
The Impact of Indoles Activating the Aryl Hydrocarbon Receptor on Androgen Receptor Activity in the 22Rv1 Prostate Cancer Cell Line


Abstract
1. Introduction
2. Results
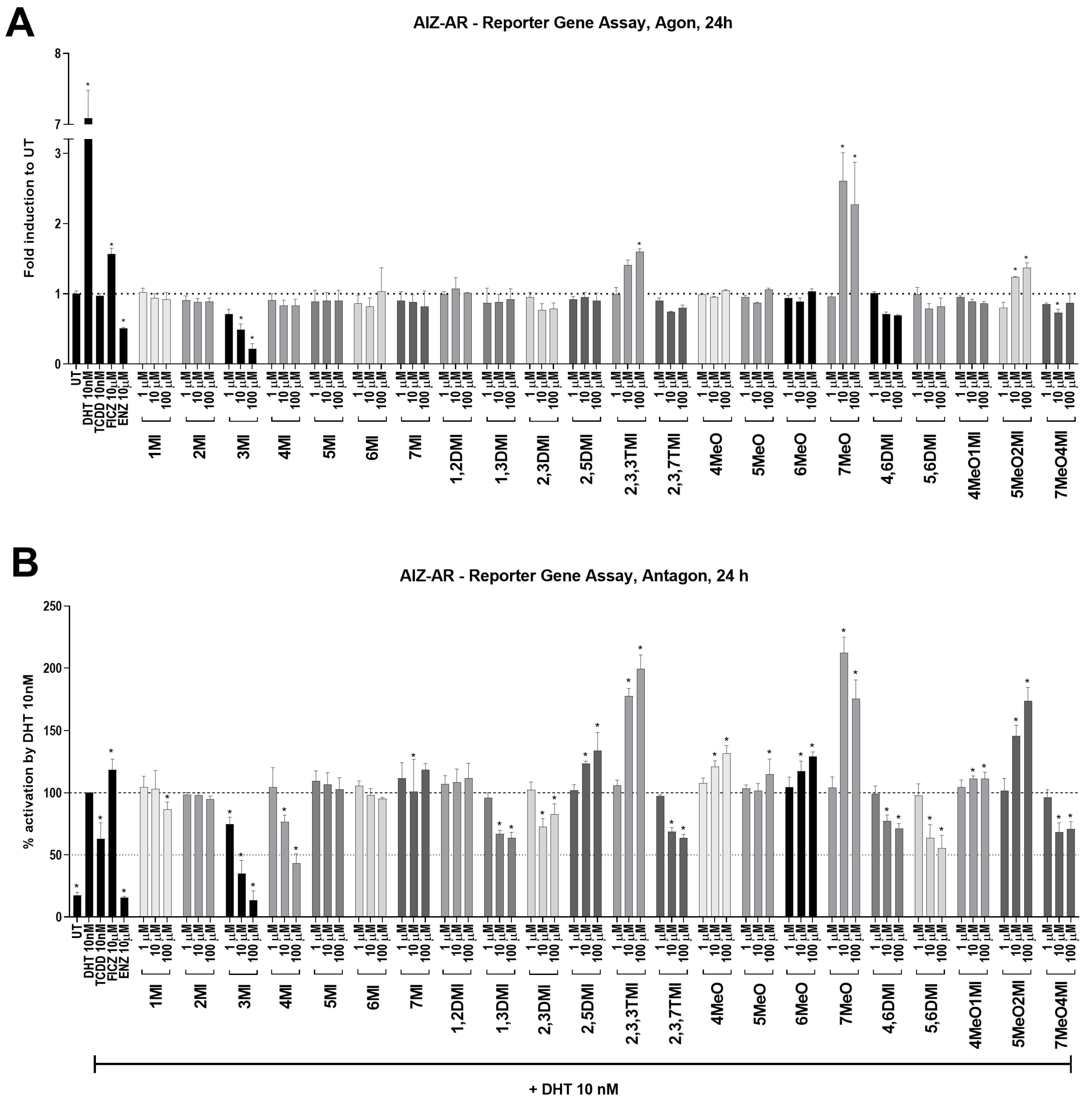
2.1. The Effects of Indoles on AhR and AR Transcription Activity
2.2. Effects of Selected Indoles on the Target Gene Expression
2.3. Binding of AR to the KLK3 Promoter after Treatment with Selected Indoles
2.4. Effects of Selected Indoles on AR-fl and AR-v7 Protein Levels
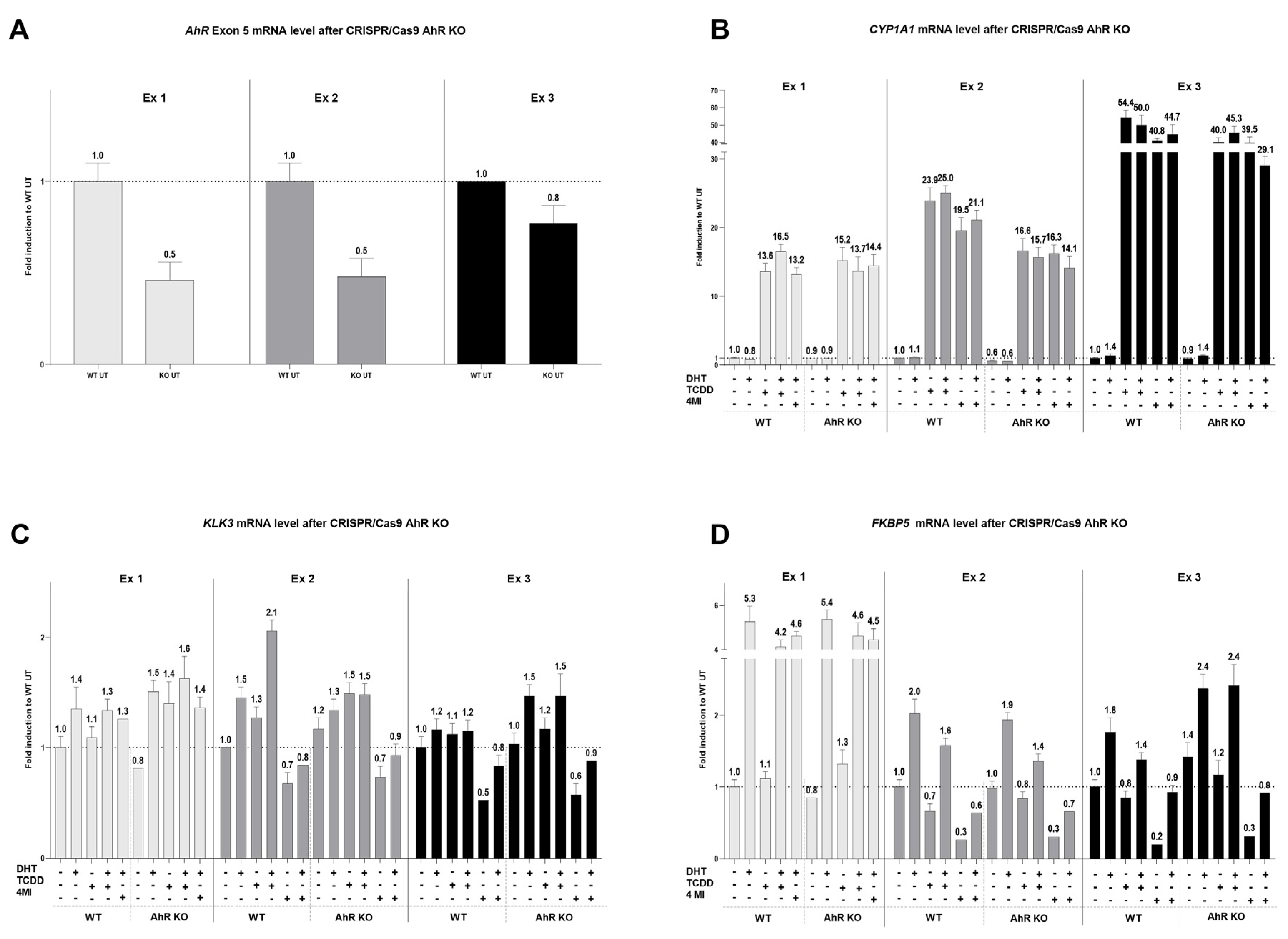
2.5. CRISPR/Cas9 AhR Knockout
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Compounds and Reagents
4.3. Reporter Gene Assay (RGA)
4.4. Quantitative Reverse Transcriptase PCR (RT-qPCR)
4.5. Western Blot
4.6. Chromatin Immunoprecipitation (ChIP)
4.7. CRISPR/Cas9 AhR Knock-Out
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dunn, M.W.; Kazer, M.W. Prostate cancer overview. Semin. Oncol. Nurs. 2011, 27, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef]
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of Intratumoral Androgens in Metastatic Prostate Cancer: A Mechanism for Castration-Resistant Tumor Growth. Cancer Res. 2008, 68, 4447–4454. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef]
- Zhang, T.; Karsh, L.I.; Nissenblatt, M.J.; Canfield, S.E. Androgen Receptor Splice Variant, AR-V7, as a Biomarker of Resistance to Androgen Axis-Targeted Therapies in Advanced Prostate Cancer. Clin. Genitourin. Cancer 2020, 18, 1–10. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Kenji, A. Androgens and Androgen Receptor: Mechanisms, Functions and Clinical Applications, 1st ed.; Springer: Boston, MA, USA, 2002; ISBN 978-1-4615-1161-8. [Google Scholar]
- Bruchovsky, N.; Wilson, J.D. The conversion of testosterone to 5-alpha-androstan-17-beta-ol-3-one by rat prostate in vivo and in vitro. J. Biol. Chem. 1968, 243, 2012–2021. [Google Scholar] [CrossRef]
- Sharifi, N.; Auchus, R.J. Steroid biosynthesis and prostate cancer. Steroids 2012, 77, 719–726. [Google Scholar] [CrossRef]
- Ghotbaddini, M.; Moultrie, V.; Powell, J.B. Constitutive Aryl Hydrocarbon Receptor Signaling in Prostate Cancer Progression. J. Cancer Treat. Diagn 2018, 2, 11–16. [Google Scholar] [CrossRef]
- Schellhammer, P.F.; Sharifi, R.; Block, N.L.; Soloway, M.S.; Venner, P.M.; Patterson, A.L.; Sarosdy, M.F.; Vogelzang, N.J.; Chen, Y.; Kolvenbag, G.J. A controlled trial of bicalutamide versus flutamide, each in combination with luteinizing hormone-releasing hormone analogue therapy, in patients with advanced prostate carcinoma. Analysis of time to progression. CASODEX Combination Study Group. Cancer 1996, 78, 2164–2169. [Google Scholar] [CrossRef]
- Poellinger, L. Mechanistic aspects--the dioxin (aryl hydrocarbon) receptor. Food Addit. Contam. 2000, 17, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Mimura, J.; Fujii-Kuriyama, Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim. Biophys. Acta 2003, 1619, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Baba, A.; Fujii-Kuriyama, Y.; Kato, S. Intrinsic AhR function underlies cross-talk of dioxins with sex hormone signalings. Biochem. Biophys. Res. Commun. 2008, 370, 541–546. [Google Scholar] [CrossRef] [PubMed]
- De Anna, J.S.; Darraz, L.A.; Painefilu, J.C.; Carcamo, J.G.; Moura-Alves, P.; Venturino, A.; Luquet, C.M. The insecticide chlorpyrifos modifies the expression of genes involved in the PXR and AhR pathways in the rainbow trout, Oncorhynchus mykiss. Pestic. Biochem. Physiol. 2021, 178, 104920. [Google Scholar] [CrossRef] [PubMed]
- Kizu, R.; Okamura, K.; Toriba, A.; Kakishima, H.; Mizokami, A.; Burnstein, K.L.; Hayakawa, K. A role of aryl hydrocarbon receptor in the antiandrogenic effects of polycyclic aromatic hydrocarbons in LNCaP human prostate carcinoma cells. Arch. Toxicol. 2003, 77, 335–343. [Google Scholar] [CrossRef]
- Fritz, W.A.; Lin, T.M.; Cardiff, R.D.; Peterson, R.E. The aryl hydrocarbon receptor inhibits prostate carcinogenesis in TRAMP mice. Carcinogenesis 2007, 28, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Baba, A.; Takada, I.; Okada, M.; Iwasaki, K.; Miki, H.; Takahashi, S.; Kouzmenko, A.; Nohara, K.; Chiba, T.; et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 2007, 446, 562–566. [Google Scholar] [CrossRef]
- Ghotbaddini, M.; Powell, J.B. The AhR Ligand, TCDD, Regulates Androgen Receptor Activity Differently in Androgen-Sensitive versus Castration-Resistant Human Prostate Cancer Cells. Int. J. Environ. Res. Public Health 2015, 12, 7506–7518. [Google Scholar] [CrossRef]
- Sanada, N.; Gotoh, Y.; Shimazawa, R.; Klinge, C.M.; Kizu, R. Repression of activated aryl hydrocarbon receptor-induced transcriptional activation by 5alpha-dihydrotestosterone in human prostate cancer LNCaP and human breast cancer T47D cells. J. Pharmacol. Sci. 2009, 109, 380–387. [Google Scholar] [CrossRef]
- Arabnezhad, M.R.; Montazeri-Najafabady, N.; Chatrabnous, N.; Ghafarian Bahreman, A.; Mohammadi-Bardbori, A. Anti-androgenic effect of 6-formylindolo[3,2-b]carbazole (FICZ) in LNCaP cells is mediated by the aryl hydrocarbon-androgen receptors cross-talk. Steroids 2020, 153, 108508. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.; Qin, C.; Smith, R., 3rd; Safe, S. Aryl hydrocarbon receptor-mediated inhibition of LNCaP prostate cancer cell growth and hormone-induced transactivation. J. Steroid Biochem. Mol. Biol. 2004, 88, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Indran, I.R.; Zhang, Z.W.; Tan, M.H.; Li, Y.; Lim, Z.L.; Hua, R.; Yang, C.; Soon, F.F.; Li, J.; et al. A novel prostate cancer therapeutic strategy using icaritin-activated arylhydrocarbon-receptor to co-target androgen receptor and its splice variants. Carcinogenesis 2015, 36, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cai, A.; Zheng, H.; Huang, H.; Sun, R.; Cui, X.; Ye, W.; Yao, Q.; Chen, R.; Kou, L. Carbidopa suppresses prostate cancer via aryl hydrocarbon receptor-mediated ubiquitination and degradation of androgen receptor. Oncogenesis 2020, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Wang, Z.; Shi, J. Chapter Seven—Pharmacological effects of icariin. In Advances in Pharmacology; Du, G., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 87, pp. 179–203. ISBN 0128201851. [Google Scholar]
- Szabo, R.; Racz, C.P.; Dulf, F.V. Bioavailability Improvement Strategies for Icariin and Its Derivates: A Review. Int. J. Mol. Sci. 2022, 23, 7519. [Google Scholar] [CrossRef]
- Seeberger, L.C.; Hauser, R.A. Levodopa/carbidopa/entacapone in Parkinson’s disease. Expert Rev. Neurother. 2009, 9, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Stepankova, M.; Bartonkova, I.; Jiskrova, E.; Vrzal, R.; Mani, S.; Kortagere, S.; Dvorak, Z. Methylindoles and Methoxyindoles are Agonists and Antagonists of Human Aryl Hydrocarbon Receptor. Mol. Pharmacol. 2018, 93, 631–644. [Google Scholar] [CrossRef]
- Bartonkova, I.; Novotna, A.; Dvorak, Z. Novel stably transfected human reporter cell line AIZ-AR as a tool for an assessment of human androgen receptor transcriptional activity. PLoS One 2015, 10, e0121316. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Ku, J.Y.; Ha, J.M.; Bae, S.S.; Lee, J.Z.; Kim, C.S.; Ha, H.K. Transcript Levels of Androgen Receptor Variant 7 and Ubiquitin-Conjugating Enzyme 2C in Hormone Sensitive Prostate Cancer and Castration-Resistant Prostate Cancer. Prostate 2017, 77, 60–71. [Google Scholar] [CrossRef]
- ATCC The Global Bioresource Center—LNCaP. Available online: https://www.atcc.org/products/crl-1740 (accessed on 20 June 2022).
- Wu, H.C.; Hsieh, J.T.; Gleave, M.E.; Brown, N.M.; Pathak, S.; Chung, L.W. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: Role of bone stromal cells. Int. J. Cancer 1994, 57, 406–412. [Google Scholar] [CrossRef]
- Tran, C.; Richmond, O.; Aaron, L.; Powell, J.B. Inhibition of constitutive aryl hydrocarbon receptor (AhR) signaling attenuates androgen independent signaling and growth in (C4-2) prostate cancer cells. Biochem. Pharmacol. 2013, 85, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Suyama, Y.; Hirayama, C. Serum indole and skatole in patients with various liver diseases. Clin. Chim. Acta 1988, 176, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nichols, W.K.; Mehta, R.; Skordos, K.; Macé, K.; Pfeifer, A.M.; Carr, B.A.; Minko, T.; Burchiel, S.W.; Yost, G.S. 3-methylindole-induced toxicity to human bronchial epithelial cell lines. Toxicol. Sci. 2003, 71, 229–236. [Google Scholar] [CrossRef]
- Novotna, A.; Pavek, P.; Dvorak, Z. Novel stably transfected gene reporter human hepatoma cell line for assessment of aryl hydrocarbon receptor transcriptional activity: Construction and characterization. Environ Sci. Technol. 2011, 45, 10133–10139. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | AhR Activation (24 h) | AR Inactivation (24 h) | |
|---|---|---|---|
| 10 µM | 100 µM | 100 µM | |
| 3MI | 2.6 fold | 1.2 fold | decrease to 13.5% |
| 4MI | 4.5 fold | 6.3 fold | decrease to 43.1% |
| 1,3DMI | 3.0 fold | 2.7 fold | decrease to 63.5% |
| 2,3DMI | 3.9 fold | 3.1 fold | decrease to 82.8% |
| 2,3,7TMI | 3.5 fold | 3.5 fold | decrease to 63.5% |
| 4,6DMI | 2.6 fold | 3.5 fold | decrease to 70.9% |
| 5,6DMI | 1.9 fold | 2.6 fold | decrease to 55.5% |
| 7MeO4MI | 4.5 fold | 4.7 fold | decrease to 70.8% |
| AhR | AR | ||
|---|---|---|---|
| Tested Compound | Model | ||
| Icaritin | 22Rv1 | ↑ CYP1A1 mRNA [24]; ↑ AhR protein level [24] | ↓ protein levels [24]; ↓ UBE2C, KLK3 mRNA [24]; ↓ recruitment to AR-target genes promoters [24] |
| LNCaP | ↑ CYP1A1 mRNA [24]; ↑ AhR protein level [24] | ↓ protein levels [24]; ↓ UBE2C, KLK3 mRNA [24]; ↓ recruitment to AR-target genes promoters [24] | |
| C4-2 | ↓ protein levels [24]; ↓ UBE2C, KLK3 mRNA [24] | ||
| TCDD | 22Rv1 | ↑ CYP1A1 mRNA | |
| LNCaP | ↑ CYP1A1, CYP1B1 mRNA [20]; ↓ AhR protein level [20] | ↓ AR protein level [20]; ↑ KLK3 mRNA [20]; ↑ AR phosphorylation [20] | |
| C4-2 | ↑ CYP1A1, CYP1B1 mRNA [20]; ↓ AhR protein level [20] | -AR protein level [20] | |
| Carbidopa | LNCaP | ↑ CYP1A1 mRNA [25]; ↑ AhR protein level [25] | ↓ AR protein level [25]; ↓ PSA mRNA [25] |
| 3 MI * | 22Rv1(this study) | ↑ AhR activity (RGA); ↑ CYP1A1 mRNA | ↓ DHT-induced AR activity (RGA); ↓ AR-target genes mRNA; ↓ AR recruitment to KLK3 promoter * ↓ AR protein level (dose-dependently) |
| 4 MI * | |||
| 1,3 DMI | |||
| 2,3 DMI | |||
| 2,3,7 TMI * | |||
| 4,6 DMI | |||
| 5,6 DMI | |||
| 7MeO4MI * |
| Compound | Abbreviation | Purity |
|---|---|---|
| 1-methylindole | 1MI | ≥97% |
| 2-methylindole | 2MI | 98% |
| 3-methylindole | 3MI | 98% |
| 4-methylindole | 4MI | 98% |
| 5-methylindole | 5MI | 99% |
| 6-methylindole | 6MI | 97% |
| 7-methylindole | 7MI | 97% |
| 1,2-methylindole | 1,2DMI | 99% |
| 1,3-methylindole | 1,3DMI | 95% |
| 2,3-methylindole | 2,3DMI | ≥97% |
| 2,5-methylindole | 2,5DMI | 97% |
| 2,3,3-trimethylindolenine | 2,3,3TMI | 98% |
| 2,3,7-trimethylindole | 2,3,7TMI | ≥97% |
| 4-methoxyindole | 4MeO | 99% |
| 5-methoxyindole | 5MeO | 99% |
| 6-methoxyindole | 6MeO | 98% |
| 7-methoxyindole | 7MeO | ≥97% |
| 4,6-dimethoxyindole | 4,6DMI | ≥98% |
| 5,6-dimethoxyindole | 5,6DMI | 99% |
| 4-methoxy-1-methylindole | 4MeO1MI | 98% |
| 5-methoxy-2-methylindole | 5MeO2MI | 99% |
| 7-methoxy-4-methylindole | 7MeO4MI | 95% |
| Target Gene | Sequence |
|---|---|
| AhR exon 5 | f: 5′ TGAATTTCAGCGTCAGCTACA 3′ |
| r: 5′ AACAGACTACTGTCTGGGGGA 3′ | |
| AhRR | f: 5′GAGATGAAAATGAGGAGCGC 3′ |
| r: 5′TTTTACTTTTGCATCCGCGG 3′ | |
| p: 5′[6FAM]AAACCCAGAGCAGACACCGCAGCCA[OQA] 3′ | |
| f: 5′TGTGTCAAAAGCGAAATGGG 3′ | |
| AR-fl | r: 5′TTCATCTCCACAGATCAGGC 3′ |
| p: 5′[6FAM]TGCGTTTGGAGACTGCCAGGGACCA[OQA] 3′ | |
| AR-v7 | f: 5′GAAATGTTAGAAGCAGGGATGACT 3′ |
| r: 5′GGTCATTTTGAGATGCTTGCAA 3′ | |
| f: 5′GGAAGTGTATCGGTGAGACC 3′ | |
| CYP1A1 | r: 5′CATAGATGGGGGTCATGTCC 3′ |
| p: 5′[6FAM]GCAACGGGTGGAATTCAGCGTGCCA[OQA] 3′ | |
| f: 5′TCCAAGACTCAGATGATGCC 3′ | |
| FKBP5 | r: 5′GGCACCCTGTAGTTATTTGC 3′ |
| p: 5′[6FAM]AAGTGTGTGTGGGGAGGGGAAGGGT[OQA] 3′ | |
| f: 5′GAAGGAAATGAATGGGCAGC 3′ | |
| GAPDH | r: 5′TCTAGGAAAAGCATCACCCG 3′ |
| p: 5′[6FAM]ACTAACCCTGCGCTCCTGCCTCGAT[OQA] 3′ | |
| f: 5′ACTGCATCAGGAACAAAAGC 3′ | |
| KLK3 | r: 5′GGAGGCTCATATCGTAGAGC 3′ |
| p: 5′[6FAM]TGGGTCGGCACAGCCTGTTTCATCC[OQA] 3′ | |
| f: 5′CCACAGTGAAGTTCCTCACG 3′ | |
| UBE2C | r: 5′GTTGGGTTCTCCTAGAAGGC 3′ |
| p: 5′[6FAM]ACCCCAACGTGGACACCCAGGGTAA[OQA] 3′ |
| Detection Format | Mono Color Hydrolysis Probe/UPL Probe | ||
|---|---|---|---|
| Reaction Volume | 10 µL | ||
| Program | Temperature | Time | Number of Cycles |
| Pre-incubation | 95 °C | 20 s | 1 |
| Amplification | 95 °C | 5 s | 45 |
| 58 °C | 30 s | ||
| Cooling | 40 °C | 30 s | 1 |
| Antibody | Type | Manufacturer | Dilution |
|---|---|---|---|
| Primary antibody | |||
| β-actin | mouse monoclonal | Santa cruz Biotechnology | 1: 2000 |
| AR | mouse monoclonal | Santa cruz Biotechnology | 1: 500 |
| Secondary antibody | |||
| Anti-mouse | IgG, HRP-linked | Santa cruz Biotechnology | 1: 2000 |
| Anti-rabbit | IgG, HRP-linked | Santa cruz Biotechnology | 1: 2000 |
| Detection Format | SYBR Green I/HRM Dye | ||
|---|---|---|---|
| Reaction Volume | 10 µL | ||
| Program | Temperature | Time | Number of Cycles |
| Pre-incubation | 95 °C | 10 min | 1 |
| Amplification | 95 °C | 15 s | 40 |
| 60 °C | 1 min | ||
| Melting curve | 95 °C | 5 s | 1 |
| 65 °C | 1 min | ||
| 97 °C | - | ||
| Cooling | 40 °C | 10 s | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zgarbová, E.; Vrzal, R. The Impact of Indoles Activating the Aryl Hydrocarbon Receptor on Androgen Receptor Activity in the 22Rv1 Prostate Cancer Cell Line. Int. J. Mol. Sci. 2023, 24, 502. https://doi.org/10.3390/ijms24010502
Zgarbová E, Vrzal R. The Impact of Indoles Activating the Aryl Hydrocarbon Receptor on Androgen Receptor Activity in the 22Rv1 Prostate Cancer Cell Line. International Journal of Molecular Sciences. 2023; 24(1):502. https://doi.org/10.3390/ijms24010502
Chicago/Turabian StyleZgarbová, Eliška, and Radim Vrzal. 2023. "The Impact of Indoles Activating the Aryl Hydrocarbon Receptor on Androgen Receptor Activity in the 22Rv1 Prostate Cancer Cell Line" International Journal of Molecular Sciences 24, no. 1: 502. https://doi.org/10.3390/ijms24010502
APA StyleZgarbová, E., & Vrzal, R. (2023). The Impact of Indoles Activating the Aryl Hydrocarbon Receptor on Androgen Receptor Activity in the 22Rv1 Prostate Cancer Cell Line. International Journal of Molecular Sciences, 24(1), 502. https://doi.org/10.3390/ijms24010502