related therapies
- MicroRNAs inhibition
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y.; et al. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation 2022, 145, e153–e639. Available online: https://pubmed.ncbi.nlm.nih.gov/35078371/ (accessed on 30 September 2022). [PubMed]
- Goyal, P.; Kwak, M.J.; al Malouf, C.; Kumar, M.; Rohant, N.; Damluji, A.A.; Denfeld, Q.E.; Bircher, K.K.; Krishnaswami, A.; Alexander, K.P.; et al. Geriatric Cardiology: Coming of Age. JACC Adv. 2022, 1, 100070. [Google Scholar] [CrossRef]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990-2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. Available online: https://pubmed.ncbi.nlm.nih.gov/33309175/ (accessed on 30 September 2022). [CrossRef] [PubMed]
- Rich, M.W.; Chyun, D.A.; Skolnick, A.H.; Alexander, K.P.; Forman, D.E.; Kitzman, D.W.; Maurer, M.S.; Mcclurken, J.B.; Resnick, B.M.; Shen, W.K.; et al. Knowledge Gaps in Cardiovascular Care of the Older Adult Population: A Scientific Statement From the American Heart Association, American College of Cardiology, and American Geriatrics Society. Circulation 2016, 133, 2103–2122. Available online: https://pubmed.ncbi.nlm.nih.gov/27067230/ (accessed on 30 September 2022). [CrossRef] [PubMed]
- Anisimov, S.V.; Boheler, K.R. Aging-associated changes in cardiac gene expression: Large scale transcriptome analysis. Adv. Gerontol. 2003, 11, 67–75. [Google Scholar] [PubMed]
- Cannatr, A.; Camparini, L.; Sinagra, G.; Giacca, M.; Loffredo, F.S. Pathways for salvage and protection of the heart under stress: Novel routes for cardiac rejuvenation. Cardiovasc. Res. 2016, 111, 142–153. Available online: https://pubmed.ncbi.nlm.nih.gov/27371745/ (accessed on 1 October 2022). [CrossRef]
- Ruiz-Meana, M.; Bou-Teen, D.; Ferdinandy, P.; Gyongyosi, M.; Pesce, M.; Perrino, C.; Schulz, R.; Sluijter, J.P.G.; Tocchetti, C.G.; Thum, T.; et al. Cardiomyocyte ageing and cardioprotection: Consensus document from the ESC working groups cell biology of the heart and myocardial function. Cardiovasc. Res. 2020, 116, 1835–1849. Available online: https://pubmed.ncbi.nlm.nih.gov/32384145/ (accessed on 1 October 2022). [CrossRef]
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part II: The aging heart in health: Links to heart disease. Circulation 2003, 107, 346–354. [Google Scholar] [CrossRef]
- Chiao, Y.A.; Rabinovitch, P.S. The aging heart. Cold Spring Harb. Perspect Med. 2015, 5, a025148. [Google Scholar] [CrossRef]
- Barger, J.L.; Kayo, T.; Vann, J.M.; Arias, E.B.; Wang, J.; Hacker, T.A.; Wang, Y.; Raederstorff, D.; Morrow, J.D.; Leeuwenburgh, C.; et al. A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice. PLoS ONE 2008, 3, e2264. [Google Scholar] [CrossRef]
- Dai, D.F.; Santana, L.F.; Vermulst, M.; Tomazela, D.M.; Emond, M.J.; MacCoss, M.J.; Gollahon, K.; Martin, G.M.; Loeb, L.A.; Ladiges, W.C.; et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 2009, 119, 2789–2797. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Plati, A.R.; Potier, M.; Schulman, Y.; Berho, M.; Banerjee, A.; Leclercq, B.; Zisman, A.; Striker, L.J.; Striker, G.E.; et al. Resistance to glomerulosclerosis in B6 mice disappears after menopause. Am. J. Pathol. 2003, 162, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, R.; Vasan, R.S. Age as a Risk Factor. Med. Clin. N. Am. 2012, 96, 87–91. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.L.; Jones, J.; Bolleddu, S.I.; Vanthenapalli, S.; Rodgers, L.E.; Shah, K.; Karia, K.; Panguluri, S.K. Cardiovascular risks associated with gender and aging. J. Cardiovasc. Dev. Dis. 2019, 6, 19. [Google Scholar] [CrossRef]
- Verjans, R.; van Bilsen, M.; Schroen, B. MiRNA Deregulation in Cardiac Aging and Associated Disorders. Int Rev Cell Mol Biol 2017, 334, 207–263. Available online: https://pubmed.ncbi.nlm.nih.gov/28838539/ (accessed on 30 November 2022).
- Seeger, T.; Boon, R.A. MicroRNAs in cardiovascular ageing. J. Physiol. 2016, 594, 2085–2094. Available online: https://pubmed.ncbi.nlm.nih.gov/26040259/ (accessed on 30 November 2022). [CrossRef]
- Vasa-Nicotera, M.; Chen, H.; Tucci, P.; Yang, A.L.; Saintigny, G.; Menghini, R.; Mahè, C.; Agostini, M.; Knight, R.A.; Melino, G.; et al. miR-146a is modulated in human endothelial cell with aging. Atherosclerosis 2011, 217, 326–330. Available online: https://pubmed.ncbi.nlm.nih.gov/21511256/ (accessed on 30 November 2022). [CrossRef]
- Yan, H.L.; Xue, G.; Mei, Q.; Wang, Y.Z.; Ding, F.X.; Liu, M.F.; Lu, M.H.; Tang, Y.; Yu, H.Y.; Sun, S.H.; et al. Repression of the miR-17-92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J. 2009, 28, 2719–2732. Available online: https://pubmed.ncbi.nlm.nih.gov/19696742/ (accessed on 1 December 2022). [CrossRef]
- Song, S.; Seo, H.H.; Lee, S.Y.; Lee, C.Y.; Lee, J.; Yoo, K.J.; Yoon, C.; Choi, E.; Hwang, K.C.; Lee, S. MicroRNA-17-mediated down-regulation of apoptotic protease activating factor 1 attenuates apoptosome formation and subsequent apoptosis of cardiomyocytes. Biochem. Biophys. Res. Commun. 2015, 465, 299–304. Available online: https://pubmed.ncbi.nlm.nih.gov/26265044/ (accessed on 1 December 2022). [CrossRef] [PubMed]
- Costantino, S.; Paneni, F.; Cosentino, F. Ageing, metabolism and cardiovascular disease. J. Physiol. 2016, 594, 2061–2073. Available online: https://pubmed.ncbi.nlm.nih.gov/26391109/ (accessed on 27 September 2022). [CrossRef] [PubMed]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. Available online: https://pubmed.ncbi.nlm.nih.gov/16051147/ (accessed on 27 September 2022). [CrossRef]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. Available online: https://pubmed.ncbi.nlm.nih.gov/22186033/ (accessed on 29 September 2022). [CrossRef] [PubMed]
- Sung, J.Y.; Woo, C.H.; Kang, Y.J.; Lee, K.Y.; Choi, H.C. AMPK induces vascular smooth muscle cell senescence via LKB1 dependent pathway. Biochem. Biophys. Res. Commun. 2011, 413, 143–148. Available online: https://pubmed.ncbi.nlm.nih.gov/21872575/ (accessed on 29 September 2022). [CrossRef] [PubMed]
- Lesniewski, L.A.; Zigler, M.C.; Durrant, J.R.; Donato, A.J.; Seals, D.R. Sustained activation of AMPK ameliorates age-associated vascular endothelial dysfunction via a nitric oxide-independent mechanism. Mech. Ageing Dev. 2012, 133, 368–371. Available online: https://pubmed.ncbi.nlm.nih.gov/22484146/ (accessed on 29 September 2022). [CrossRef] [PubMed]
- El Messaoudi, S.; Rongen, G.A.; de Boer, R.A.; Riksen, N.P. The cardioprotective effects of metformin. Curr. Opin. Lipidol. 2011, 22, 445–453. Available online: https://pubmed.ncbi.nlm.nih.gov/21897229/ (accessed on 29 September 2022). [CrossRef]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007, 100, 1512–1521. Available online: https://pubmed.ncbi.nlm.nih.gov/17446436/ (accessed on 29 September 2022). [CrossRef]
- Sin, T.K.; Yu, A.P.; Yung, B.Y.; Yip, S.P.; Chan, L.W.; Wong, C.S.; Ying, M.; Rudd, J.A.; Siu, P.M. Modulating effect of SIRT1 activation induced by resveratrol on Foxo1-associated apoptotic signalling in senescent heart. J. Physiol. 2014, 592, 2535–2548. Available online: https://pubmed.ncbi.nlm.nih.gov/24639483/ (accessed on 29 September 2022). [CrossRef]
- Stein, S.; Matter, C.M. Protective roles of SIRT1 in atherosclerosis. Cell Cycle 2011, 10, 640–647. Available online: https://pubmed.ncbi.nlm.nih.gov/21293192/ (accessed on 29 September 2022). [CrossRef]
- Mattagajasingh, I.; Kim, C.S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. Available online: https://pubmed.ncbi.nlm.nih.gov/17785417/ (accessed on 29 September 2022). [CrossRef] [PubMed]
- Pillarisetti, S. A review of Sirt1 and Sirt1 modulators in cardiovascular and metabolic diseases. Recent Pat. Cardiovasc. Drug Discov. 2008, 3, 156–164. Available online: https://pubmed.ncbi.nlm.nih.gov/18991791/ (accessed on 29 September 2022). [CrossRef] [PubMed]
- Tas, S.; Vervoordeldonk, M.; Tak, P. Gene therapy targeting nuclear factor-kappaB: Towards clinical application in inflammatory diseases and cancer. Curr. Gene Ther. 2009, 9, 160–170. Available online: https://pubmed.ncbi.nlm.nih.gov/19519361/ (accessed on 30 September 2022). [CrossRef] [PubMed]
- Paneni, F.; Osto, E.; Costantino, S.; Mateescu, B.; Briand, S.; Coppolino, G.; Perna, E.; Mocharla, P.; Akhmedov, A.; Kubant, R.; et al. Deletion of the activated protein-1 transcription factor JunD induces oxidative stress and accelerates age-related endothelial dysfunction. Circulation 2013, 127, 1229–1240. Available online: https://pubmed.ncbi.nlm.nih.gov/23410942/ (accessed on 1 October 2022). [CrossRef]
- Yang, Z.; Ming, X.F. mTOR signalling: The molecular interface connecting metabolic stress, aging and cardiovascular diseases. Obes. Rev. 2012, 13 (Suppl. 2), 58–68. Available online: https://pubmed.ncbi.nlm.nih.gov/23107260/ (accessed on 1 October 2022). [CrossRef]
- Ungvari, Z.; Kaley, G.; de Cabo, R.; Sonntag, W.E.; Csiszar, A. Mechanisms of vascular aging: New perspectives. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 1028–1041. Available online: https://pubmed.ncbi.nlm.nih.gov/20576649/ (accessed on 1 October 2022). [CrossRef]
- Kritchevsky, S.B.; Cesari, M.; Pahor, M. Inflammatory markers and cardiovascular health in older adults. Cardiovasc. Res. 2005, 66, 265–275. Available online: https://academic.oup.com/cardiovascres/article/66/2/265/270123 (accessed on 3 October 2022). [CrossRef]
- Cesari, M.; Penninx, B.W.J.H.; Newman, A.B.; Kritchevsky, S.B.; Nicklas, B.J.; Sutton-Tyrrell, K.; Rubin, S.M.; Ding, J.; Simonsick, E.M.; Harris, T.B.; et al. Inflammatory markers and onset of cardiovascular events: Results from the Health ABC study. Circulation 2003, 108, 2317–2322. Available online: https://pubmed.ncbi.nlm.nih.gov/14568895/ (accessed on 1 October 2022). [CrossRef]
- Harris, T.B.; Ferrucci, L.; Tracy, R.P.; Corti, M.C.; Wacholder, S.; Ettinger, W.H.; Heimovitz, H.; Cohen, H.J.; Wallace, R. Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am. J. Med. 1999, 106, 506–512. Available online: https://pubmed.ncbi.nlm.nih.gov/10335721/ (accessed on 1 October 2022). [CrossRef]
- Kannel, W.B.; Wolf, P.A.; Castelli, W.P.; D’agostino, R.B. Fibrinogen and Risk of Cardiovascular Disease: The Framingham Study. JAMA 1987, 258, 1183–1186. Available online: https://jamanetwork.com/journals/jama/fullarticle/367933 (accessed on 3 October 2022). [CrossRef]
- de Yébenes, V.G.; Briones, A.M.; Martos-Folgado, I.; Mur, S.M.; Oller, J.; Bilal, F.; González-Amor, M.; Méndez-Barbero, N.; Silla-Castro, J.C.; Were, F.; et al. Aging-Associated miR-217 Aggravates Atherosclerosis and Promotes Cardiovascular Dysfunction. Arter. Thromb. Vasc. Biol. 2020, 40, 2408–2424. Available online: https://pubmed.ncbi.nlm.nih.gov/32847388/ (accessed on 1 October 2022). [CrossRef] [PubMed]
- Yamamoto, K.; Takeshita, K.; Kojima, T.; Takamatsu, J.; Saito, H. Aging and plasminogen activator inhibitor-1 (PAI-1) regulation: Implication in the pathogenesis of thrombotic disorders in the elderly. Cardiovasc. Res. 2005, 66, 276–285. Available online: https://academic.oup.com/cardiovascres/article/66/2/276/270209 (accessed on 1 October 2022). [CrossRef] [PubMed]
- Durante, A.; Peretto, G.; Laricchia, A.; Ancona, F.; Spartera, M.; Mangieri, A.; Cianflone, D. Role of the renin-angiotensin-aldosterone system in the pathogenesis of atherosclerosis. Curr. Pharm. Des. 2012, 18, 981–1004. Available online: https://pubmed.ncbi.nlm.nih.gov/22283771/ (accessed on 6 October 2022). [CrossRef] [PubMed]
- Larocca, T.J.; Henson, G.D.; Thorburn, A.; Sindler, A.L.; Pierce, G.L.; Seals, D.R. Translational evidence that impaired autophagy contributes to arterial ageing. J. Physiol. 2012, 590, 3305–3316. Available online: https://pubmed.ncbi.nlm.nih.gov/22570377/ (accessed on 1 October 2022). [CrossRef] [PubMed]
- Yu, H.T.; Park, S.; Shin, E.C.; Lee, W.W. T cell senescence and cardiovascular diseases. Clin. Exp. Med. 2016, 16, 257–263. Available online: https://pubmed.ncbi.nlm.nih.gov/26188489/ (accessed on 1 October 2022). [CrossRef]
- Alfaro Leon, M.L.; Zuckerman, S.H. Gamma interferon: A central mediator in atherosclerosis. Inflamm. Res. 2005, 54, 395–411. Available online: https://pubmed.ncbi.nlm.nih.gov/16283107/ (accessed on 1 October 2022). [CrossRef]
- Johnson, J.L. Matrix metalloproteinases: Influence on smooth muscle cells and atherosclerotic plaque stability. Expert Rev. Cardiovasc. Ther. 2007, 5, 265–282. Available online: https://pubmed.ncbi.nlm.nih.gov/17338671/ (accessed on 1 October 2022). [CrossRef]
- Hosseini, L.; Vafaee, M.S.; Mahmoudi, J.; Badalzadeh, R. Nicotinamide adenine dinucleotide emerges as a therapeutic target in aging and ischemic conditions. Biogerontology 2019, 20, 381–395. Available online: https://pubmed.ncbi.nlm.nih.gov/30838484/ (accessed on 1 October 2022). [CrossRef]
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda di Fagagna, F. Telomere dysfunction in ageing and age-related diseases. Nat. Cell Biol. 2022, 24, 135–147. Available online: https://www.nature.com/articles/s41556-022-00842-x (accessed on 3 October 2022). [CrossRef]
- Boniewska-Bernacka, E.; Pańczyszyn, A.; Klinger, M. Telomeres and telomerase in risk assessment of cardiovascular diseases. Exp. Cell Res. 2020, 397, 112361. Available online: https://pubmed.ncbi.nlm.nih.gov/33171154/ (accessed on 3 October 2022). [CrossRef]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492. [Google Scholar] [CrossRef] [PubMed]
- Dookun, E.; Walaszczyk, A.; Redgrave, R.; Palmowski, P.; Tual-Chalot, S.; Suwana, A.; Chapman, J.; Jirkovsky, E.; Donastorg-Sosa, L.; Gill, E.; et al. Clearance of senescent cells during cardiac ischemia-reperfusion injury improves recovery. Aging Cell 2020, 19, e13249. Available online: https://pubmed.ncbi.nlm.nih.gov/32996233/ (accessed on 3 October 2022). [CrossRef] [PubMed]
- Edo, M.D.; Andrés, V. Aging, telomeres, and atherosclerosis. Cardiovasc. Res. 2005, 66, 213–221. Available online: https://pubmed.ncbi.nlm.nih.gov/15820190/ (accessed on 3 October 2022). [CrossRef] [PubMed]
- Samani, N.J.; Boultby, R.; Butler, R.; Thompson, J.R.; Goodall, A.H. Telomere shortening in atherosclerosis. Lancet 2001, 358, 472–473. Available online: https://pubmed.ncbi.nlm.nih.gov/11513915/ (accessed on 3 October 2022). [CrossRef] [PubMed]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: Effects of telomerase and oxidative stress. Circ. Res. 2006, 99, 156–164. Available online: https://pubmed.ncbi.nlm.nih.gov/16794190/ (accessed on 3 October 2022). [CrossRef]
- Triposkiadis, F.; Xanthopoulos, A.; Parissis, J.; Butler, J.; Farmakis, D. Pathogenesis of chronic heart failure: Cardiovascular aging, risk factors, comorbidities, and disease modifiers. Heart Fail. Rev. 2022, 27, 337–344. Available online: https://pubmed.ncbi.nlm.nih.gov/32524327/ (accessed on 3 October 2022). [CrossRef]
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part I: Aging arteries: A ‘set up’ for vascular disease. Circulation 2003, 107, 139–146. Available online: https://pubmed.ncbi.nlm.nih.gov/12515756/ (accessed on 3 October 2022). [CrossRef]
- Forman, D.E.; Ahmed, A.; Fleg, J.L. Heart failure in very old adults. Curr. Heart Fail. Rep. 2013, 10, 387–400. Available online: https://pubmed.ncbi.nlm.nih.gov/24091808/ (accessed on 3 October 2022). [CrossRef]
- Roh, J.D.; Hobson, R.; Chaudhari, V.; Quintero, P.; Yeri, A.; Benson, M.; Xiao, C.; Zlotoff, D.; Bezzerides, V.; Houstis, N.; et al. Activin type II receptor signaling in cardiac aging and heart failure. Sci. Transl. Med. 2019, 11, eaau8680. [Google Scholar] [CrossRef]
- Uchmanowicz, I.; Łoboz-Rudnicka, M.; Szeląg, P.; Jankowska-Polańska, B.; Łoboz-Grudzień, K. Frailty in heart failure. Curr. Heart Fail. Rep. 2014, 11, 266–273. Available online: https://pubmed.ncbi.nlm.nih.gov/24733407/ (accessed on 3 October 2022). [CrossRef]
- Chen, L.H.; Chiou, G.Y.; Chen, Y.W.; Li, H.Y.; Chiou, S.H. MicroRNA and aging: A novel modulator in regulating the aging network. Ageing Res. Rev. 2010, 9 (Suppl. 1), S59–S66. Available online: https://pubmed.ncbi.nlm.nih.gov/20708718/ (accessed on 3 October 2022). [CrossRef] [PubMed]
- Schroen, B.; Heymans, S. Small but smart—microRNAs in the centre of inflammatory processes during cardiovascular diseases, the metabolic syndrome, and ageing. Cardiovasc. Res. 2012, 93, 605–613. Available online: https://academic.oup.com/cardiovascres/article/93/4/605/433375 (accessed on 3 October 2022). [CrossRef] [PubMed]
- O’Neill, L.A.; Sheedy, F.J.; McCoy, C.E. MicroRNAs: The fine-tuners of Toll-like receptor signalling. Nat. Rev. Immunol. 2011, 11, 163–175. Available online: https://pubmed.ncbi.nlm.nih.gov/21331081/ (accessed on 3 October 2022). [CrossRef] [PubMed]
- Ferreira, J.P.; Ouwerkerk, W.; Santema, B.T.; van Veldhuisen, D.J.; Lang, C.C.; Ng, L.L.; Anker, S.D.; Dickstein, K.; Metra, M.; Cleland, J.G.F.; et al. Differences in biomarkers and molecular pathways according to age for patients with HFrEF. Cardiovasc. Res. 2021, 117, 2228–2236. Available online: https://academic.oup.com/cardiovascres/article/117/10/2228/5917017 (accessed on 3 October 2022). [CrossRef] [PubMed]
- Pugliese, N.R.; Pellicori, P.; Filidei, F.; De Biase, N.; Maffia, P.; Guzik, T.J.; Masi, S.; Taddei, S.; Cleland, J.G. Inflammatory pathways in heart failure with preserved left ventricular ejection fraction: Implications for future interventions. Cardiovasc. Res. 2022, cvac133. [Google Scholar] [CrossRef] [PubMed]
- Chugh, S.S.; Jui, J.; Gunson, K.; Stecker, E.C.; John, B.T.; Thompson, B.; Ilias, N.; Vickers, C.; Dogra, V.; Daya, M.; et al. Current burden of sudden cardiac death: Multiple source surveillance versus retrospective death certificate-based review in a large U. S. community. J. Am. Coll. Cardiol. 2004, 44, 1268–1275. [Google Scholar] [CrossRef]
- Piccini, J.P.; Hammill, B.G.; Sinner, M.F.; Jensen, P.N.; Hernandez, A.F.; Heckbert, S.R.; Benjamin, E.J.; Curtis, L.H. Incidence and prevalence of atrial fibrillation and associated mortality among medicare beneficiaries: 1993–2007. Circ. Cardiovasc. Qual. Outcomes 2012, 5, 85–93. [Google Scholar] [CrossRef]
- Roger, V.L.; Go, A.S.; Lloyd-Jones, D.M.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. AHA Statistical Update Heart Disease and Stroke Statistics-2012 Update A Report From the American Heart Association. Circulation 2012, 125, e2–e220. [Google Scholar]
- Strait, J.B.; Lakatta, E.G. Aging-Associated Cardiovascular Changes and Their Relationship to Heart Failure. Heart Fail. Clin. 2012, 8, 143–164. [Google Scholar] [CrossRef]
- Fuster, V.; Rydén, L.E.; Cannom, D.S.; Crijns, H.J.; Curtis, A.B.; Ellenbogen, K.A.; Halperin, J.L.; Kay, G.N.; Le-Huezey, J.Y.; Lowe, J.E.; et al. 2011 ACCF/AHA/HRS Focused Updates Incorporated Into the ACC/AHA/ESC 2006 Guidelines for the Management of Patients With Atrial Fibrillation. Circulation 2011, 123, 1161–1167. [Google Scholar] [CrossRef]
- ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A Report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death). J. Am. Coll. Cardiol. 2006, 48, e247–e346.
- Gregoratos, G.; Abrams, J.; Epstein, A.E.; Freedman, R.A.; Hayes, D.L.; Hlatky, M.A.; Kerber, R.E.; Naccarelli, G.V.; Schoenfeld, M.H.; Silka, M.J.; et al. American College of Cardiology/American Heart Association Task Force on Practice Guidelines/North American Society for Pacing and Electrophysiology Committee to Update the 1998 Pacemaker Guidelines. ACC/AHA/NASPE 2002 guideline update for implantation of cardiac pacemakers and antiarrhythmia devices: Summary article: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/NASPE Committee to Update the 1998 Pacemaker Guidelines). Circulation 2002, 106, 2145–2161. [Google Scholar] [CrossRef] [PubMed]
- Rosenqvist, M.; Obel, I.W.P. Atrial Pacing and the Risk for AV Block: Is There a Time for Change in Attitude? Pacing Clin. Electrophysiol. 1989, 12, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Chow, G.V.; Marine, J.E.; Fleg, J.L. Epidemiology of arrhythmias and conduction disorders in older adults. Clin Geriatr. Med. 2012, 28, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Furberg, C.D.; Psaty, B.M.; Siscovick, D. Physical Activity and Incidence of Atrial Fibrillation in Older Adults. Circulation 2008, 118, 800–807. [Google Scholar] [CrossRef]
- Kim, M.H.; Johnston, S.S.; Chu, B.C.; Dalal, M.R.; Schulman, K.L. Estimation of total incremental health care costs in patients with atrial fibrillation in the United States. Circ. Cardiovasc. Qual. Outcomes 2011, 4, 313–320. [Google Scholar] [CrossRef]
- Miyasaka, Y.; Barnes, M.E.; Gersh, B.J.; Cha, S.S.; Bailey, K.R.; Abhayaratna, W.P.; Seward, J.B.; Tsang, T.S.M. Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation 2006, 114, 119–125. [Google Scholar] [CrossRef]
- Mirza, M.; Strunets, A.; Shen, W.K.; Jahangir, A. Mechanisms of Arrhythmias and Conduction Disorders in Older Adults. Clin. Geriatr. Med. 2012, 28, 555–573. [Google Scholar] [CrossRef]
- Kistler, P.M.; Sanders, P.; Fynn, S.P.; Stevenson, I.H.; Spence, S.J.; Vohra, J.K.; Sparks, P.B.; Kalman, J.M. Electrophysiologic and electroanatomic changes in the human atrium associated with age. J. Am. Coll. Cardiol. 2004, 44, 109–116. [Google Scholar] [CrossRef]
- Preston, C.C.; Oberlin, A.S.; Holmuhamedov, E.L.; Gupta, A.; Sagar, S.; Syed, R.H.K.; Siddiqui, S.A.; Raghavakaimal, S.; Terzic, A.; Jahangir, A. Aging-induced alterations in gene transcripts and functional activity of mitochondrial oxidative phosphorylation complexes in the heart. Mech. Ageing Dev. 2008, 129, 304–312. [Google Scholar] [CrossRef]
- Roberts-Thomson, K.C.; Kistler, P.M.; Sanders, P.; Morton, J.B.; Haqqani, H.M.; Stevenson, I.; Vohra, J.K.; Sparks, P.B.; Kalman, J.M. Fractionated atrial electrograms during sinus rhythm: Relationship to age, voltage, and conduction velocity. Heart Rhythm. 2009, 6, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Olivetti, G.; Melissari, M.; Capasso, J.M.; Anversa, P. Cardiomyopathy of the aging human heart. Myocyte Loss React. Cell. Hypertrophy. Circ. Res. 1991, 68, 1560–1568. [Google Scholar]
- Rahlf, G. Das Herz des älteren Menschen. Pathologie: Makroskopische und lichtmikroskopische Befunde am Herzen [The heart in the elderly. Pathology: Macroscopic and light microscopy findings in the heart]. Z. Kardiol. 1985, 74 (Suppl. 7), 9–16. (In German) [Google Scholar] [PubMed]
- Lakatta, E.G. Cardiovascular regulatory mechanisms in advanced age. Physiol. Rev. 1993, 73, 413–465. [Google Scholar] [CrossRef] [PubMed]
- Tellez, J.O.; Maczewski, M.; Yanni, J.; Sutyagin, P.; Mackiewicz, U.; Atkinson, A.; Inada, S.; Beresewicz, A.; Billeter, R.; Dobrzynski, H.; et al. Ageing-dependent remodelling of ion channel and Ca2+ clock genes underlying sino-atrial node pacemaking. Exp. Physiol. 2011, 96, 1163–1178. [Google Scholar] [CrossRef] [PubMed]
- Dun, W.; Boyden, P.A. Aged atria: Electrical remodeling conducive to atrial fibrillation. J. Interv. Card. Electrophysiol. 2009, 25, 9–18. [Google Scholar] [CrossRef]
- Fleg, J.L.; O’Connor, F.; Gerstenblith, G.; Becker, L.C.; Clulow, J.; Schulman, S.P.; Lakatta, E.G. Impact of age on the cardiovascular response to dynamic upright exercise in healthy men and women. J. Appl. Physiol. 1995, 78, 890–900. [Google Scholar] [CrossRef]
- Epstein, A.E.; DiMarco, J.P.; Ellenbogen, K.A.; Estes, N.A.M.; Freedman, R.A.; Gettes, L.S.; Gillinov, A.M.; Gregoratos, G.; Hammill, S.C.; Hayes, D.L.; et al. ACC/AHA/HRS 2008 Guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices) Developed in Collaboration With the American Association for Thoracic Surgery and Society of Thoracic Surgeons. J. Am. Coll. Cardiol. 2008, 51, e1–e62. [Google Scholar]
- Tresch, D.D. Evaluation and management of cardiac arrhythmias in the elderly. Med. Clin. N. Am. 2001, 85, 527–550. [Google Scholar] [CrossRef] [PubMed]
- Lie, J.T.; Hammond, P.I. Pathology of the Senescent Heart: Anatomic Observations on 237 Autopsy Studies of Patients 90 to 105 Years Old. Mayo Clin. Proc. 1988, 63, 552–564. [Google Scholar] [CrossRef]
- Cheitlin, M.D. Cardiovascular Physiology—Changes With Aging. Am. J. Geriatr. Cardiol. 2003, 12, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Curtis, A.B.; Karki, R.; Hattoum, A.; Sharma, U.C. Arrhythmias in Patients ≥80 Years of Age: Pathophysiology, Management, and Outcomes. J. Am. Coll. Cardiol. 2018, 71, 2041–2057. [Google Scholar] [CrossRef]
- Mymin, D.; Mathewson, F.A.L.; Tate, R.B.; Manfreda, J. The Natural History of Primary First-Degree Atrioventricular Heart Block. N. Engl. J. Med. 1986, 315, 1183–1187. [Google Scholar] [CrossRef]
- Fisch, G.R.; Zipes, D.P.; Fisch, C. Bundle branch block and sudden death. Prog. Cardiovasc. Dis. 1980, 23, 187–224. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial paradigm for degenerative diseases and ageing. Novartis Found Symp. 2001, 235, 247–266. [Google Scholar] [PubMed]
- Volkova, M.; Garg, R.; Dick, S.; Boheler, K.R. Aging-associated changes in cardiac gene expression. Cardiovasc. Res. 2005, 66, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Moghaddas, S.; Tandler, B.; Kerner, J.; Hoppel, C.L. Mitochondrial dysfunction in cardiac disease: Ischemia—Reperfusion, aging, and heart failure. J. Mol. Cell Cardiol. 2001, 33, 1065–1089. [Google Scholar] [CrossRef]
- Ames, B.N. Delay in mitochondrial decay of aging. Ann. N. Y. Acad. Sci. 2004, 1019, 406–411. [Google Scholar] [CrossRef]
- Bodyak, N.; Kang, P.M.; Hiromura, M.; Sulijoadikusumo, I.; Horikoshi, N.; Khrapko, K.; Usheva, A. Gene expression profiling of the aging mouse cardiac myocytes. Nucleic Acids Res. 2002, 30, 3788–3794. [Google Scholar] [CrossRef]
- Lee, C.K.; Allison, D.B.; Brand, J.; Weindruch, R.; Prolla, T.A. Transcriptional profiles associated with aging and middle age-onset caloric restriction in mouse hearts. Proc. Natl. Acad. Sci. USA 2002, 99, 14988–14993. [Google Scholar] [CrossRef]
- Huang, C.; Ding, W.; Li, L.; Zhao, D. Differences in the aging-associated trends of the monophasic action potential duration and effective refractory period of the right and left atria of the rat. Circ. J. 2006, 70, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.K.; Jahangir, A.; Terzic, A.; Gersh, B.J.; Hammill, S.C.; Shen, W.K. Age- and sex-related atrial electrophysiologic and structural changes. Am. J. Cardiol. 2004, 94, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Spach, M.S.; Heidlage, J.F.; Dolber, P.C.; Barr, R.C. Mechanism of origin of conduction disturbances in aging human atrial bundles: Experimental and model study. Heart Rhythm. 2007, 4, 175–185. [Google Scholar] [CrossRef]
- Josephson, I.R.; Guia, A.; Stern, M.D.; Lakatta, E.G. Alterations in properties of L-type Ca channels in aging rat heart. J. Mol. Cell Cardiol. 2002, 34, 297–308. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Maltsev, V.A.; Vinogradova, T.M. A Coupled SYSTEM of Intracellular Ca2+ Clocks and Surface Membrane Voltage Clocks Controls the Timekeeping Mechanism of the Heart’s Pacemaker. Circ. Res. 2010, 106, 659–673. [Google Scholar] [CrossRef]
- Muraleva, N.A.; Devyatkin, V.A.; Kolosova, N.G. Phosphorylation of αB-crystallin in the myocardium: Analysis of relations with aging and cardiomyopathy. Exp. Gerontol. 2017, 95, 26–33. [Google Scholar] [CrossRef]
- Walker, K.E.; Lakatta, E.G.; Houser, S.R. Age associated changes in membrane currents in rat ventricular myocytes. Cardiovasc. Res. 1993, 27, 1968–1977. [Google Scholar] [CrossRef] [PubMed]
- Janczewski, A.M.; Spurgeon, H.A.; Lakatta, E.G. Action potential prolongation in cardiac myocytes of old rats is an adaptation to sustain youthful intracellular Ca2+ regulation. J. Mol. Cell Cardiol. 2002, 34, 641–648. [Google Scholar] [CrossRef]
- Zhou, Y.Y.; Lakatta, E.G.; Xiao, R.P. Age-Associated Alterations in Calcium Current and its Modulation in Cardiac Myocytes. Drugs Aging 1998, 13, 2. [Google Scholar] [CrossRef]
- Morita, N.; Lee, J.H.; Bapat, A.; Fishbein, M.C.; Mandel, W.J.; Chen, P.S.; Weiss, J.N.; Karagueuzian, H.S. Glycolytic inhibition causes spontaneous ventricular fibrillation in aged hearts. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, 180–191. [Google Scholar] [CrossRef]
- Xiao, R.P.; Zhu, W.; Zheng, M.; Cao, C.; Zhang, Y.; Lakatta, E.G.; Han, Q. Subtype-specific α1- and β-adrenoceptor signaling in the heart. Trends Pharm. Sci. 2006, 27, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Jahangir, A.; Terzic, A. KATP channel therapeutics at the bedside. J. Mol. Cell Cardiol. 2005, 39, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Toro, L.; Marijic, J.; Nishimaru, K.; Tanaka, Y.; Song, M.; Stefani, E. Aging, ion channel expression, and vascular function. Vasc. Pharmacol. 2002, 38, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.; Terentyev, D. Altered Intracellular Calcium Homeostasis and Arrhythmogenesis in the Aged Heart. Int. J. Mol. Sci. 2019, 20, 2386. [Google Scholar] [CrossRef] [PubMed]
- Rich, M.W.; Shen, W.K. Cardiac rhythm disorders in older adults. Clin. Geriatr. Med. 2012, 28, xiii–xiv. [Google Scholar] [CrossRef] [PubMed]
- Taffet, G.E.; Tate, C.A. CaATPase content is lower in cardiac sarcoplasmic reticulum isolated from old rats. Am. J. Physiol. Circ. Physiol. 1993, 264, H1609–H1614. [Google Scholar] [CrossRef] [PubMed]
- Lompre, A.M.; Lambert, F.; Lakatta, E.G.; Schwartz, K. Expression of sarcoplasmic reticulum Ca(2+)-ATPase and calsequestrin genes in rat heart during ontogenic development and aging. Circ. Res. 1991, 69, 1380–1388. [Google Scholar] [CrossRef]
- Kawamura, S.; Takahashi, M.; Ishihara, T.; Uchino, F. Incidence and distribution of isolated atrial amyloid: Histologic and immunohistochemical studies of 100 aging hearts. Pathol. Int. 1995, 45, 335–342. [Google Scholar] [CrossRef]
- Anyukhovsky, E.P.; Sosunov, E.A.; Chandra, P.; Rosen, T.S.; Boyden, P.A.; Danilo, P.; Rosen, M.R. Age-associated changes in electrophysiologic remodeling: A potential contributor to initiation of atrial fibrillation. Cardiovasc. Res. 2005, 66, 353–363. [Google Scholar] [CrossRef]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Xu, A.; Narayanan, N. Effects of aging on sarcoplasmic reticulum Ca2+-cycling proteins and their phosphorylation in rat myocardium. Am. J. Physiol. Heart Circ. Physiol. 1998, 275, H2087–H2094. [Google Scholar] [CrossRef] [PubMed]
- Koban, M. Expressional analysis of the cardiac Na–Ca exchanger in rat development and senescence. Cardiovasc. Res. 1998, 37, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, K.; Monfredi, O.J.; Sirenko-Tagirova, S.G.; Maltseva, L.A.; Bychkov, R.; Kim, M.S.; Ziman, B.D.; Tarasov, K.V.; Tarasova, Y.S.; Zhang, J.; et al. A coupled-clock system drives the automaticity of human sinoatrial nodal pacemaker cells. Sci. Signal. 2018, 11, eaap7608. [Google Scholar] [CrossRef] [PubMed]
- Jahangir, A.; Ozcan, C.; Holmuhamedov, E.L.; Terzic, A. Increased calcium vulnerability of senescent cardiac mitochondria: Protective role for a mitochondrial potassium channel opener. Mech. Ageing Dev. 2001, 122, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Jahangir, A.; Sagar, S.; Terzic, A. Aging and cardioprotection. J. Appl. Physiol. 2007, 103, 2120–2128. [Google Scholar] [CrossRef]
- Nakou, E.S.; Parthenakis, F.I.; Kallergis, E.M.; Marketou, M.E.; Nakos, K.S.; Vardas, P.E. Healthy aging and myocardium: A complicated process with various effects in cardiac structure and physiology. Int. J. Cardiol. 2016, 209, 167–175. [Google Scholar] [CrossRef]
- Shah, P.M.; Abelmann, W.H.; Gersh, B.J. Cardiomyopathies in the elderly. J. Am. Coll Cardiol. 1987, 10, 77A–79A. [Google Scholar] [CrossRef][Green Version]
- Alashi, A.; Smedira, N.G.; Popovic, Z.B.; Fava, A.; Thamilarasan, M.; Kapadia, S.R.; Wierup, P.; Lever, H.M.; Desai, M.Y. Characteristics and Outcomes of Elderly Patients With Hypertrophic Cardiomyopathy. J. Am. Heart Assoc. 2021, 10, e018527. [Google Scholar] [CrossRef]
- Knickelbine, T.; Lesser, J.R.; Haas, T.S.; Brandenburg, E.R.; Gleason-Han, B.K.; Flygenring, B.; Longe, T.F.; Schwartz, R.S.; Maron, B.J. Identification of Unexpected Nonatherosclerotic Cardiovascular Disease With Coronary CT Angiography. JACC Cardiovasc. Imaging 2009, 2, 1085–1092. [Google Scholar] [CrossRef][Green Version]
- Maron, M.S.; Maron, B.J.; Harrigan, C.; Buros, J.; Gibson, C.M.; Olivotto, I.; Biller, L.; Lesser, J.R.; Udelson, J.E.; Manning, W.J.; et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J. Am. Coll Cardiol. 2009, 54, 220–228. [Google Scholar] [CrossRef]
- Olcum, M.; Cheedipudi, S.M.; Rouhi, L.; Fan, S.; Jeong, H.H.; Zhao, Z.; Gurha, P.; Marian, A.J. The WNT/β-catenin pathway regulates expression of the genes involved in cell cycle progression and mitochondrial oxidative phosphorylation in the postmitotic cardiac myocytes. J. Cardiovasc. Aging 2022, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Söti, C.; Csermely, P. Molecular chaperones and the aging process. Biogerontology 2000, 1, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Bakthisaran, R.; Tangirala, R.; Rao, C.M. Small heat shock proteins: Role in cellular functions and pathology. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2015, 1854, 291–319. [Google Scholar] [CrossRef]
- Clark, J.I. Functional sequences in human alphaB crystallin. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2016, 1860, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, R.; Budde, H.; Zhazykbayeva, S.; Herwig, M.; Sieme, M.; Delalat, S.; Mostafi, N.; Gömöri, K.; Tangos, M.; Jarkas, M.; et al. Stress activated signalling impaired protein quality control pathways in human hypertrophic cardiomyopathy. Int. J. Cardiol. 2021, 344, 160–169. [Google Scholar] [CrossRef]
- Triposkiadis, F.; Xanthopoulos, A.; Butler, J. Cardiovascular Aging and Heart Failure: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 804–813. [Google Scholar] [CrossRef]
- Bullard, B.; Ferguson, C.; Minajeva, A.; Leake, M.C.; Gautel, M.; Labeit, D.; Ding, L.; Labeit, S.; Horwitz, J.; Leonard, K.R.; et al. Association of the Chaperone αB-crystallin with Titin in Heart Muscle. J. Biol. Chem. 2004, 279, 7917–7924. [Google Scholar] [CrossRef]
- Braun, N.; Zacharias, M.; Peschek, J.; Kastenmüller, A.; Zou, J.; Hanzlik, M.; Haslbeck, M.; Rappsilber, J.; Buchner, J.; Weinkauf, S.; et al. Multiple molecular architectures of the eye lens chaperone αB-crystallin elucidated by a triple hybrid approach. Proc. Natl. Acad. Sci. USA 2011, 108, 20491–20496. [Google Scholar] [CrossRef]
- Taneike, M.; Yamaguchi, O.; Nakai, A.; Hikoso, S.; Takeda, T.; Mizote, I.; Oka, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 2010, 6, 600–606. [Google Scholar] [CrossRef]
- Cheedipudi, S.M.; Fan, S.; Rouhi, L.; Marian, A.J. Pharmacological suppression of the WNT signaling pathway attenuates age-dependent expression of the phenotype in a mouse model of arrhythmogenic cardiomyopathy. J. Cardiovasc. Aging 2021, 1, 3. [Google Scholar] [CrossRef]
- Kubo, T.; Matsumura, Y.; Kitaoka, H.; Okawa, M.; Hirota, T.; Hamada, T.; Hitomi, N.; Hoshikawa, E.; Hayato, K.; Shimizu, Y.; et al. Improvement in prognosis of dilated cardiomyopathy in the elderly over the past 20 years. J. Cardiol. 2008, 52, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.C.Y.; Chang, A.C.H.; Kirillova, A.; Sasagawa, K.; Su, W.; Weber, G.; Lin, J.; Termglinchan, V.; Karakikes, I.; Seeger, T.; et al. Telomere shortening is a hallmark of genetic cardiomyopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 9276–9281. Available online: https://pubmed.ncbi.nlm.nih.gov/30150400/ (accessed on 3 October 2022). [CrossRef] [PubMed]
- Chatterjee, S.; de Gonzalo-Calvo, D.; Derda, A.A.; Schimmel, K.; Sonnenschein, K.; Bavendiek, U.; Bauersachs, J.; Bär, C.; Thum, T. Leukocyte telomere length correlates with hypertrophic cardiomyopathy severity. Sci. Rep. 2018, 8, 11227. Available online: https://pubmed.ncbi.nlm.nih.gov/30046139/ (accessed on 3 October 2022). [CrossRef] [PubMed]
- Foglieni, C.; Lombardi, M.; Lazzeroni, D.; Zerboni, R.; Lazzarini, E.; Bertoli, G.; Pisano, A.; Girolami, F.; Andolfo, A.; Magagnotti, C.; et al. Myosins and MyomiR Network in Patients with Obstructive Hypertrophic Cardiomyopathy. Biomedicines 2022, 10, 2180. Available online: https://pubmed.ncbi.nlm.nih.gov/36140281/ (accessed on 6 October 2022). [CrossRef]
- Peretto, G.; Sala, S.; Rizzo, S.; de Luca, G.; Campochiaro, C.; Sartorelli, S.; Benedetti, G.; Palmisano, A.; Esposito, A.; Tresoldi, M.; et al. Arrhythmias in myocarditis: State of the art. Heart Rhythm. 2019, 16, 793–801. Available online: https://pubmed.ncbi.nlm.nih.gov/30476544/ (accessed on 27 March 2022). [CrossRef]
- Peretto, G.; Sala, S.; Benedetti, S.; di Resta, C.; Gigli, L.; Ferrari, M.; Bella, P. della. Updated clinical overview on cardiac laminopathies: An electrical and mechanical disease. Nucleus 2018, 9, 380–391. Available online: https://pubmed.ncbi.nlm.nih.gov/29929425/ (accessed on 6 October 2022). [CrossRef]
- Garcia-Pavia, P.; Rapezzi, C.; Adler, Y.; Arad, M.; Basso, C.; Brucato, A.; Burazor, I.; Caforio, A.L.P.; Damy, T.; Eriksson, U.; et al. Diagnosis and treatment of cardiac amyloidosis. A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. J. Heart Fail. 2021, 23, 512–526. Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/ejhf.2140 (accessed on 3 October 2022). [CrossRef]
- Porcari, A.; Fontana, M.; Gillmore, J.D. Transthyretin cardiac amyloidosis. Cardiovasc. Res. 2022, cvac119. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Buxbaum, J.N.; Reixach, N. Age-related oxidative modifications of transthyretin modulate its amyloidogenicity. Biochemistry 2013, 52, 1913–1926. Available online: https://pubmed.ncbi.nlm.nih.gov/23414091/ (accessed on 3 October 2022). [CrossRef]
- Visseren, F.L.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2021, 42, 3227–3337. Available online: https://pubmed.ncbi.nlm.nih.gov/34458905/ (accessed on 30 September 2022). [CrossRef]
- Forman, D.E.; de Lemos, J.A.; Shaw, L.J.; Reuben, D.B.; Lyubarova, R.; Peterson, E.D.; Spertus, J.A.; Zieman, S.; Salive, M.E.; Rich, M.W. Cardiovascular Biomarkers and Imaging in Older Adults: JACC Council Perspectives. J. Am. Coll. Cardiol. 2020, 76, 1577–1594. Available online: https://pubmed.ncbi.nlm.nih.gov/32972536/ (accessed on 1 October 2022). [CrossRef] [PubMed]
- Lettino, M.; Mascherbauer, J.; Nordaby, M.; Ziegler, A.; Collet, J.P.; Derumeaux, G.; Hohnloser, S.H.; Leclercq, C.; O’neill, D.E.; Visseren, F.; et al. Cardiovascular disease in the elderly: Proceedings of the European Society of Cardiology-Cardiovascular Round Table. Eur. J. Prev. Cardiol. 2022, 29, 1412–1424. Available online: https://pubmed.ncbi.nlm.nih.gov/35167666/ (accessed on 30 September 2022). [CrossRef] [PubMed]
- Hadley, E.C.; Lakatta, E.G.; Morrison-Bogorad, M.; Warner, H.R.; Hodes, R.J. The future of aging therapies. Cell 2005, 120, 557–567. Available online: http://www.cell.com/article/S0092867405001042/fulltext (accessed on 30 September 2022). [CrossRef] [PubMed]
- McNeil, J.J.; Wolfe, R.; Woods, R.L.; Tonkin, A.M.; Donnan, G.A.; Nelson, M.R.; Reid, C.M.; Lockery, J.E.; Kirpach, B.; Storey, E.; et al. Effect of Aspirin on Cardiovascular Events and Bleeding in the Healthy Elderly. N. Engl. J. Med. 2018, 379, 1509–1518. Available online: https://pubmed.ncbi.nlm.nih.gov/30221597/ (accessed on 30 September 2022). [CrossRef]
- Collaboration SO working group and EC risk. SCORE2-OP risk prediction algorithms: Estimating incident cardiovascular event risk in older persons in four geographical risk regions. Eur. Heart J. 2021, 42, 2455. [Google Scholar] [CrossRef]
- Castellano, J.M.; Pocock, S.J.; Bhatt, D.L.; Quesada, A.J.; Owen, R.; Fernandez-Ortiz, A.; Sanchez, P.L.; Marin Ortuño, F.; Vazquez Rodriguez, J.M.; Domingo-Fernández, A.; et al. Polypill Strategy in Secondary Cardiovascular Prevention. N. Engl. J. Med. 2022, 387, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Ruff, C.T.; Giugliano, R.P.; Braunwald, E.; Hoffman, E.B.; Deenadayalu, N.; Ezekowitz, M.D.; Camm, A.J.; Weitz, J.I.; Lewis, B.S.; Parkhomenko, A.; et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: A meta-analysis of randomised trials. Lancet 2014, 383, 955–962. Available online: https://pubmed.ncbi.nlm.nih.gov/24315724/ (accessed on 30 September 2022). [CrossRef]
- Shah, S.J.; Singer, D.E.; Fang, M.C.; Reynolds, K.; Go, A.S.; Eckman, M.H. Net Clinical Benefit of Oral Anticoagulation Among Older Adults With Atrial Fibrillation. Circ. Cardiovasc. Qual. Outcomes. 2019, 12, e006212. [Google Scholar] [CrossRef]
- Pfisterer, M. Trial of invasive versus medical therapy in elderly patients with chronic symptomatic coronary-artery disease (TIME): A randomised trial. Lancet 2001, 358, 951–957. Available online: https://pubmed.ncbi.nlm.nih.gov/11583747/ (accessed on 30 September 2022).
- Stone, G.W.; Lindenfeld, J.; Abraham, W.T.; Kar, S.; Lim, D.S.; Mishell, J.M.; Whisenant, B.; Grayburn, P.A.; Rinaldi, M.; Kapadia, S.R.; et al. Transcatheter Mitral-Valve Repair in Patients with Heart Failure. N. Engl. J. Med. 2018, 379, 2307–2318. Available online: https://pubmed.ncbi.nlm.nih.gov/30280640/ (accessed on 30 September 2022). [CrossRef]
- Leon, M.B.; Smith, C.R.; Mack, M.; Miller, D.C.; Moses, J.W.; Svensson, L.G.; Tuzcu, E.M.; Webb, J.G.; Fontana, G.P.; Makkar, R.R.; et al. Transcatheter aortic-valve implantation for aortic stenosis in patients who cannot undergo surgery. N. Engl. J. Med. 2010, 363, 1597–1607. Available online: https://pubmed.ncbi.nlm.nih.gov/20961243/ (accessed on 30 September 2022). [CrossRef] [PubMed]
- Kumar, S.; McDaniel, M.; Samady, H.; Forouzandeh, F. Contemporary Revascularization Dilemmas in Older Adults. J. Am. Heart Assoc. 2020, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Arisha, M.J.; Ibrahim, D.A.; Abouarab, A.A.; Rahouma, M.; Kamel, M.K.; Baudo, M.; Mehta, K.; Gaudino, M.F.L. Percutaneous coronary intervention in the elderly: Current updates and trends. Vessel Plus 2018, 2, 14. [Google Scholar] [CrossRef][Green Version]
- Bangalore, S.; Maron, D.J.; Stone, G.W.; Hochman, J.S. Routine Revascularization Versus Initial Medical Therapy for Stable Ischemic Heart Disease: A Systematic Review and Meta-Analysis of Randomized Trials. Circulation 2020, 143, e809–e810. [Google Scholar] [CrossRef] [PubMed]
- Kilic, Y.; Smer, A.; Goldstein, N. The Importance of Palliative Care in Cardiology: Differences Between Countries. JACC Case Rep. 2020, 2, 326–329. Available online: https://pubmed.ncbi.nlm.nih.gov/34317235/ (accessed on 30 September 2022). [CrossRef] [PubMed]
- Rogers, J.G.; Patel, C.B.; Mentz, R.J.; Granger, B.B.; Steinhauser, K.E.; Fiuzat, M.; Adams, P.A.; Speck, A.; Johnson, K.S.; Krishnamoorthy, A.; et al. Palliative Care in Heart Failure: The PAL-HF Randomized, Controlled Clinical Trial. J. Am. Coll. Cardiol. 2017, 70, 331–341. Available online: https://pubmed.ncbi.nlm.nih.gov/28705314/ (accessed on 30 September 2022). [CrossRef]
- Infante, B.; Bellanti, F.; Correale, M.; Pontrelli, P.; Franzin, R.; Leo, S.; Calvaruso, M.; Mercuri, S.; Netti, G.S.; Ranieri, E.; et al. mTOR inhibition improves mitochondria function/biogenesis and delays cardiovascular aging in kidney transplant recipients with chronic graft dysfunction. Aging 2021, 13, 8026–8039. [Google Scholar] [CrossRef]
- Kofman, A.E.; McGraw, M.R.; Payne, C.J. Rapamycin increases oxidative stress response gene expression in adult stem cells. Aging 2012, 4, 279–289. [Google Scholar] [CrossRef][Green Version]
- Cao, R.Y.; Zheng, Y.; Zhang, Y.; Jiang, L.; Li, Q.; Sun, W.; Gu, W.; Cao, W.; Zhou, L.; Zheng, H.Y.J. Berberine on the Prevention and Management of Cardiometabolic Disease: Clinical Applications and Mechanisms of Action. Am. J. Chin. Med. 2021, 49, 1645–1666. [Google Scholar] [CrossRef]
- Most, J.; Gilmore, L.A.; Smith, S.R.; Han, H.; Ravussin, E.; Redman, L.M. Significant improvement in cardiometabolic health in healthy nonobese individuals during caloric restriction-induced weight loss and weight loss maintenance. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E396–E405. [Google Scholar] [CrossRef]
- Belsky, D.W.; Huffman, K.M.; Pieper, C.F.; Shalev, I.; Kraus, W.E.; Anderson, R. Change in the rate of biological aging in response to caloric restriction: Calerie Biobank analysis. J. Gerontol.—Ser. A Biol. Sci. Med. Sci. 2018, 73, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Gogulamudi, V.R.; Cai, J.; Lesniewski, L.A. Reversing Age-Associated Arterial Dysfunction: Insight from Preclinical Models. J. Appl. Physiol. 2018, 84148, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Suzuki, K.; Hattori, S.; Kasai, K. Metformin inhibits cytokine-induced nuclear factor κB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension 2006, 47, 1183–1188. [Google Scholar] [CrossRef]
- Brandauer, J.; Andersen, M.A.; Kellezi, H.; Risis, S.; Frøsig, C.; Vienberg, S.G.; Treebak, J.T. AMP-activated protein kinase controls exercise training- and AICAR-induced increases in SIRT3 and MnSOD. Front. Physiol. 2015, 6, 1–16. [Google Scholar] [CrossRef]
- Hattori, Y.; Nakano, Y.; Hattori, S.; Tomizawa, A.; Inukai, K.; Kasai, K. High molecular weight adiponectin activates AMPK and suppresses cytokine-induced NF-κB activation in vascular endothelial cells. FEBS Lett. 2008, 582, 1719–1724. [Google Scholar] [CrossRef]
- Minor, R.K.; Baur, J.A.; Gomes, A.P.; Ward, T.M.; Csiszar, A.; Mercken, E.M.; Abdelmohsen, K.; Shin, Y.K.; Canto, C.; Scheibye-Knudsen, M.; et al. SRT1720 improves survival and healthspan of obese mice. Sci. Rep. 2011, 1, 70. [Google Scholar] [CrossRef] [PubMed]
- Gano, L.B.; Donato, A.J.; Pasha, H.M.; Hearon, C.M.; Sindler, A.L.; Seals, D.R. The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1754–H1763. [Google Scholar] [CrossRef]
- Huang, Y.; Lu, J.; Zhan, L.; Wang, M.; Shi, R.; Yuan, X.; Gao, X.; Liu, X.; Zang, J.; Liu, W.; et al. Resveratrol-induced Sirt1 phosphorylation by LKB1 mediates mitochondrial metabolism. J. Biol. Chem. 2021, 297, 100929. [Google Scholar] [CrossRef]
- Boccardi, V.; Herbig, U. Telomerase gene therapy: A novel approach to combat aging. EMBO Mol. Med. 2012, 4, 685–687. [Google Scholar] [CrossRef]
- Yeh, J.K.; Lin, M.H.; Wang, C.Y. Telomeres as Therapeutic Targets in Heart Disease. JACC Basic Transl. Sci. 2019, 4, 855–865. [Google Scholar] [CrossRef]
- Martin-Ruiz, C.M.; Gussekloo, J.; van Heemst, D.; von Zglinicki, T.; Westendorp, R.G.J. Telomere length in white blood cells is not associated with morbidity or mortality in the oldest old: A population-based study. Aging Cell 2005, 4, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Franzon, K.; Byberg, L.; Sjögren, P.; Zethelius, B.; Cederholm, T.; Kilander, L. Predictors of Independent Aging and Survival: A 16-Year Follow-Up Report in Octogenarian Men. J. Am. Geriatr. Soc. 2017, 65, 1953–1960. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Belcher, M.; Van Der Harst, P. Healthy aging and disease: Role for telomere biology? Clin. Sci. 2011, 120, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Tomás-Loba, A.; Flores, I.; Fernández-Marcos, P.J.; Cayuela, M.L.; Maraver, A.; Tejera, A.; Borrás, C.; Matheu, A.; Klatt, P.; Flores, J.M.; et al. Telomerase Reverse Transcriptase Delays Aging in Cancer-Resistant Mice. Cell 2008, 135, 609–622. [Google Scholar] [CrossRef]







| Molecular Mechanisms and Intracellular Modifications Underlying Heart Failure | Studies |
|---|---|
| - Degeneration of elastin fibers and increase in collagen | [57] |
| - Clustering and hypertrophy of smooth muscle cells | [57] |
| - Endothelial dysfunction, which affects the production of NO and other peptides | [57] |
| - Myocardial interstitial fibrosis, calcium deposition, and amyloid accumulations | [8] |
| - Upregulation of the activin/ActRII pathway and TLR signaling | [59,62,63] |
| Molecular Mechanisms and Intracellular Modifications Underlying Cardiomyopathies | Studies |
|---|---|
| - Dysfunction in the physiological protein quality control system, which includes heat shock proteins, the autophagy/lysosomal pathway, and the ubiquitin-proteasome system (HCM) | [128] |
| - WNT pathway, due to its correlation with cardiac dilatation and myocardial fibrosis (ACM) | [69,140] |
| - Telomere shortening (HCM and DCM) | [142] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazzeroni, D.; Villatore, A.; Souryal, G.; Pili, G.; Peretto, G. The Aging Heart: A Molecular and Clinical Challenge. Int. J. Mol. Sci. 2022, 23, 16033. https://doi.org/10.3390/ijms232416033
Lazzeroni D, Villatore A, Souryal G, Pili G, Peretto G. The Aging Heart: A Molecular and Clinical Challenge. International Journal of Molecular Sciences. 2022; 23(24):16033. https://doi.org/10.3390/ijms232416033
Chicago/Turabian StyleLazzeroni, Davide, Andrea Villatore, Gaia Souryal, Gianluca Pili, and Giovanni Peretto. 2022. "The Aging Heart: A Molecular and Clinical Challenge" International Journal of Molecular Sciences 23, no. 24: 16033. https://doi.org/10.3390/ijms232416033
APA StyleLazzeroni, D., Villatore, A., Souryal, G., Pili, G., & Peretto, G. (2022). The Aging Heart: A Molecular and Clinical Challenge. International Journal of Molecular Sciences, 23(24), 16033. https://doi.org/10.3390/ijms232416033