
Spectrum of Genes for Non-GJB2-Related Non-Syndromic Hearing Loss in the Russian Population Revealed by a Targeted Deafness Gene Panel

 , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Results
2.1. Genetic Diagnoses
2.2. Analysis of Causative Variants
2.3. Genetic Heterogeneity of NSHL
3. Discussion
4. Materials and Methods
4.1. Clinical Data
4.2. Genetic Testing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smith, R.J.; Van Camp, G. Non-syndromic hearing impairment: Gene linkage and cloning. Int. J. Pediatr. Otorhinolaryngol. 1999, 49 (Suppl. 1), S159–S163. [Google Scholar] [CrossRef] [PubMed]
- Morton, C.C.; Nance, W.E. Newborn hearing screening--a silent revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Tavartkiladze, G.A.; Markova, T.G.; Chibisova, S.S.; Al-Sharjabi, E.; Tsygankova, E.R. The Russian and international experience with the implementation of the programs of universal audiological screening of the newborn infants. Vestn. Otorinolaringol. 2016, 81, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Abou Tayoun, A.N.; Al Turki, S.H.; Oza, A.M.; Bowser, M.J.; Hernandez, A.L.; Funke, B.H.; Rehm, H.L.; Amr, S.S. Improving hearing loss gene testing: A systematic review of gene evidence toward more efficient next-generation sequencing-based diagnostic testing and interpretation. Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 18, 545–553. [Google Scholar] [CrossRef]
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J.H. Hereditary Hearing Loss and Deafness Overview. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Petersen, M.B.; Willems, P.J. Non-syndromic, autosomal-recessive deafness. Clin. Genet. 2006, 69, 371–392. [Google Scholar] [CrossRef]
- Lingala, H.B.; Penagaluru, P.R. Role of connexin 26 (GJB2) & mitochondrial small ribosomal RNA (mt 12S rRNA) genes in sporadic & aminoglycoside-induced non syndromic hearing impairment. Indian J. Med. Res. 2009, 130, 369–378. [Google Scholar]
- Bliznets, E.A.; Galkina, V.A.; Matiushchenko, G.N.; Kisina, A.G.; Markova, T.G.; Poliakov, A.V. Changes in the connexin 26 (GJB2) gene in Russian patients with hearing disorders: Results of long-term molecular diagnostics of hereditary nonsyndromic deafness. Genetika 2012, 48, 112–124. [Google Scholar]
- Alford, R.L.; Arnos, K.S.; Fox, M.; Lin, J.W.; Palmer, C.G.; Pandya, A.; Rehm, H.L.; Robin, N.H.; Scott, D.A.; Yoshinaga-Itano, C. American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genet. Med. Off. J. Am. Coll. Med. Genet. 2014, 16, 347–355. [Google Scholar] [CrossRef]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef]
- Baux, D.; Vache, C.; Blanchet, C.; Willems, M.; Baudoin, C.; Moclyn, M.; Faugere, V.; Touraine, R.; Isidor, B.; Dupin-Deguine, D.; et al. Combined genetic approaches yield a 48% diagnostic rate in a large cohort of French hearing-impaired patients. Sci. Rep. 2017, 7, 16783. [Google Scholar] [CrossRef]
- Garcia-Garcia, G.; Berzal-Serrano, A.; Garcia-Diaz, P.; Villanova-Aparisi, R.; Juarez-Rodriguez, S.; de Paula-Vernetta, C.; Cavalle-Garrido, L.; Jaijo, T.; Armengot-Carceller, M.; Millan, J.M.; et al. Improving the Management of Patients with Hearing Loss by the Implementation of an NGS Panel in Clinical Practice. Genes 2020, 11, 1467. [Google Scholar] [CrossRef] [PubMed]
- Posukh, O.L. Genetic etiology of hearing loss in Russia. Hum. Genet. 2022, 141, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Markova, T.G.; Alekseeva, N.N.; Mironovich, O.L.; Galeeva, N.M.; Lalayants, M.R.; Bliznetz, E.A.; Chibisova, S.S.; Polyakov, A.V.; Tavartkiladze, G.A. Clinical features of hearing loss caused by STRC gene deletions/mutations in Russian population. Int. J. Pediatr. Otorhinolaryngol. 2020, 138, 110247. [Google Scholar] [CrossRef] [PubMed]
- Markova, T.G.; Lalayants, M.R.; Alekseeva, N.N.; Ryzhkova, O.P.; Shatokhina, O.L.; Galeeva, N.M.; Bliznetz, E.A.; Weener, M.E.; Belov, O.A.; Chibisova, S.S.; et al. Early audiological phenotype in patients with mutations in the USH2A gene. Int. J. Pediatr. Otorhinolaryngol. 2022, 157, 111140. [Google Scholar] [CrossRef] [PubMed]
- Lalayants, M.R.; Mironovich, O.L.; Bliznets, E.A.; Markova, T.G.; Polyakov, A.V.; Tavartkiladze, G.A. OTOF-related auditory neuropathy spectrum disorder. Vestn. Otorinolaringol. 2020, 85, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef]
- Markova, S.P.; Brozkova, D.S.; Lassuthova, P.; Meszarosova, A.; Krutova, M.; Neupauerova, J.; Raskova, D.; Trkova, M.; Stanek, D.; Seeman, P. STRC Gene Mutations, Mainly Large Deletions, are a Very Important Cause of Early-Onset Hereditary Hearing Loss in the Czech Population. Genet. Test. Mol. Biomark. 2018, 22, 127–134. [Google Scholar] [CrossRef]
- Nishio, S.Y.; Moteki, H.; Usami, S.I. Simple and efficient germline copy number variant visualization method for the Ion AmpliSeq custom panel. Mol. Genet. Genom. Med. 2018, 6, 678–686. [Google Scholar] [CrossRef]
- Smith, R.J.H. Pendred Syndrome/Nonsyndromic Enlarged Vestibular Aqueduct. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Lentz, J.; Keats, B. Usher Syndrome Type II. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Verpy, E.; Masmoudi, S.; Zwaenepoel, I.; Leibovici, M.; Hutchin, T.P.; Del Castillo, I.; Nouaille, S.; Blanchard, S.; Laine, S.; Popot, J.L.; et al. Mutations in a new gene encoding a protein of the hair bundle cause non-syndromic deafness at the DFNB16 locus. Nat. Genet. 2001, 29, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Van Heurck, R.; Carminho-Rodrigues, M.T.; Ranza, E.; Stafuzza, C.; Quteineh, L.; Gehrig, C.; Hammar, E.; Guipponi, M.; Abramowicz, M.; Senn, P.; et al. Benefits of Exome Sequencing in Children with Suspected Isolated Hearing Loss. Genes 2021, 12, 1277. [Google Scholar] [CrossRef]
- van Wijk, E.; Pennings, R.J.; te Brinke, H.; Claassen, A.; Yntema, H.G.; Hoefsloot, L.H.; Cremers, F.P.; Cremers, C.W.; Kremer, H. Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am. J. Hum. Genet. 2004, 74, 738–744. [Google Scholar] [CrossRef]
- Baux, D.; Larrieu, L.; Blanchet, C.; Hamel, C.; Ben Salah, S.; Vielle, A.; Gilbert-Dussardier, B.; Holder, M.; Calvas, P.; Philip, N.; et al. Molecular and in silico analyses of the full-length isoform of usherin identify new pathogenic alleles in Usher type II patients. Hum. Mutat. 2007, 28, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.; Abu Rayan, A.; Abu Sa’ed, J.; Shahin, H.; Shepshelovich, J.; Lee, M.K.; Hirschberg, K.; Tekin, M.; Salhab, W.; Avraham, K.B.; et al. Genomic analysis of a heterogeneous Mendelian phenotype: Multiple novel alleles for inherited hearing loss in the Palestinian population. Hum. Genom. 2006, 2, 203–211. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Park, H.J.; Shaukat, S.; Liu, X.Z.; Hahn, S.H.; Naz, S.; Ghosh, M.; Kim, H.N.; Moon, S.K.; Abe, S.; Tukamoto, K.; et al. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: Global implications for the epidemiology of deafness. J. Med. Genet. 2003, 40, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.E.; Trubilin, V.N.; Atarshchikov, D.S.; Demchinsky, A.M.; Strelnikov, V.V.; Tanas, A.S.; Orlova, O.M.; Machalov, A.S.; Overchenko, K.V.; Markova, T.V.; et al. Genetic screening of Russian Usher syndrome patients toward selection for gene therapy. Ophthalmic Genet. 2018, 39, 706–713. [Google Scholar] [CrossRef]
- Bliznetz, E.A.; Lalayants, M.R.; Markova, T.G.; Balanovsky, O.P.; Balanovska, E.V.; Skhalyakho, R.A.; Pocheshkhova, E.A.; Nikitina, N.V.; Voronin, S.V.; Kudryashova, E.K.; et al. Update of the GJB2/DFNB1 mutation spectrum in Russia: A founder Ingush mutation del(GJB2-D13S175) is the most frequent among other large deletions. J. Hum. Genet. 2017, 62, 789–795. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kimberling, W.J.; Hildebrand, M.S.; Shearer, A.E.; Jensen, M.L.; Halder, J.A.; Trzupek, K.; Cohn, E.S.; Weleber, R.G.; Stone, E.M.; Smith, R.J. Frequency of Usher syndrome in two pediatric populations: Implications for genetic screening of deaf and hard of hearing children. Genet. Med. Off. J. Am. Coll. Med. Genet. 2010, 12, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Barashkov, N.A.; Klarov, L.A.; Teryutin, F.M.; Solovyev, A.V.; Pshennikova, V.G.; Konnikova, E.E.; Romanov, G.P.; Tobokhov, A.V.; Morozov, I.V.; Bondar, A.A.; et al. A novel pathogenic variant c.975G>A (p.Trp325*) in the POU3F4 gene in Yakut family (Eastern Siberia, Russia) with the X-linked deafness-2 (DFNX2). Int. J. Pediatr. Otorhinolaryngol. 2018, 104, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Bademci, G.; Foster, J., 2nd; Mahdieh, N.; Bonyadi, M.; Duman, D.; Cengiz, F.B.; Menendez, I.; Diaz-Horta, O.; Shirkavand, A.; Zeinali, S.; et al. Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort. Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 18, 364–371. [Google Scholar] [CrossRef]
- Yan, D.; Tekin, D.; Bademci, G.; Foster, J., 2nd; Cengiz, F.B.; Kannan-Sundhari, A.; Guo, S.; Mittal, R.; Zou, B.; Grati, M.; et al. Spectrum of DNA variants for non-syndromic deafness in a large cohort from multiple continents. Hum. Genet. 2016, 135, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Churbanov, A.Y.; Karafet, T.M.; Morozov, I.V.; Mikhalskaia, V.Y.; Zytsar, M.V.; Bondar, A.A.; Posukh, O.L. Whole Exome Sequencing Reveals Homozygous Mutations in RAI1, OTOF, and SLC26A4 Genes Associated with Nonsyndromic Hearing Loss in Altaian Families (South Siberia). PLoS ONE 2016, 11, e0153841. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
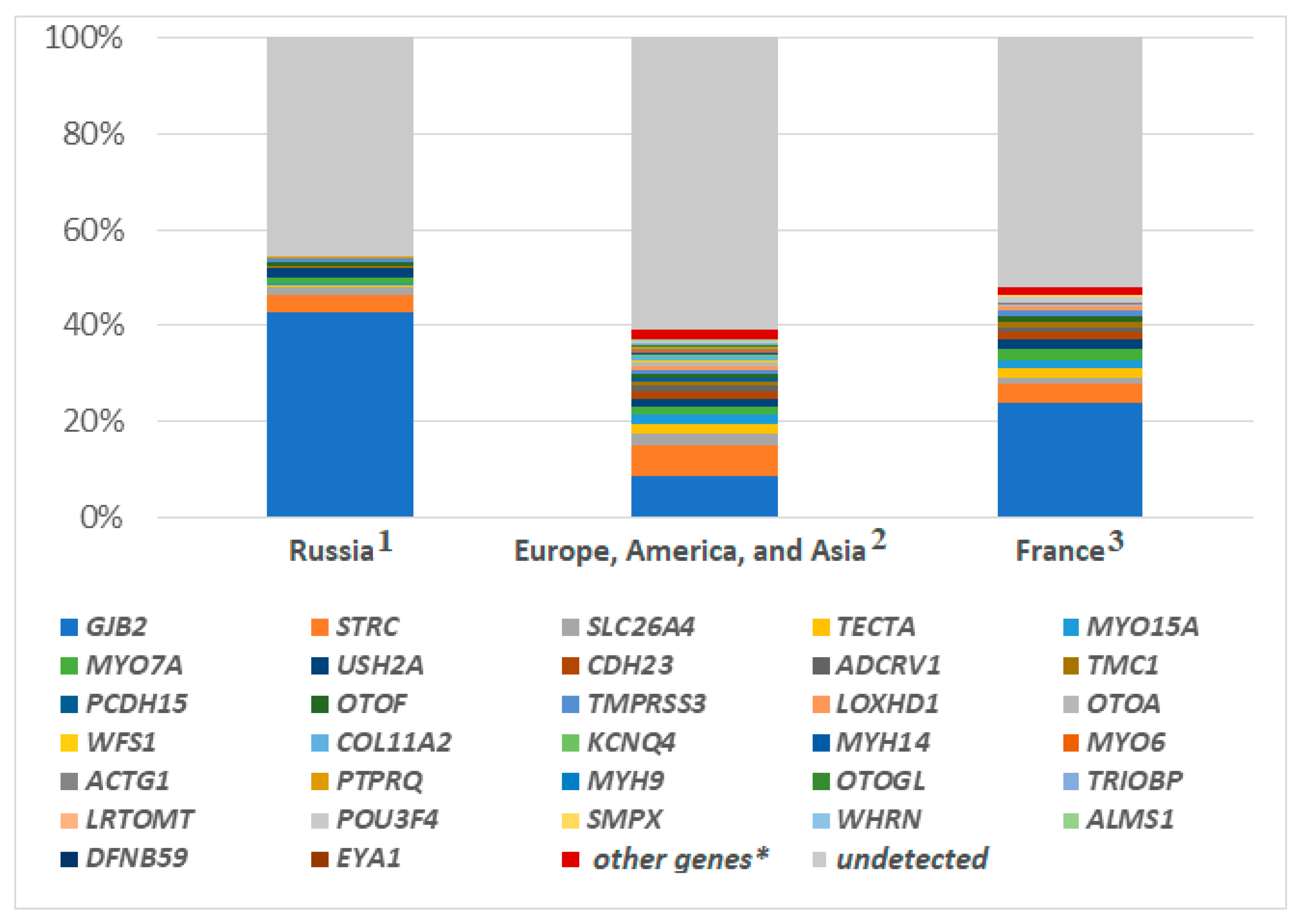{kind=link}
| Gene | Variant | Pathogenicity | Criteria a | The ClinVar Accession Numbers |
|---|---|---|---|---|
| ADGRV1 | c.6610C>T (p.Gln2204*) | Likely pathogenic | PVS1, PM2 | SCV002754430 |
| ADGRV1 | c.10198C>T (p.Gln3400*) | Likely pathogenic | PVS1, PM2 | SCV002754431 |
| MYO15A | c.8910del (p.Val2971fs*63) | Likely pathogenic | PVS1, PM2 | SCV002754432 |
| MYO7A | c.1738_1745del (p.Val581Leufs*28) | Likely pathogenic | PVS1, PM2 | SCV002754433 |
| MYO7A | c.3893G>A (p.Gly1298Glu) | Likely pathogenic | PM2, PM3, PM5, PP3 | SCV002754434 |
| MYO7A | c.3612_3615del (p.Ser1205Argfs*26) | Likely pathogenic | PVS1, PM2 | SCV002754435 |
| MYO7A | c.4528G>A (p.Glu1510Lys) | Likely pathogenic | PM2, PM3, PP2, PP3 | SCV002754436 |
| OTOA | c.562_569dup (p.Phe191fs*48) | Likely pathogenic | PVS1, PM2 | SCV002754437 |
| OTOF | c.2656del (p.Val886Serfs*114) b | Likely pathogenic | PVS1, PM2 | SCV002754438 |
| OTOF | c.5169_5170del (p.Ile1724Leufs*19) b | Likely pathogenic | PVS1, PM2 | SCV002754439 |
| OTOF | c.4903A>T (p.Arg1635*) b | Likely pathogenic | PVS1, PM2 | SCV002754440 |
| OTOF | c.2214+5G>C b | Likely pathogenic | PM2, PM3, PP3, PP4 | SCV002754441 |
| POU3F4 | c.983A>G (p.Asn328Ser) | Likely pathogenic | PS1, PM2, PM5, PP3 | SCV002754443 |
| PTPRQ | c.1291C>T (p.Arg431*) | Likely pathogenic | PVS1, PM2 | SCV002754445 |
| SLC26A4 | c.107A>C (p.His36Pro) | Likely pathogenic | PM2, PM3, PP3, PP4 | SCV002754447 |
| SLC26A4 | c.208C>T (p.Pro70Ser) | Likely pathogenic | PM2, PM3, PP4, PP5 | SCV002754448 |
| STRC | g.(?_43906612)_(43906674_)?del (delSTRC, ex5) b | Pathogenic | PVS1, PM2, PM3 | SCV002754455 |
| TECTA | c.2458A>T (p.Lys820*) | Likely pathogenic | PVS1, PM2 | SCV002754449 |
| TMC1 | c.1750C>T *p.Gln584*) | Likely pathogenic | PVS1, PM2 | SCV002754450 |
| USH2A | g.(?_216108034)_(216108100_?)del (delUSH2A, ex38) b | Likely pathogenic | PVS1, PM2 | SCV002754452 |
| USH2A | g. (?_216462679)_(216462739_?)del (delUSH2A, ex11) | Likely pathogenic | PVS1, PM2 | SCV002754453 |
| Gene | Phenotype | Number of Probands | The Proportion of Probands with Established Genes of Hearing Loss | p Value * | |
|---|---|---|---|---|---|
| Among the 48 Cases of This Study (95%CI) | In Europe, America and Asia * | ||||
| STRC | DFNB16 | 14 | 29% (17–43%) | 30% | 0.963 |
| USH2A | Usher syndrome | 8 | 17% (7–30%) | 8% | 0.114 |
| SLC26A4 | DFNB4, Pendred syndrome | 6 | 12% (3–22%) | 12% | 0.906 |
| MYO7A | DFNB2, Usher syndrome/DFNA11 | 5 (3/2) | 10% (3–22%) | 8% | 0.878 |
| OTOF | DFNB9 | 4 | 8% (2–20%) | 4% | 0.324 |
| TECTA | DFNB21/DFNA8/12 | 2 (1/1) | 4% (1–17%) | 10% | 0.625 |
| POU3F4 | DFNX2 | 2 | 4% (0.5–14%) | 1% | 0.269 |
| TMPRSS3 | DFNB8/10 | 2 | 4% (0.5–14%) | 4% | 0.769 |
| ADGRV1 | Usher syndrome | 1 | 2% (0.05–11%) | 5% | 0.598 |
| MYO15A | DFNB3 | 1 | 2% (0.05–11%) | 9% | 0.190 |
| OTOA | DFNB22 | 1 | 2% (0.05–11%) | 3% | 0.986 |
| PTPRQ | DFNB84 | 1 | 2% (0.05–11%) | 2% | 0.679 |
| TMC1 | DFNB7/11 | 1 | 2% (0.05–11%) | 4% | 0.769 |
| Gene | RefSeq | Gene | RefSeq |
|---|---|---|---|
| ACTG1 | NM_001614 | OTOF | NM_194248 |
| ADGRV1 | NM_032119 | OTOGL | NM_173591 |
| ALMS1 | NM_015120 | PCDH15 | NM_033056 |
| CDH23 | NM_022124 | POU3F4 | NM_000307 |
| CLDN14 | NM_144492 | PTPRQ | NM_001145026 |
| COL11A2 | NM_080680 | SLC26A4 | NM_000441 |
| PJVK | NM_001042702 | SMPX | NM_014332 |
| EYA1 | NM_000503 | STRC | NM_153700 |
| KCNQ4 | NM_004700 | TECTA | NM_005422 |
| LOXHD1 | NM_144612 | TMC1 | NM_138691 |
| LRTOMT | NM_001145308 | TMPRSS3 | NM_024022 |
| MYH14 | NM_024729 | TPRN | NM_001128228 |
| MYH9 | NM_002473 | TRIOBP | NM_001039141 |
| MYO15A | NM_016239 | USH2A | NM_206933 |
| MYO6 | NM_004999 | WFS1 | NM_006005 |
| MYO7A | NM_000260 | WHRN | NM_015404 |
| OTOA | NM_144672 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shatokhina, O.; Galeeva, N.; Stepanova, A.; Markova, T.; Lalayants, M.; Alekseeva, N.; Tavarkiladze, G.; Markova, T.; Bessonova, L.; Petukhova, M.; et al. Spectrum of Genes for Non-GJB2-Related Non-Syndromic Hearing Loss in the Russian Population Revealed by a Targeted Deafness Gene Panel. Int. J. Mol. Sci. 2022, 23, 15748. https://doi.org/10.3390/ijms232415748
Shatokhina O, Galeeva N, Stepanova A, Markova T, Lalayants M, Alekseeva N, Tavarkiladze G, Markova T, Bessonova L, Petukhova M, et al. Spectrum of Genes for Non-GJB2-Related Non-Syndromic Hearing Loss in the Russian Population Revealed by a Targeted Deafness Gene Panel. International Journal of Molecular Sciences. 2022; 23(24):15748. https://doi.org/10.3390/ijms232415748
Chicago/Turabian StyleShatokhina, Olga, Nailya Galeeva, Anna Stepanova, Tatiana Markova, Maria Lalayants, Natalia Alekseeva, George Tavarkiladze, Tatiana Markova, Liudmila Bessonova, Marina Petukhova, and et al. 2022. "Spectrum of Genes for Non-GJB2-Related Non-Syndromic Hearing Loss in the Russian Population Revealed by a Targeted Deafness Gene Panel" International Journal of Molecular Sciences 23, no. 24: 15748. https://doi.org/10.3390/ijms232415748
APA StyleShatokhina, O., Galeeva, N., Stepanova, A., Markova, T., Lalayants, M., Alekseeva, N., Tavarkiladze, G., Markova, T., Bessonova, L., Petukhova, M., Guseva, D., Anisimova, I., Polyakov, A., Ryzhkova, O., & Bliznetz, E. (2022). Spectrum of Genes for Non-GJB2-Related Non-Syndromic Hearing Loss in the Russian Population Revealed by a Targeted Deafness Gene Panel. International Journal of Molecular Sciences, 23(24), 15748. https://doi.org/10.3390/ijms232415748