
Rare Amyloid Precursor Protein Point Mutations Recapitulate Worldwide Migration and Admixture in Healthy Individuals: Implications for the Study of Neurodegeneration


Abstract
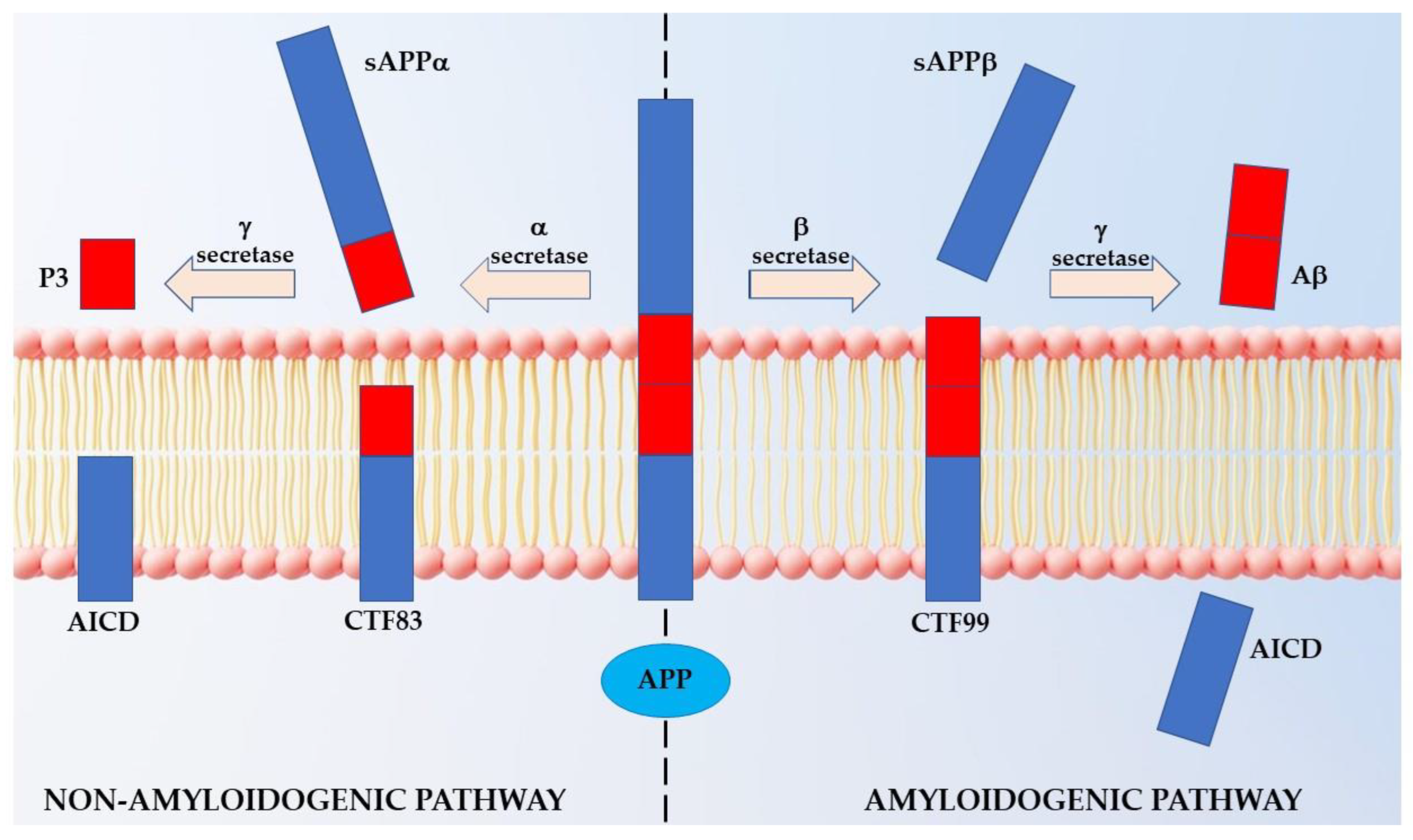
1. Introduction
2. Results
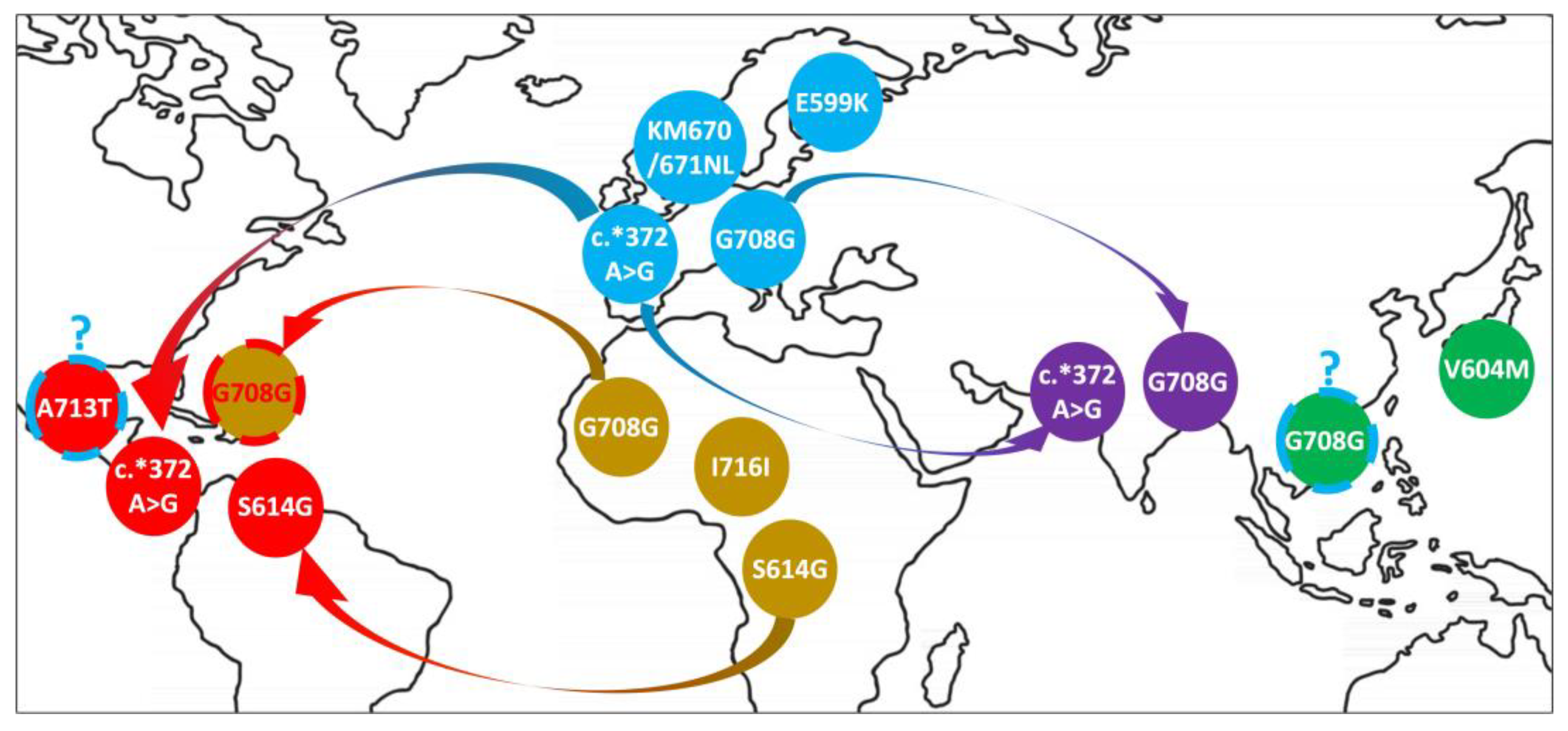
2.1. Distribution of APP Point Mutations
2.2. Segment Sharing across Individuals
3. Discussion
4. Materials and Methods
4.1. Population Data Recovery
4.2. APP Gene Point Mutations
4.3. Dataset Quality Control
4.4. Data Phasing and Haplotype Estimation
4.5. Segment Detection and Statistical Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kirkitadze, M.D.; Kowalska, A. Molecular mechanisms initiating amyloid beta-fibril formation in Alzheimer’s disease. Acta Biochim. Pol. 2005, 52, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Kojro, E.; Fahrenholz, F. The Non-Amyloidogenic Pathway: Structure and Function of α-Secretases. Alzheimer’s Dis. 2005, 38, 105–127. [Google Scholar] [CrossRef]
- Sanchez, M.I.G.L.; Van Wijngaarden, P.; Trounce, I.A. Amyloid precursor protein-mediated mitochondrial regulation and Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2018, 176, 3464–3474. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Hartley, D.M.; Cahill, C.M.; Lahiri, D.K.; Chattopadhyay, N.; Rogers, J.T. Interleukin-1α stimulates non-amyloidogenic pathway by α-secretase (ADAM-10 and ADAM-17) cleavage of APP in human astrocytic cells involving p38 MAP kinase. J. Neurosci. Res. 2006, 84, 106–118. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J. Targeting amyloid clearance in Alzheimer’s disease as a therapeutic strategy. Br. J. Pharmacol. 2019, 176, 3447–3463. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Tcw, J.; Goate, A.M. Genetics of β-Amyloid Precursor Protein in Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2016, 7, a024539. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.F.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Kero, M.; Paetau, A.; Polvikoski, T.; Tanskanen, M.; Sulkava, R.; Jansson, L.; Myllykangas, L.; Tienari, P.J. Amyloid precursor protein (APP) A673T mutation in the elderly Finnish population. Neurobiol. Aging 2013, 34, 1518.e1–1518.e3. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-W.; He, Y.-H.; Zhang, Y.-X.; Cai, W.-W.; Yang, L.-Q.; Xu, L.-Y.; Kong, Q.-P. Absence of A673T variant in APP gene indicates an alternative protective mechanism contributing to longevity in Chinese individuals. Neurobiol. Aging 2014, 35, 935.e11–935.e12. [Google Scholar] [CrossRef] [PubMed]
- Abondio, P.; Sazzini, M.; Garagnani, P.; Boattini, A.; Monti, D.; Franceschi, C.; Luiselli, D.; Giuliani, C. The Genetic Variability of APOE in Different Human Populations and Its Implications for Longevity. Genes 2019, 10, 222. [Google Scholar] [CrossRef]
- Adams, H.H.H.; Evans, T.E.; Terzikhan, N. The Uncovering Neurodegenerative Insights Through Ethnic Diversity consortium. Lancet Neurol. 2019, 18, 915. [Google Scholar] [CrossRef]
- Mills, M.C.; Rahal, C. A scientometric review of genome-wide association studies. Commun. Biol. 2019, 2, 9. [Google Scholar] [CrossRef]
- Sirugo, G.; Williams, S.M.; Tishkoff, S.A. The Missing Diversity in Human Genetic Studies. Cell 2019, 177, 26–31. [Google Scholar] [CrossRef]
- Raman, R.; Quiroz, Y.T.; Langford, O.; Choi, J.; Ritchie, M.; Baumgartner, M.; Rentz, D.; Aggarwal, N.T.; Aisen, P.; Sperling, R.; et al. Disparities by Race and Ethnicity Among Adults Recruited for a Preclinical Alzheimer Disease Trial. JAMA Netw. Open 2021, 4, e2114364. [Google Scholar] [CrossRef]
- Sassi, C.; Guerreiro, R.; Gibbs, R.; Ding, J.; Lupton, M.K.; Troakes, C.; Al-Sarraj, S.; Niblock, M.; Gallo, J.-M.; Adnan, J.; et al. Investigating the role of rare coding variability in Mendelian dementia genes (APP, PSEN1, PSEN2, GRN, MAPT, and PRNP) in late-onset Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2881.e1–2881.e6. [Google Scholar] [CrossRef]
- Schulte, E.C.; Fukumori, A.; Mollenhauer, B.; Hor, H.; Arzberger, T.; Perneczky, R.; Kurz, A.; Diehl-Schmid, J.; Hüll, M.; Lichtner, P.; et al. Rare variants in β-Amyloid precursor protein (APP) and Parkinson’s disease. Eur. J. Hum. Genet. 2015, 23, 1328–1333. [Google Scholar] [CrossRef]
- Nicolas, G.; Wallon, D.; Charbonnier, C.; Quenez, O.; Rousseau, S.; Richard, A.-C.; Rovelet-Lecrux, A.; Coutant, S.; Le Guennec, K.; Bacq, D.; et al. Screening of dementia genes by whole-exome sequencing in early-onset Alzheimer disease: Input and lessons. Eur. J. Hum. Genet. 2015, 24, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.; Pimenova, A.A.; Hayes, K.; Villa, J.A.; Rosene, M.J.; Jere, M.; Goate, A.M.; Karch, C.M. Systematic validation of variants of unknown significance in APP, PSEN1 and PSEN2. Neurobiol. Dis. 2020, 139, 104817. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Wallon, D.; Goupil, C.; Richard, A.-C.; Pottier, C.; Dorval, V.; Sarov-Rivière, M.; Riant, F.; Hervé, D.; Amouyel, P.; et al. Mutation in the 3’untranslated region of APP as a genetic determinant of cerebral amyloid angiopathy. Eur. J. Hum. Genet. 2015, 24, 92–98. [Google Scholar] [CrossRef]
- Abrahamson, M.; Gustafson, L.; Nilsson, K.; Brun, A.; Grubb, A. A novel mutation in the β-protein coding region of the amyloid ?-protein precursor (APP) gene. Qual. Life Res. 1992, 89, 580–582. [Google Scholar] [CrossRef]
- Lilius, L.; Lannfelt, L. No amyloid precursor protein 708 mutation in 50 Swedish Alzheimer’s disease patients. Qual. Life Res. 1994, 93, 227–228. [Google Scholar] [CrossRef]
- Jia, L.; Fu, Y.; Shen, L.; Zhang, H.; Zhu, M.; Qiu, Q.; Wang, Q.; Yan, X.; Kong, C.; Hao, J.; et al. PSEN1, PSEN2, and APP mutations in 404 Chinese pedigrees with familial Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 178–191. [Google Scholar] [CrossRef]
- Lee, J.H.; Kahn, A.; Cheng, R.; Reitz, C.; Vardarajan, B.; Lantigua, R.; Medrano, M.; Jiménez-Velázquez, I.Z.; Williamson, J.; Nagy, P.; et al. Disease-related mutations among Caribbean Hispanics with familial dementia. Mol. Genet. Genom. Med. 2014, 2, 430–437. [Google Scholar] [CrossRef]
- Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N–terminus of β–amyloid. Nat. Genet. 1992, 1, 345–347. [Google Scholar] [CrossRef]
- Zhou, B.; Lu, J.G.; Siddu, A.; Wernig, M.; Südhof, T.C. Synaptogenic effect of APP-Swedish mutation in familial Alzheimer’s disease. Sci. Transl. Med. 2022, 14, eabn9380. [Google Scholar] [CrossRef]
- Carter, D.; Desmarais, E.; Bellis, M.; Campion, D.; Clerget-Darpoux, F.; Brice, A.; Agid, Y.; Jaillard-Serradt, A.; Mallet, J. More missense in amyloid gene. Nat. Genet. 1992, 2, 255–256. [Google Scholar] [CrossRef]
- Lanoiselée, H.-M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.-C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.; Boada, M.; Rey, M.; Vidal, N.; Ferrer, I. Familial Alzheimer disease associated with A713T mutation in APP. Neurosci. Lett. 2004, 370, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Barber, I.S.; García-Cárdenas, J.M.; Sakdapanichkul, C.; Deacon, C.; Erazo, G.Z.; Guerreiro, R.; Bras, J.; Hernandez, D.; Singleton, A.; Guetta-Baranes, T.; et al. Screening exons 16 and 17 of the amyloid precursor protein gene in sporadic early-onset Alzheimer’s disease. Neurobiol. Aging 2015, 39, 220.e1–220.e7. [Google Scholar] [CrossRef] [PubMed]
- Suarez, M.C.F.; Brusco, I.; Damasso, C.; Olivar, N.; Morelli, L.; Russo, G. Heterozygous app a713t mutation carrier with inflammatoy amyloid angiopathy and family history of alzheimer´s disease. First case in Argentina. J. Neurol. Stroke 2019, 9, 86–89. [Google Scholar] [CrossRef]
- Bruno, F.; Laganà, V.; Di Lorenzo, R.; Bruni, A.C.; Maletta, R. Calabria as a Genetic Isolate: A Model for the Study of Neurodegenerative Diseases. Biomedicines 2022, 10, 2288. [Google Scholar] [CrossRef]
- Rossi, G.; Giaccone, G.; Maletta, R.; Morbin, M.; Capobianco, R.; Mangieri, M.; Giovagnoli, A.R.; Bizzi, A.; Tomaino, C.; Perri, M.; et al. A family with Alzheimer disease and strokes associated with A713T mutation of the APP gene. Neurology 2004, 63, 910–912. [Google Scholar] [CrossRef]
- Bernardi, L.; Geracitano, S.; Colao, R.; Puccio, G.; Gallo, M.; Anfossi, M.; Frangipane, F.; Curcio, S.A.; Mirabelli, M.; Tomaino, C.; et al. AβPP A713T Mutation in Late Onset Alzheimer’s Disease with Cerebrovascular Lesions. J. Alzheimer’s Dis. 2009, 17, 383–389. [Google Scholar] [CrossRef]
- Conidi, M.E.; Bernardi, L.; Puccio, G.; Smirne, N.; Muraca, M.G.; Curcio, S.A.; Colao, R.; Piscopo, P.; Gallo, M.; Anfossi, M.; et al. Homozygous carriers of APP A713T mutation in an autosomal dominant Alzheimer disease family. Neurology 2015, 84, 2266–2273. [Google Scholar] [CrossRef]
- Abondio, P.; Sarno, S.; Giuliani, C.; Laganà, V.; Maletta, R.; Bernardi, L.; Bruno, F.; Colao, R.; Puccio, G.; Frangipane, F.; et al. Amyloid Precursor Protein A713T Mutation in Calabrian Patients with Alzheimer’s Disease: A Population Genomics Approach to Estimate Inheritance from a Common Ancestor. Biomedicines 2021, 10, 20. [Google Scholar] [CrossRef]
- Smith, P.H.; Green, J.N. Modern Latin America, 9th ed.; Oxford University Press: New York, NY, USA, 2019; ISBN 978-0-19-067465-6. [Google Scholar]
- Global Goods and the Spanish Empire, 1492–1824: Circulation, Resistance and Diversity; Aram, B., Yun Casalilla, B., Eds.; Palgrave Macmillan: Basingstoke, UK, 2014; ISBN 978-1-137-32405-4. [Google Scholar]
- The Spanish Empire: A Historical Encyclopedia; Tarver Denova, H.M., Slape, E., Eds.; Empires of the world; ABC-CLIO: Santa Barbara, CA, USA, 2016; ISBN 978-1-61069-421-6. [Google Scholar]
- The Italian Diaspora: Migration across the Globe; Pozzetta, G.E., Ed.; Studies in Ethnic and immigration history; Multicultural History Society: Toronto, ON, Canada, 1992; ISBN 978-0-919045-59-0. [Google Scholar]
- Italian Mobilities, 1st ed.; Ben-Ghiat, R., Hom, S.M., Eds.; Routledge, Taylor & Francis Group: London, UK; New York, NY, USA, 2015; ISBN 978-1-138-77814-6. [Google Scholar]
- Galante, J.S. On the Other Shore: The Atlantic Worlds of Italians in South America during the Great War; University of Nebraska Press: Lincoln, NE, USA, 2022; ISBN 978-1-4962-0791-3. [Google Scholar]
- Van Giau, V.; Senanarong, V.; Bagyinszky, E.; Limwongse, C.; An, S.S.A.; Kim, S. Identification of a novel mutation in APP gene in a Thai subject with early-onset Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2018, 14, 3015–3023. [Google Scholar] [CrossRef]
- Van Giau, V.; Senanarong, V.; Bagyinszky, E.; An, S.S.A.; Kim, S. Analysis of 50 Neurodegenerative Genes in Clinically Diagnosed Early-Onset Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 1514. [Google Scholar] [CrossRef] [PubMed]
- Van Giau, V.; Bagyinszky, E.; Youn, Y.C.; An, S.S.A.; Kim, S. APP, PSEN1, and PSEN2 Mutations in Asian Patients with Early-Onset Alzheimer Disease. Int. J. Mol. Sci. 2019, 20, 4757. [Google Scholar] [CrossRef] [PubMed]
- Tagore, D.; Aghakhanian, F.; Naidu, R.; Phipps, M.E.; Basu, A. Insights into the demographic history of Asia from common ancestry and admixture in the genomic landscape of present-day Austroasiatic speakers. BMC Biol. 2021, 19, 61. [Google Scholar] [CrossRef]
- The East Asian Maritime World 1400–1800: Its Fabrics of Power and Dynamics of Exchanges; Schottenhammer, A., Ed.; East Asian Economic and Socio-Cultural Studies. East Asian Maritime History; Harrassowitz: Wiesbaden, Germany, 2007; ISBN 978-3-447-05474-4. [Google Scholar]
- Akamine, M. The Ryukyu Kingdom: Cornerstone of East Asia; Huey, R.N., Ed.; Paperback edition; University of Hawaii Press: Honolulu, HI, USA, 2019; ISBN 978-0-8248-5517-8. [Google Scholar]
- East Asia in the World: Twelve Events That Shaped the Modern International Order; Haggard, S., Kang, D.C., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2020; ISBN 978-1-108-80740-1. [Google Scholar]
- Winius, G.D. Studies on Portuguese Asia, 1495–1689; Variorum Collected Studies Series; Ashgate: Aldershot, UK; Burlington, MA, USA, 2001; ISBN 978-0-86078-872-0. [Google Scholar]
- Subrahmanyam, S. The Portuguese Empire in Asia, 1500–1700: A Political and Economic History, 2nd ed.; Wiley: Hoboken, NJ, USA, 2012; ISBN 978-1-118-27402-6. [Google Scholar]
- Lawson, P. The East India Company: A History; Studies in Modern History; Longman: London, UK; New York, NY, USA, 1993; ISBN 978-0-582-07386-9. [Google Scholar]
- PITMAN, F.W. Development of the British West Indies: 1700–1763; Routledge: Oxfordshire, UK, 2020; ISBN 978-0-367-14259-9. [Google Scholar]
- Collins, J.K. Tracing British West Indian Slavery Laws: A Comparative Analysis of Legal Transplants; Routledge Studies in Comparative Legal History; Routledge: Oxfordshire, UK; New York, NY, USA, 2021; ISBN 978-1-00-051567-1. [Google Scholar]
- Delle, J.A. The Colonial Caribbean: Landscapes of Power in Jamaica’s Plantation System; Cambridge University Press: New York, NY, USA, 2014; ISBN 978-1-139-02404-4. [Google Scholar]
- The Colonial Landscape of the British Caribbean; Leech, R., Leech, P., Eds.; The Society for Post-Medieval Archaeology Monograph; The Boydell Press: Woodbridge, CT, USA, 2021; ISBN 978-1-78327-565-6. [Google Scholar]
- Akinyemi, R.O.; Yaria, J.; Ojagbemi, A.; Guerchet, M.; Okubadejo, N.; Njamnshi, A.K.; Sarfo, F.S.; Akpalu, A.; Ogbole, G.; Ayantayo, T.; et al. Dementia in Africa: Current evidence, knowledge gaps, and future directions. Alzheimer’s Dement. 2021, 18, 790–809. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-H.; Dang, Y.; Zhou, Z.; Wu, C.; Zhao, F.; Sachs, M.S.; Liu, Y. Codon Usage Influences the Local Rate of Translation Elongation to Regulate Co-translational Protein Folding. Mol. Cell 2015, 59, 744–754. [Google Scholar] [CrossRef]
- Qian, W.; Yang, J.-R.; Pearson, N.M.; Maclean, C.; Zhang, J. Balanced Codon Usage Optimizes Eukaryotic Translational Efficiency. PLoS Genet. 2012, 8, e1002603. [Google Scholar] [CrossRef]
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2017, 19, 20–30. [Google Scholar] [CrossRef]
- Liu, Y. A code within the genetic code: Codon usage regulates co-translational protein folding. Cell Commun. Signal. 2020, 18, 145. [Google Scholar] [CrossRef]
- Arella, D.; Dilucca, M.; Giansanti, A. Codon usage bias and environmental adaptation in microbial organisms. Mol. Genet. Genom. 2021, 296, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Emery, L.R.; Zeng, K. Forces that influence the evolution of codon bias. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 1203–1212. [Google Scholar] [CrossRef]
- Bornelöv, S.; Selmi, T.; Flad, S.; Dietmann, S.; Frye, M. Codon usage optimization in pluripotent embryonic stem cells. Genome Biol. 2019, 20, 119. [Google Scholar] [CrossRef] [PubMed]
- The 1000 Genomes Project Consortium; Consortium, G.P.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Herrero, J.; Muffato, M.; Beal, K.; Fitzgerald, S.; Gordon, L.; Pignatelli, M.; Vilella, A.J.; Searle, S.M.J.; Amode, R.; Brent, S.; et al. Ensembl comparative genomics resources. Database 2016, 2016, bav096. [Google Scholar] [CrossRef]
- Delaneau, O.; Marchini, J.; Zagury, J.-F. A linear complexity phasing method for thousands of genomes. Nat. Methods 2011, 9, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Delaneau, O.; Zagury, J.F.; Marchini, J. Improved whole-chromosome phasing for disease and population genetic studies. Nat. Methods 2013, 10, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Zeberg, H.; Pääbo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 2020, 587, 610–612. [Google Scholar] [CrossRef]
- Zeberg, H.; Dannemann, M.; Sahlholm, K.; Tsuo, K.; Maricic, T.; Wiebe, V.; Hevers, W.; Robinson, H.P.; Kelso, J.; Pääbo, S. A Neanderthal Sodium Channel Increases Pain Sensitivity in Present-Day Humans. Curr. Biol. 2020, 30, 3465–3469.e4. [Google Scholar] [CrossRef]
- Zhang, X.; Witt, K.E.; Bañuelos, M.M.; Ko, A.; Yuan, K.; Xu, S.; Nielsen, R.; Huerta-Sanchez, E. The history and evolution of the Denisovan-EPAS1 haplotype in Tibetans. Proc. Natl. Acad. Sci. USA 2021, 118, e2020803118. [Google Scholar] [CrossRef]
- Sazzini, M.; Abondio, P.; Sarno, S.; Gnecchi-Ruscone, G.A.; Ragno, M.; Giuliani, C.; De Fanti, S.; Ojeda-Granados, C.; Boattini, A.; Marquis, J.; et al. Genomic history of the Italian population recapitulates key evolutionary dynamics of both Continental and Southern Europeans. BMC Biol. 2020, 18, 51. [Google Scholar] [CrossRef]
- Marnetto, D.; Pankratov, V.; Mondal, M.; Montinaro, F.; Pärna, K.; Vallini, L.; Molinaro, L.; Saag, L.; Loog, L.; Montagnese, S.; et al. Ancestral genomic contributions to complex traits in contemporary Europeans. Curr. Biol. 2022, 32, 1412–1419.e3. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Protein Mutation | Location | Recorded Codon Change | dbSNP_ID | Position (hg19) | Macroareas 2 |
|---|---|---|---|---|---|
| c.*372 A>G | 3′ UTR | Noncoding region | rs187940037 | 27253609 | AMR, EUR, SAS |
| I716I 1 | Exon 17 | ATC to ATA or ATT | rs145564988 | 27264097 | AFR |
| A713T | Exon 17 | GCG to ACG | rs63750066 | 27264108 | AMR |
| G708G | Exon 17 | GGC to GGT | rs148888161 | 27264121 | AFR, EAS, EUR, SAS |
| KM670/671NL | Exon 16 | AAG.ATG to AAT.CTG | rs281865161 | 27269938 | EUR |
| S614G | Exon 14 | AGC to GGC | rs112263157 | 27284122 | AFR, AMR |
| V604M | Exon 14 | GTG to ATG | rs199887707 | 27284152 | EAS |
| E599K | Exon 14 | GAA to AAA | rs140304729 | 27284167 | EUR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abondio, P.; Bruno, F.; Bruni, A.C.; Luiselli, D. Rare Amyloid Precursor Protein Point Mutations Recapitulate Worldwide Migration and Admixture in Healthy Individuals: Implications for the Study of Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 15871. https://doi.org/10.3390/ijms232415871
Abondio P, Bruno F, Bruni AC, Luiselli D. Rare Amyloid Precursor Protein Point Mutations Recapitulate Worldwide Migration and Admixture in Healthy Individuals: Implications for the Study of Neurodegeneration. International Journal of Molecular Sciences. 2022; 23(24):15871. https://doi.org/10.3390/ijms232415871
Chicago/Turabian StyleAbondio, Paolo, Francesco Bruno, Amalia Cecilia Bruni, and Donata Luiselli. 2022. "Rare Amyloid Precursor Protein Point Mutations Recapitulate Worldwide Migration and Admixture in Healthy Individuals: Implications for the Study of Neurodegeneration" International Journal of Molecular Sciences 23, no. 24: 15871. https://doi.org/10.3390/ijms232415871
APA StyleAbondio, P., Bruno, F., Bruni, A. C., & Luiselli, D. (2022). Rare Amyloid Precursor Protein Point Mutations Recapitulate Worldwide Migration and Admixture in Healthy Individuals: Implications for the Study of Neurodegeneration. International Journal of Molecular Sciences, 23(24), 15871. https://doi.org/10.3390/ijms232415871