
Macrophage-Colony-Stimulating Factor Receptor Enhances Prostate Cancer Cell Growth and Aggressiveness In Vitro and In Vivo and Increases Osteopontin Expression
 ,
,  and
and
Abstract
1. Introduction
2. Results
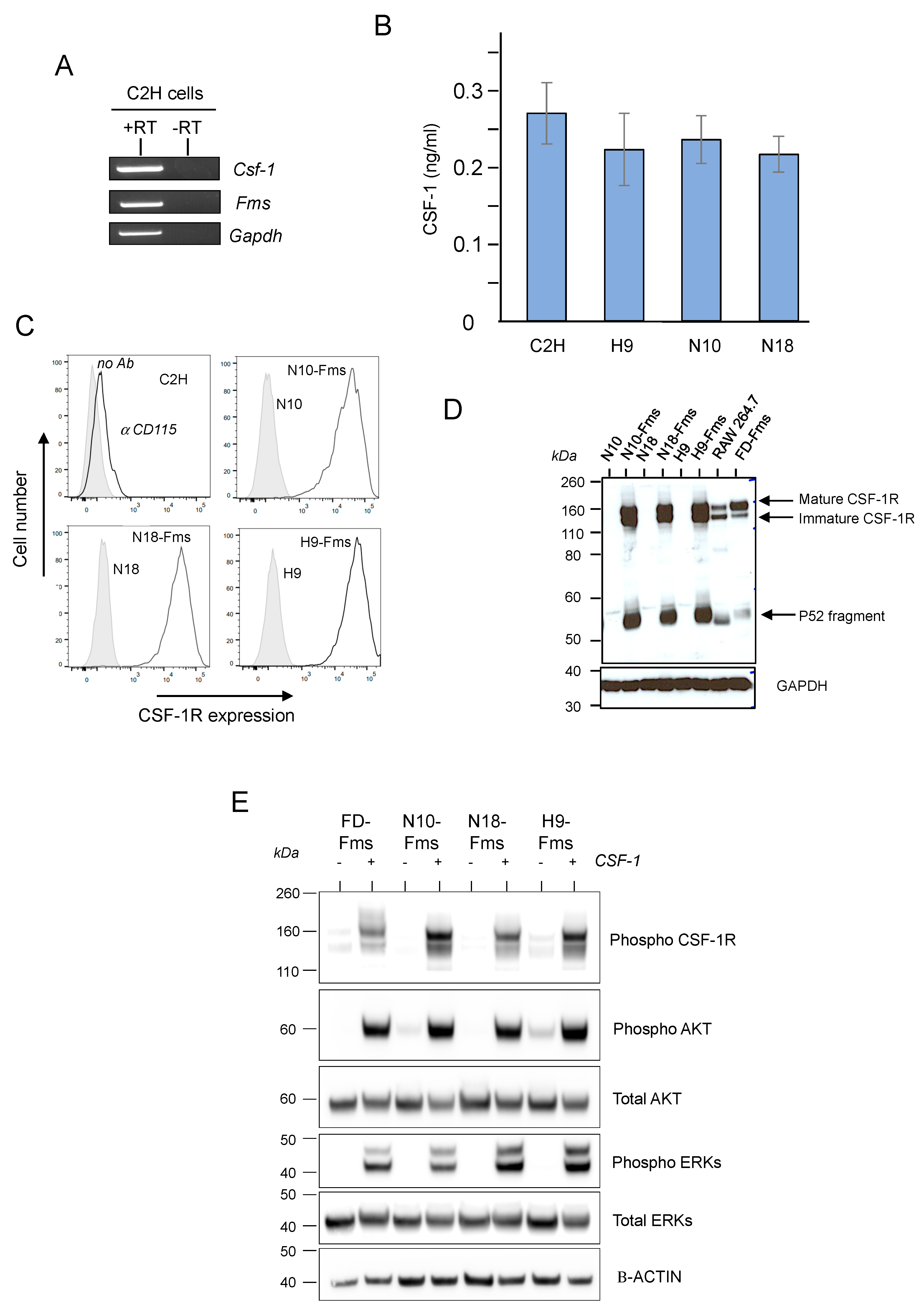
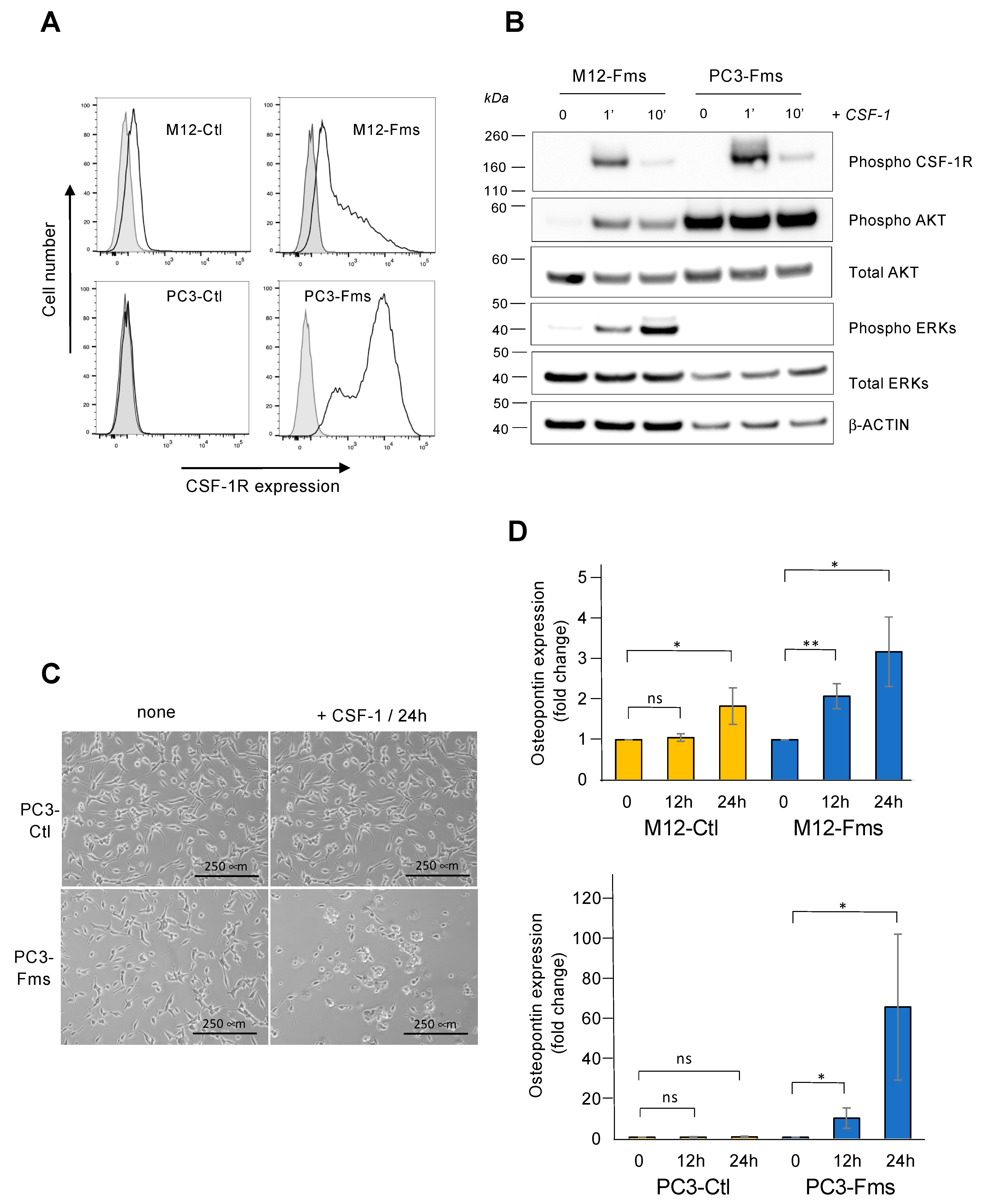
2.1. CSF-1 Receptor Is Functional in Murine Prostate Cancer Cells
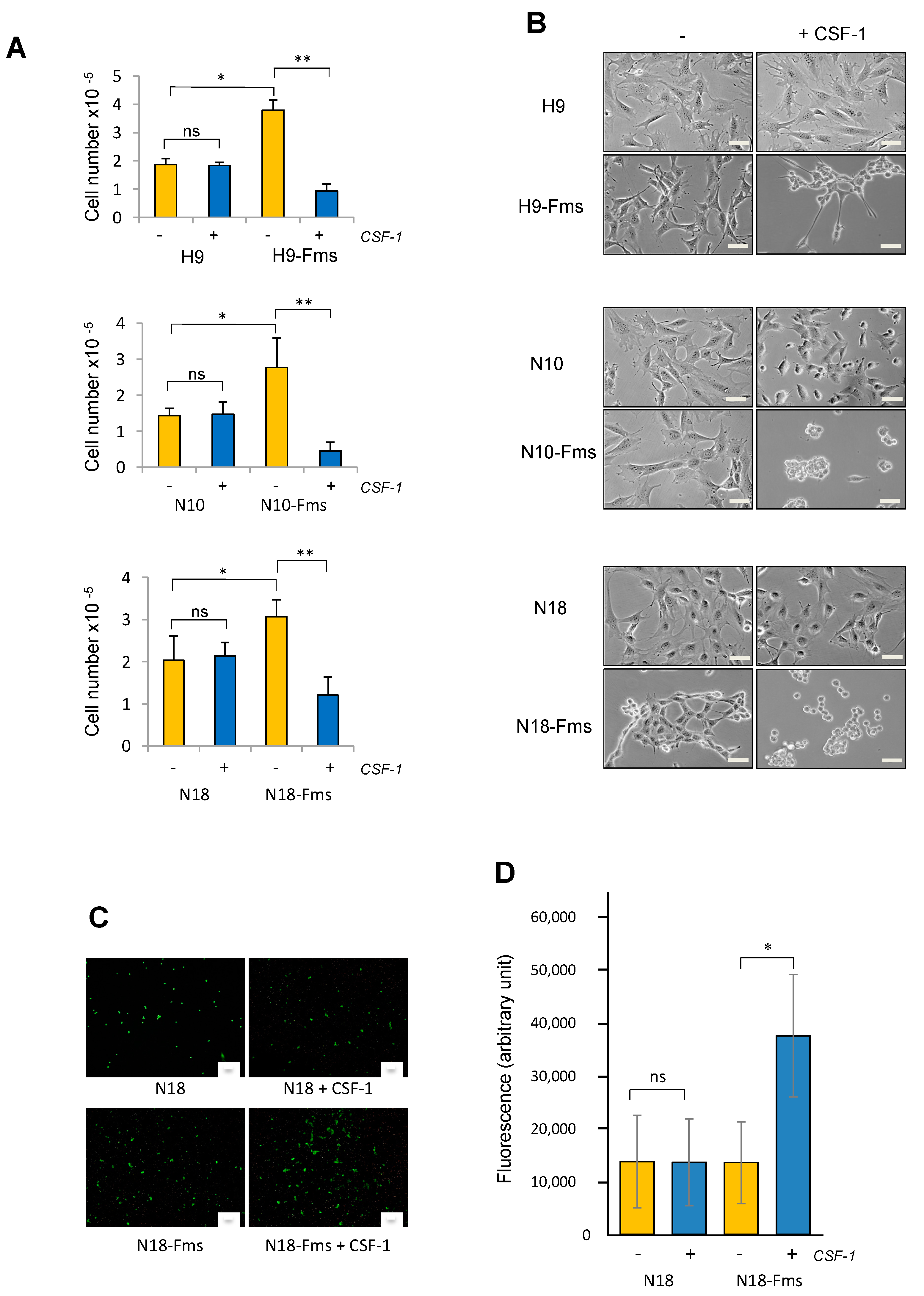
2.2. CSF-1 Receptor Expression Promotes Prostate C2H Cell Growth and Invasion
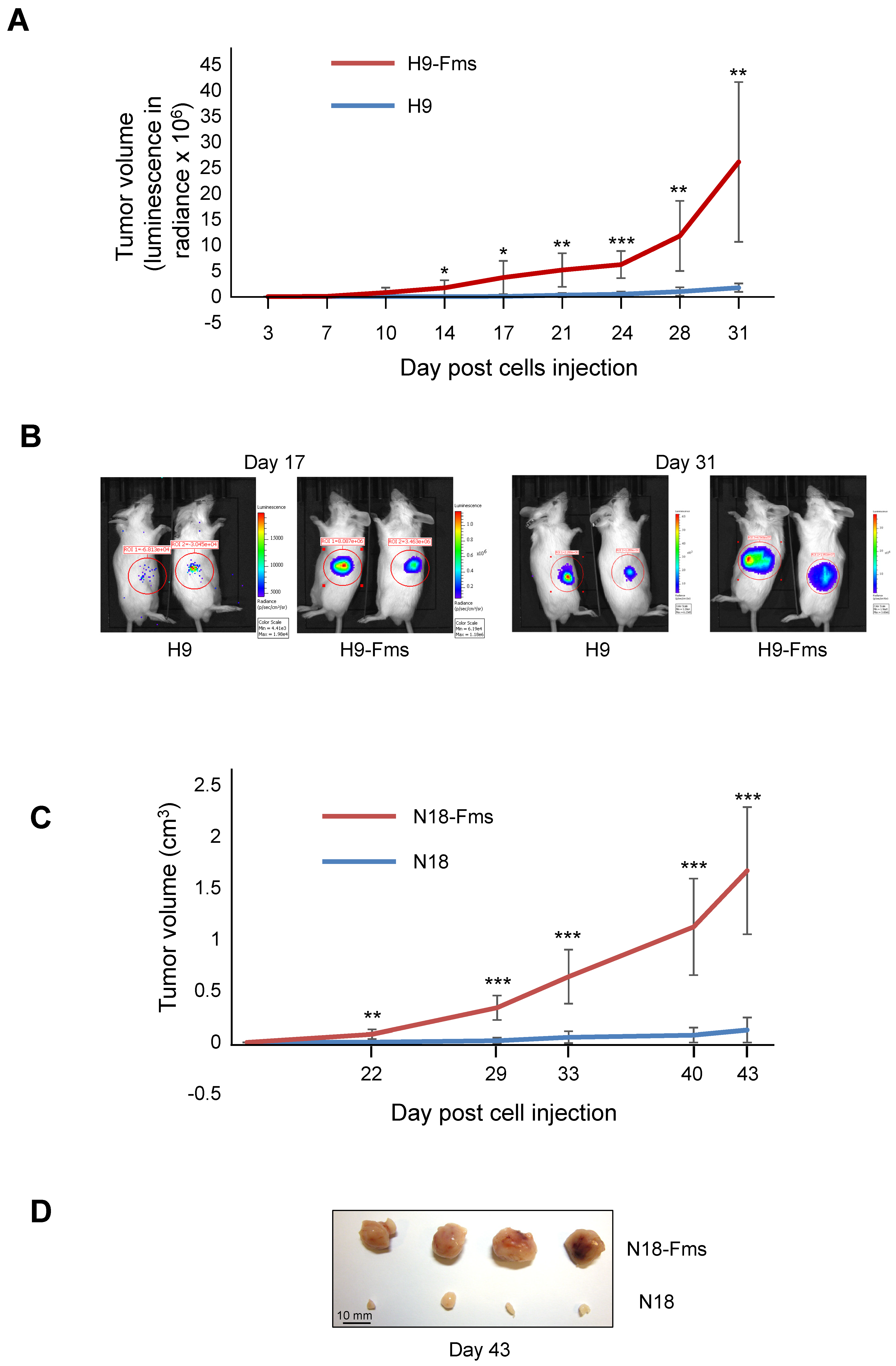
2.3. CSF-1 Receptor Expression Increased Tumor Cell Growth In Vivo
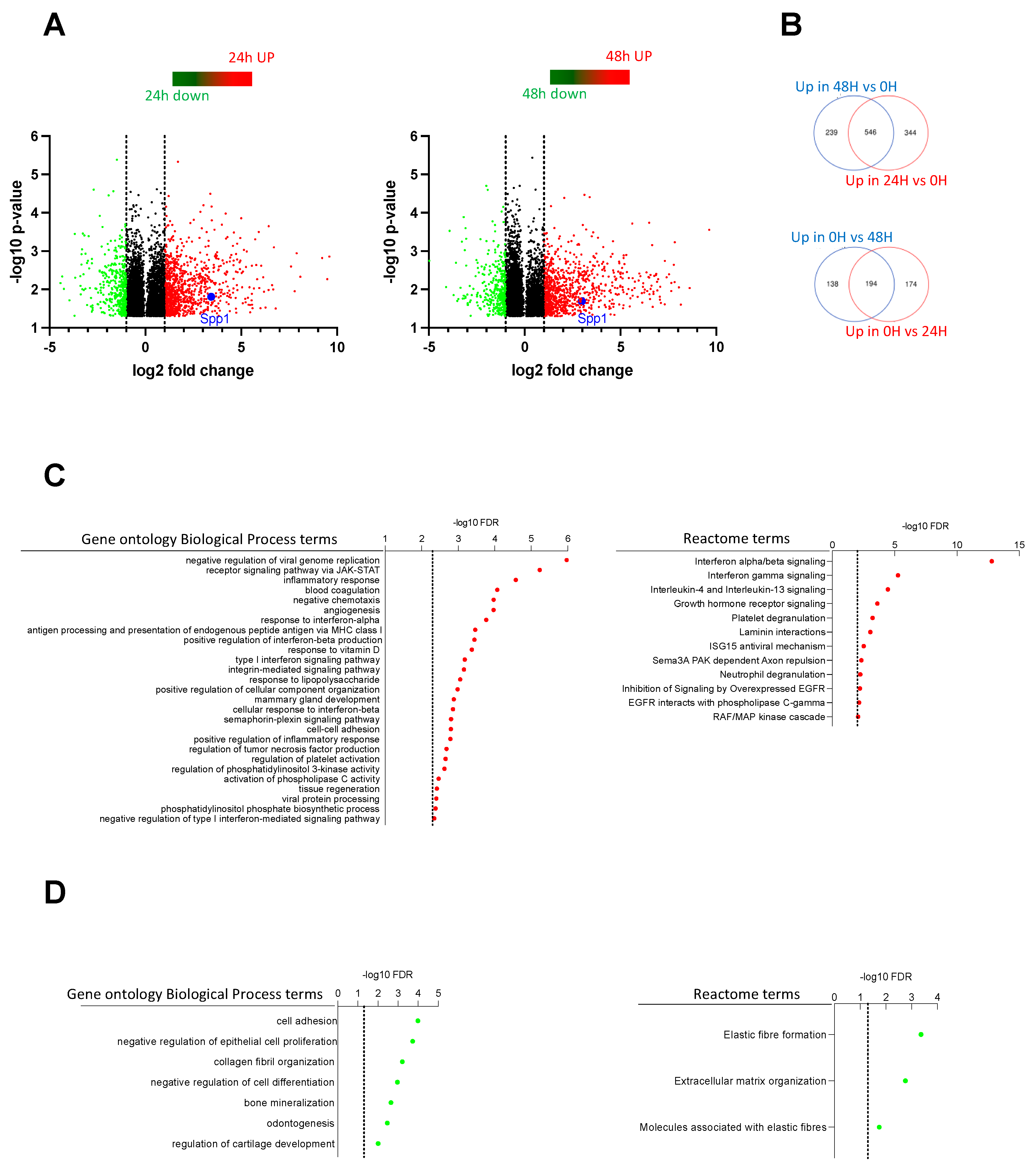
2.4. Transcriptome Analysis Discovered Key Functions Regulated by CSF-1R in Murine Prostate Cancer Cells
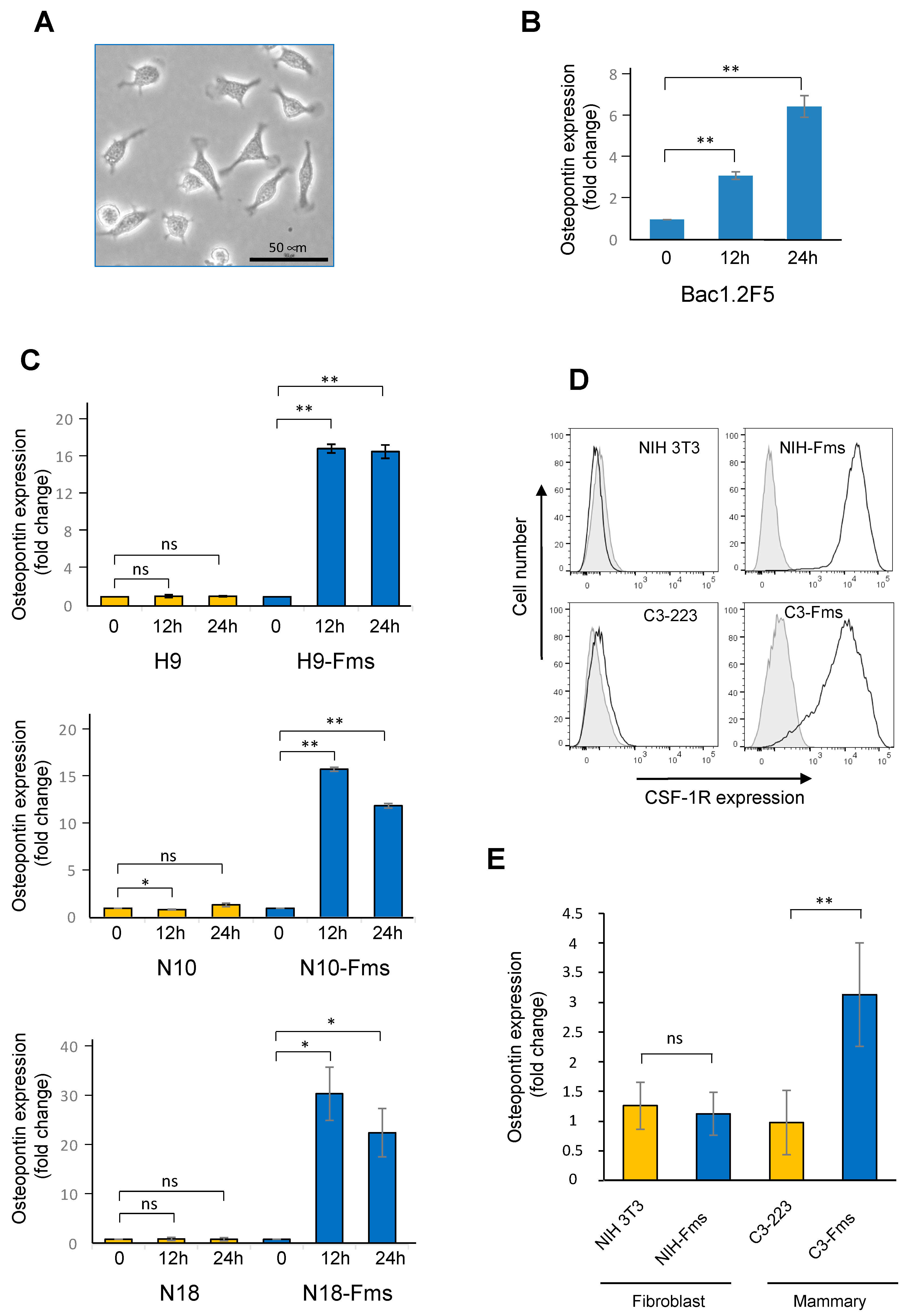
2.5. Spp1 Gene Encoding Osteopontin Is a Target of CSF-1R in Murine Prostate Cancer Cells
2.6. CSF-1R Is Functional in Human Prostate Cancer Cells and Increases Osteopontin Gene Expression
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. CSF-1R Transduction
4.3. Growth Studies
4.4. Invasion Assay
4.5. ELISA
4.6. RT-PCR
4.7. Microarray Analysis
4.8. Immunoblotting and Antibodies
4.9. Tumor Xenograft Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Reverse (5′ → 3′) | Forward (5′ → 3′) |
|---|---|---|
| Mouse CSF-1 | ACTTGCTGATCCTCCTTCC | CATCTCCATTCCCTAAATCAAC |
| Mouse CSF-1R | GTGCACCAGTTGGCATAGTAAATGTAGAGGCT | GACTGGAGAGGAGAGACCAGGACTATG |
| Human CSF-1 | CATGCTCTTCATAATCCTTG | GCTGTTGTTGGTCTGTCTC |
| Human CSF-1R | GGCTCCAGCTCCGAGGGTCTT | TGCTGCTCCTGCTGCTATTG |
| Mouse GAPDH | GCCTTCTCCATGGTGGTGAA | GCACAGTCAAGGCCGAGAAT |
| Human RPLP0 | GATGACCAGCCCAAAGGAGA | GTGATGTGCAGCTGATCAAGACT |
| Mouse RPLP38 | TGACAGACTTGGCATCCTTCC | GGTTCTCATCGCTGTGCGG |
| Mouse OPN | CTAGTTGACCTCAGAAGATGAACTC | GGACCTCACCTCTCACATGAAGAGC |
| Human OPN | CATCAGACTGGTGAGAATCATC | ATCTCCTAGCCCCACAGAAT |
References
- Culp, M.B.; Soerjomataram, I.; Efstathiou, J.A.; Bray, F.; Jemal, A. Recent Global Patterns in Prostate Cancer Incidence and Mortality Rates. Eur. Urol. 2020, 77, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate cancer. Nat. Rev. Dis. Primers. 2021, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Ghosh, S.; Marrocco, I.; Yarden, Y. Roles for receptor tyrosine kinases in tumor progression and implications for cancer treatment. Adv. Cancer Res. 2020, 147, 1–57. [Google Scholar]
- Batth, I.S.; Huang, S.B.; Villarreal, M.; Gong, J.; Chakravarthy, D.; Keppler, B.; Jayamohan, S.; Osmulski, P.; Xie, J.; Rivas, P.; et al. Evidence for 2-Methoxyestradiol-Mediated Inhibition of Receptor Tyrosine Kinase RON in the Management of Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 1852. [Google Scholar] [CrossRef]
- Sherr, C.J.; Rettenmier, C.W.; Sacca, R.; Roussel, M.F.; Look, A.T.; Stanley, E.R. The c-fms proto-oncogene product is related to the receptor for the mononuclear phagocyte growth factor, CSF-1. Cell 1985, 41, 665–676. [Google Scholar] [CrossRef]
- Wolford, J.; Rothwell, V.; Rohrschneider, L.R. Characterization of the human c-fms gene product and its expression in cells of the monocyte-macrophage lineage. Mol. Cell. Biol. 1985, 5, 3458–3466. [Google Scholar]
- Grassot, J.; Gouy, M.; Perrière, G.; Mouchiroud, G. Origin and molecular evolution of receptor tyrosine kinases with immunoglobulin-like domains. Mol. Biol. Evol. 2006, 23, 1232–1241. [Google Scholar] [CrossRef]
- Lin, H.; Lee, E.; Hestir, K.; Leo, C.; Huang, M.; Bosch, E.; Halenbeck, R.; Wu, G.; Zhou, A.; Behrens, D.; et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 2008, 320, 807–811. [Google Scholar] [CrossRef]
- Chihara, T.; Suzu, S.; Hassan, R.; Chutiwitoonchai, N.; Hiyoshi, M.; Motoyoshi, K.; Kimura, F.; Okada, S. IL-34 and M-CSF share the receptor Fms but are not identical in biological activity and signal activation. Cell Death Differ. 2010, 17, 1917–1927. [Google Scholar] [CrossRef] [PubMed]
- Wiktor-Jedrzejczak, W.; Bartocci, A.; Ferrante, A.W., Jr.; Ahmed-Ansari, A.; Sell, K.W.; Pollard, J.W.; Stanley, E.R. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc. Natl. Acad. Sci. USA 1990, 87, 4828–4832. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.M.; Ryan, G.R.; Hapel, A.J.; Dominguez, M.G.; Russell, R.G.; Kapp, S.; Sylvestre, V.; Stanley, E.R. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 2002, 99, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A.; MacDonald, K.P. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood 2012, 119, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Silakari, O.; Singh, R.K. Recent advances in colony stimulating factor-1 receptor/c-FMS as an emerging target for various therapeutic implications. Biomed. Pharmacother. 2018, 103, 662–679. [Google Scholar] [CrossRef]
- Bourette, R.P.; Rohrschneider, L.R. Early events in M-CSF receptor signaling. Growth Factors 2000, 17, 155–166. [Google Scholar] [CrossRef]
- Mouchemore, K.A.; Pixley, F.J. CSF-1 signaling in macrophages: Pleiotrophy through phosphotyrosine-based signaling pathways. Crit. Rev. Clin. Lab. Sci. 2012, 49, 49–61. [Google Scholar] [CrossRef]
- Chitu, V.; Stanley, E.R. Regulation of Embryonic and Postnatal Development by the CSF-1 Receptor. Curr. Top. Dev. Biol. 2017, 123, 229–275. [Google Scholar]
- Hu, B.; Duan, S.; Wang, Z.; Li, X.; Zhou, Y.; Zhang, X.; Zhang, Y.W.; Xu, H.; Zheng, H. Insights into the Role of CSF1R in the Central Nervous System and Neurological Disorders. Front. Aging Neurosci. 2021, 13, 789834. [Google Scholar] [CrossRef]
- Pollard, J.W.; Hennighausen, L. Colony stimulating factor 1 is required for mammary gland development during pregnancy. Proc. Natl. Acad. Sci. USA 1994, 91, 9312–9316. [Google Scholar] [CrossRef]
- Patsialou, A.; Wyckoff, J.; Wang, Y.; Goswami, S.; Stanley, E.R.; Condeelis, J.S. Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Res. 2009, 69, 9498–9506. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Burugu, S.; Cheng, A.S.; Leung, S.C.Y.; Gao, D.; Nielsen, T.O. Prognostic Significance of CSF-1R Expression in Early Invasive Breast Cancer. Cancers 2021, 13, 5769. [Google Scholar] [CrossRef] [PubMed]
- Ide, H.; Seligson, D.B.; Memarzadeh, S.; Xin, L.; Horvath, S.; Dubey, P.; Flick, M.B.; Kacinski, B.M.; Palotie, A.; Witte, O.N. Expression of colony-stimulating factor 1 receptor during prostate development and prostate cancer progression. Proc. Natl. Acad. Sci. USA 2002, 99, 14404–14409. [Google Scholar] [CrossRef] [PubMed]
- Savarese, D.M.; Valinski, H.; Quesenberry, P.; Savarese, T. Expression and function of colony-stimulating factors and their receptors in human prostate carcinoma cell lines. Prostate 1998, 34, 80–91. [Google Scholar] [CrossRef]
- Wrobel, C.N.; Debnath, J.; Lin, E.; Beausoleil, S.; Roussel, M.F.; Brugge, J.S. Autocrine CSF-1R activation promotes Src-dependent disruption of mammary epithelial architecture. J. Cell. Biol. 2004, 165, 263–273. [Google Scholar] [CrossRef]
- Niehus, S.E.; Tran, D.D.H.; Mischak, M.; Koch, A. Colony-stimulating factor-1 receptor provides a growth advantage in epithelial cancer cell line A431 in the presence of epidermal growth factor receptor inhibitor gefitinib. Cell. Signal. 2018, 51, 191–198. [Google Scholar] [CrossRef]
- Greenberg, N.M.; DeMayo, F.; Finegold, M.J.; Medina, D.; Tilley, W.D.; Aspinall, J.O.; Cunha, G.R.; Donjacour, A.A.; Matusik, R.J.; Rosen, J.M. Prostate cancer in a transgenic mouse. Proc. Natl. Acad. Sci. USA 1995, 92, 3439–3443. [Google Scholar] [CrossRef]
- Foster, B.A.; Gingrich, J.R.; Kwon, E.D.; Madias, C.; Greenberg, N.M. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 1997, 57, 3325–3330. [Google Scholar]
- Bourette, R.P.; Myles, G.M.; Carlberg, K.; Chen, A.R.; Rohrschneider, L.R. Uncoupling of the proliferation and differentiation signals mediated by the murine macrophage colony-stimulating factor receptor expressed in myeloid FDC-P1 cells. Cell Growth Differ. 1995, 6, 631–645. [Google Scholar]
- Downing, J.R.; Roussel, M.F.; Sherr, C.J. Ligand and protein kinase C downmodulate the colony-stimulating factor 1 receptor by independent mechanisms. Mol. Cell. Biol. 1989, 9, 2890–2896. [Google Scholar]
- Takahashi, F.; Takahashi, K.; Maeda, K.; Tominaga, S.; Fukuchi, Y. Osteopontin is induced by nitric oxide in RAW 264.7 cells. IUBMB Life 2000, 49, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Bae, V.L.; Jackson-Cook, C.K.; Maygarden, S.J.; Plymate, S.R.; Chen, J.; Ware, J.L. Metastatic sublines of an SV40 large T antigen immortalized human prostate epithelial cell line. Prostate 1998, 34, 275–282. [Google Scholar] [CrossRef]
- Zhao, X.; Deng, R.; Wang, Y.; Zhang, H.; Dou, J.; Li, L.; Du, Y.; Chen, R.; Cheng, J.; Yu, J. Twist1/Dnmt3a and miR186 establish a regulatory circuit that controls inflammation-associated prostate cancer progression. Oncogenesis 2017, 6, e315. [Google Scholar] [CrossRef]
- Kaighn, M.E.; Narayan, K.S.; Ohnuki, Y.; Lechner, J.F.; Jones, L.W. Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Investig. Urol. 1979, 17, 16–23. [Google Scholar]
- Lee, J.T.; Steelman, L.S.; Chappell, W.H.; McCubrey, J.A. Akt inactivates ERK causing decreased response to chemotherapeutic drugs in advanced CaP cells. Cell Cycle 2008, 7, 631–636. [Google Scholar] [PubMed]
- Weiss, F.; Lauffenburger, D.; Friedl, P. Towards targeting of shared mechanisms of cancer metastasis and therapy resistance. Nat. Rev. Cancer 2022, 22, 157–173. [Google Scholar] [CrossRef]
- Guan, W.; Li, F.; Zhao, Z.; Zhang, Z.; Hu, J.; Zhang, Y. Tumor-Associated Macrophage Promotes the Survival of Cancer Cells upon Docetaxel Chemotherapy via the CSF1/CSF1R-CXCL12/CXCR4 Axis in Castration-Resistant Prostate Cancer. Genes 2021, 12, 773. [Google Scholar] [CrossRef]
- Krisnawan, V.E.; Stanley, J.A.; Schwarz, J.K.; DeNardo, D.G. Tumor Microenvironment as a Regulator of Radiation Therapy: New Insights into Stromal-Mediated Radioresistance. Cancers 2020, 12, 2916. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Mun, S.H.; Park, P.S.U.; Park-Min, K.H. The M-CSF receptor in osteoclasts and beyond. Exp. Mol. Med. 2020, 52, 1239–1254. [Google Scholar] [CrossRef]
- Autio, K.A.; Klebanoff, C.A.; Schaer, D.; Kauh, J.S.W.; Slovin, S.F.; Adamow, M.; Blinder, V.S.; Brahmachary, M.; Carlsen, M.; Comen, E.; et al. Immunomodulatory Activity of a Colony-stimulating Factor-1 Receptor Inhibitor in Patients with Advanced Refractory Breast or Prostate Cancer: A Phase I Study. Clin. Cancer Res. 2020, 26, 5609–5620. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Lai, Y.S.; Hsuuw, Y.D.; Chang, K.T. Withholding of M-CSF Supplement Reprograms Macrophages to M2-Like via Endogenous CSF-1 Activation. Int. J. Mol. Sci. 2021, 22, 3532. [Google Scholar] [CrossRef] [PubMed]
- Menke, J.; Kriegsmann, J.; Schimanski, C.C.; Schwartz, M.M.; Schwarting, A.; Kelley, V.R. Autocrine CSF-1 and CSF-1 receptor coexpression promotes renal cell carcinoma growth. Cancer Res. 2012, 72, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.H.; László, C.F.; Greco, S.; Chambers, S.K. Regulation of colony stimulating factor-1 expression and ovarian cancer cell behavior in vitro by miR-128 and miR-152. Mol. Cancer 2012, 11, 58. [Google Scholar] [CrossRef]
- Shi, X.; Kaller, M.; Rokavec, M.; Kirchner, T.; Horst, D.; Hermeking, H. Characterization of a p53/miR-34a/CSF1R/STAT3 Feedback Loop in Colorectal Cancer. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 391–418. [Google Scholar] [CrossRef]
- Lee, Y.H.; Lee, M.M.; De Silva, D.M.; Roy, A.; Wright, C.E.; Wong, T.K.; Costello, R.; Olaku, O.; Grubb, R.L., 3rd; Agarwal, P.K.; et al. Autocrine signaling by receptor tyrosine kinases in urothelial carcinoma of the bladder. PLoS ONE 2021, 16, e0241766. [Google Scholar] [CrossRef]
- Cioce, M.; Canino, C.; Goparaju, C.; Yang, H.; Carbone, M.; Pass, H.I. Autocrine CSF-1R signaling drives mesothelioma chemoresistance via AKT activation. Cell Death Dis. 2014, 5, e1167. [Google Scholar] [CrossRef]
- Giricz, O.; Mo, Y.; Dahlman, K.B.; Cotto-Rios, X.M.; Vardabasso, C.; Nguyen, H.; Matusow, B.; Bartenstein, M.; Polishchuk, V.; Johnson, D.B.; et al. The RUNX1/IL-34/CSF-1R axis is an autocrinally regulated modulator of resistance to BRAF-V600E inhibition in melanoma. JCI Insight 2018, 3, e120422. [Google Scholar] [CrossRef]
- Kluger, H.M.; Kluger, Y.; Gilmore-Hebert, M.; DiVito, K.; Chang, J.T.; Rodov, S.; Mironenko, O.; Kacinski, B.M.; Perkins, A.S.; Sapi, E. cDNA microarray analysis of invasive and tumorigenic phenotypes in a breast cancer model. Lab. Investig. 2004, 84, 320–331. [Google Scholar] [CrossRef]
- Knowlton, M.L.; Selfors, L.M.; Wrobel, C.N.; Gu, T.L.; Ballif, B.A.; Gygi, S.P.; Polakiewicz, R.; Brugge, J.S. Profiling Y561-dependent and -independent substrates of CSF-1R in epithelial cells. PLoS ONE 2010, 5, e13587. [Google Scholar] [CrossRef]
- Morandi, A.; Barbetti, V.; Riverso, M.; Dello Sbarba, P.; Rovida, E. The colony-stimulating factor-1 (CSF-1) receptor sustains ERK1/2 activation and proliferation in breast cancer cell lines. PLoS ONE 2011, 6, e27450. [Google Scholar] [CrossRef]
- Zwaenepoel, O.; Tzenaki, N.; Vergetaki, A.; Makrigiannakis, A.; Vanhaesebroeck, B.; Papakonstanti, E.A. Functional CSF-1 receptors are located at the nuclear envelope and activated via the p110δ isoform of PI 3-kinase. FASEB J. 2012, 26, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Barbetti, V.; Morandi, A.; Tusa, I.; Digiacomo, G.; Riverso, M.; Marzi, I.; Cipolleschi, M.G.; Bessi, S.; Giannini, A.; Di Leo, A.; et al. Chromatin-associated CSF-1R binds to the promoter of proliferation-related genes in breast cancer cells. Oncogene 2014, 33, 4359–4364. [Google Scholar] [CrossRef] [PubMed]
- Kai, K.; Iwamoto, T.; Zhang, D.; Shen, L.; Takahashi, Y.; Rao, A.; Thompson, A.; Sen, S.; Ueno, N.T. CSF-1/CSF-1R axis is associated with epithelial/mesenchymal hybrid phenotype in epithelial-like inflammatory breast cancer. Sci. Rep. 2018, 8, 9427. [Google Scholar] [CrossRef]
- Patsialou, A.; Wang, Y.; Pignatelli, J.; Chen, X.; Entenberg, D.; Oktay, M.; Condeelis, J.S. Autocrine CSF1R signaling mediates switching between invasion and proliferation downstream of TGFβ in claudin-low breast tumor cells. Oncogene 2015, 34, 2721–2731. [Google Scholar] [CrossRef] [PubMed]
- Kluger, H.M.; Dolled-Filhart, M.; Rodov, S.; Kacinski, B.M.; Camp, R.L.; Rimm, D.L. Macrophage colony-stimulating factor-1 receptor expression is associated with poor outcome in breast cancer by large cohort tissue microarray analysis. Clin. Cancer Res. 2004, 10, 173–177. [Google Scholar] [CrossRef]
- Achkova, D.; Maher, J. Role of the colony-stimulating factor (CSF)/CSF-1 receptor axis in cancer. Biochem. Soc. Trans. 2016, 44, 333–341. [Google Scholar] [CrossRef]
- Richardsen, E.; Uglehus, R.D.; Due, J.; Busch, C.; Busund, L.T. The prognostic impact of M-CSF, CSF-1 receptor, CD68 and CD3 in prostatic carcinoma. Histopathology 2008, 53, 30–38. [Google Scholar] [CrossRef]
- Gobert Gosse, S.; Bourgin, C.; Liu, W.Q.; Garbay, C.; Mouchiroud, G. M-CSF stimulated differentiation requires persistent MEK activity and MAPK phosphorylation independent of Grb2-Sos association and phosphatidylinositol 3-kinase activity. Cell. Signal. 2005, 17, 1352–1362. [Google Scholar] [CrossRef]
- Filderman, A.E.; Bruckner, A.; Kacinski, B.M.; Deng, N.; Remold, H.G. Macrophage colony-stimulating factor (CSF-1) enhances invasiveness in CSF-1 receptor-positive carcinoma cell lines. Cancer Res. 1992, 52, 3661–6366. [Google Scholar]
- Dwyer, A.R.; Greenland, E.L.; Pixley, F.J. Promotion of Tumor Invasion by Tumor-Associated Macrophages: The Role of CSF-1-Activated Phosphatidylinositol 3 Kinase and Src Family Kinase Motility Signaling. Cancers 2017, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Linde, N.; Casanova-Acebes, M.; Sosa, M.S.; Mortha, A.; Rahman, A.; Farias, E.; Harper, K.; Tardio, E.; Reyes Torres, I.; Jones, J.; et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat. Commun. 2018, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.; Sahai, E.; Wyckoff, J.B.; Cammer, M.; Cox, D.; Pixley, F.J.; Stanley, E.R.; Segall, J.E.; Condeelis, J.S. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005, 65, 5278–5283. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.J.; Zhang, B.; Jia, D.; Zhang, L.; Wei, X.; Zhou, Z.; Liu, D.; Huynh, K.T.; Zhang, K.; Zhang, Y.; et al. Elevated expression of the colony-stimulating factor 1 (CSF1) induces prostatic intraepithelial neoplasia dependent of epithelial-Gp130. Oncogene 2022, 41, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Castello, L.M.; Raineri, D.; Salmi, L.; Clemente, N.; Vaschetto, R.; Quaglia, M.; Garzaro, M.; Gentilli, S.; Navalesi, P.; Cantaluppi, V.; et al. Osteopontin at the Crossroads of Inflammation and Tumor Progression. Mediators Inflamm. 2017, 2017, 4049098. [Google Scholar] [CrossRef]
- Moorman, H.R.; Poschel, D.; Klement, J.D.; Lu, C.; Redd, P.S.; Liu, K. Osteopontin: A Key Regulator of Tumor Progression and Immunomodulation. Cancers 2020, 12, 3379. [Google Scholar] [CrossRef] [PubMed]
- Marcondes, M.C.; Poling, M.; Watry, D.D.; Hall, D.; Fox, H.S. In vivo osteopontin-induced macrophage accumulation is dependent on CD44 expression. Cell. Immunol. 2008, 254, 56–62. [Google Scholar] [CrossRef]
- Kahles, F.; Findeisen, H.M.; Bruemmer, D. Osteopontin: A novel regulator at the cross roads of inflammation, obesity and diabetes. Mol. Metab. 2014, 3, 384–393. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, J.; Xu, D.; Gao, X.M.; Zhang, Z.; Hsu, J.L.; Li, C.W.; Lim, S.O.; Sheng, Y.Y.; Zhang, Y.; et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut 2019, 68, 1653–1666. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, Q.; Alam, A.; Cui, J.; Suen, K.C.; Soo, A.P.; Eguchi, S.; Gu, J.; Ma, D. The role of osteopontin in the progression of solid organ tumour. Cell Death Dis. 2018, 9, 356. [Google Scholar] [CrossRef]
- Khodavirdi, A.C.; Song, Z.; Yang, S.; Zhong, C.; Wang, S.; Wu, H.; Pritchard, C.; Nelson, P.S.; Roy-Burman, P. Increased expression of osteopontin contributes to the progression of prostate cancer. Cancer Res. 2006, 66, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Xie, R.; Zhang, Z.; Liu, Q.; Wu, S.; Cui, Y. Identification of SPP1 as an Extracellular Matrix Signature for Metastatic Castration-Resistant Prostate Cancer. Front. Oncol. 2019, 9, 924. [Google Scholar] [CrossRef] [PubMed]
- Messex, J.K.; Byrd, C.J.; Thomas, M.U.; Liou, G.Y. Macrophages Cytokine Spp1 Increases Growth of Prostate Intraepithelial Neoplasia to Promote Prostate Tumor Progression. Int. J. Mol. Sci. 2022, 23, 4247. [Google Scholar] [CrossRef] [PubMed]
- Green, J.E.; Shibata, M.A.; Yoshidome, K.; Liu, M.L.; Jorcyk, C.; Anver, M.R.; Wigginton, J.; Wiltrout, R.; Shibata, E.; Kaczmarczyk, S.; et al. The C3(1)/SV40 T-antigen transgenic mouse model of mammary cancer: Ductal epithelial cell targeting with multistage progression to carcinoma. Oncogene 2000, 19, 1020–1027. [Google Scholar] [CrossRef]
- Maroulakou, I.G.; Anver, M.; Garrett, L.; Green, J.E. Prostate and mammary adenocarcinoma in transgenic mice carrying a rat C3(1) simian virus 40 large tumor antigen fusion gene. Proc. Natl. Acad. Sci. USA 1994, 91, 11236–11240. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mougel, A.; Adriaenssens, E.; Guyot, B.; Tian, L.; Gobert, S.; Chassat, T.; Persoons, P.; Hannebique, D.; Bauderlique-Le Roy, H.; Vicogne, J.; et al. Macrophage-Colony-Stimulating Factor Receptor Enhances Prostate Cancer Cell Growth and Aggressiveness In Vitro and In Vivo and Increases Osteopontin Expression. Int. J. Mol. Sci. 2022, 23, 16028. https://doi.org/10.3390/ijms232416028
Mougel A, Adriaenssens E, Guyot B, Tian L, Gobert S, Chassat T, Persoons P, Hannebique D, Bauderlique-Le Roy H, Vicogne J, et al. Macrophage-Colony-Stimulating Factor Receptor Enhances Prostate Cancer Cell Growth and Aggressiveness In Vitro and In Vivo and Increases Osteopontin Expression. International Journal of Molecular Sciences. 2022; 23(24):16028. https://doi.org/10.3390/ijms232416028
Chicago/Turabian StyleMougel, Alexandra, Eric Adriaenssens, Boris Guyot, Lu Tian, Stéphanie Gobert, Thierry Chassat, Philippe Persoons, David Hannebique, Hélène Bauderlique-Le Roy, Jérôme Vicogne, and et al. 2022. "Macrophage-Colony-Stimulating Factor Receptor Enhances Prostate Cancer Cell Growth and Aggressiveness In Vitro and In Vivo and Increases Osteopontin Expression" International Journal of Molecular Sciences 23, no. 24: 16028. https://doi.org/10.3390/ijms232416028
APA StyleMougel, A., Adriaenssens, E., Guyot, B., Tian, L., Gobert, S., Chassat, T., Persoons, P., Hannebique, D., Bauderlique-Le Roy, H., Vicogne, J., Le Bourhis, X., & Bourette, R. P. (2022). Macrophage-Colony-Stimulating Factor Receptor Enhances Prostate Cancer Cell Growth and Aggressiveness In Vitro and In Vivo and Increases Osteopontin Expression. International Journal of Molecular Sciences, 23(24), 16028. https://doi.org/10.3390/ijms232416028